

Introduction
The prenylation of proteins, including several in the visual transduction pathway, is one of the most recently discovered modifications of eukaryotic cell proteins.1–4 Some prenylated proteins, including rhodopsin kinase, the γ subunit of transducin, the α subunit of retinal cGMP phosphodiesterase, Ras, and lamin B, are initially produced with a C-terminal sequence motif CaaX (C is cysteine, a is usually but not necessarily an aliphatic residue, and X is typically M, S, or Q). Prenylation occurs by enzyme-catalyzed attachment of a 15-carbon farnesyl group to cysteine via a thioether linkage. After prenylation, aaX is released by a membrane-bound endoprotease,5–8 and the newly exposed S-farnesylcysteine is methylated on its α-carboxyl group by a membrane-bound methyltransferase.9,10 Other prenylated proteins, including the γ subunits of many heterotrimeric G proteins, the β subunit of retinal cGMP phosphodiesterase, and a subset of small GTP-binding proteins, contain a CaaX motif (X = L, F) and are modified by the attachment of the 20-carbon geranylgeranyl group. Removal of aaX and C-terminal methylation occurs as for farnesylated proteins. Finally, the subset of GTP-binding proteins termed Rab contains two cysteines near or at the C terminus (CCXX, XXCC, CXC), and both cysteine sulfhydryls contain a thioether-linked geranylgeranyl group.11 The attachment of prenyl groups to these three classes of proteins is catalyzed by three distinct protein prenyltranferases.12,13
Structures of protein prenyl groups have been rigorously established by releasing the protein-bound lipids with Raney nickel (to cleave reductively the bond between cysteine S and C-1 of the prenyl group) and analyzing the released hydrocarbons by combined gas chromatography–mass spectrometry versus authentic standards.14–17 Protein prenyl groups can also be released by treatment with methyl iodide to produce farnesol and geranylgeraniol along with isomerized prenols.16,18 This method has been used when relatively small amounts of prenylated protein are available, typically from tissue culture cells that have been grown in the presence of radiolabeled mevalonic acid (the precursor of prenyl groups) or its lactone in the presence of statins, which block the production of endogenous mevalonic acid. The radiolabeled prenol is analyzed versus standards by reversed phase high-performance liquid chromatography (HPLC).16 While this approach does not establish the chemical structure of the prenyl group, it provides strong circumstantial evidence for the presence of protein-bound farnesyl and geranylgeranyl groups. However, in our hands, we have experienced difficulties with methyl iodide cleavage, in that yields are highly variable and can be quite low in some cases (<10% cleavage). Work in our laboratory has shown that the Raney nickel cleavage method is superior to methyl iodide treatment, and the details of this procedure, combined with HPLC analysis of the released radiolabeled hydrocarbons, are described in this chapter. This method has been used to analyze protein prenyl groups from protein extracted from sodium dodecyl sulfate (SDS)–polyacrylamide gel slices. Also described here is the use of Raney nickel cleavage followed by combined gas chromatography–mass spectrometry to determine the structure of prenyl groups released from delipidated total cell protein.
The rigorous structural analysis of aaX removal and C-terminal methylation has been carried out by fast atom bombardment mass spectrometry of C-terminal peptides from prenylated proteins.11,19 A less rigorous approach has been used to explore C-terminal methylation: HPLC analysis of the product of oxidative scission of the prenyl–cysteine linkage combined with amide bond hydrolysis to yield cysteic acid methyl ester.20 In the present chapter, a method for electrospray mass spectrometric analysis of protein C-terminal peptides is presented, which has been applied to the analysis of prenylated peptides in a complex mixture (a tryptic digest of partially purified bovine rhodopsin kinase). This method is facilitated by the availability of prenylated peptide standards, and a convenient synthesis of peptides and peptide methyl esters radiolabeled with high specific activity farnesyl and geranylgeranyl groups is described.
Analysis of Protein Prenyl Groups with Raney Nickel Cleavage
Preparation of Radioprenylated Proteins, Protein Delipidation, and Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis
To radiolabel protein prenyl groups, cells are cultured with 3H- or 14C-labeled mevalonolactone or mevalonic acid [New England Nuclear (Boston, MA) or American Radiolabeled Chemicals (St. Louis, MO)] in the presence of an inhibitor of hydroxymethylglutaryl-CoA reductase (such as simvastatin or lovastatin) to suppress production of endogenous mevalonic acid.16,17 Statins such as lovastatin and simvastatin require conversion of the lactone form to the carboxylate salt as described.17 Mevalonolactone tends to be more cell permeable than mevalonic acid but must undergo intracellular hydrolytic ring opening prior to incorporation into the isoprenoid pathway. Thus, it is best to carry out small-scale radiolabeling experiments with both mevalonic acid and its lactone to gauge the level of incorporation into proteins (determined by fluorography of SDS–polyacrylamide gels containing delipidated cell protein; see below). A disadvantage of this method for the analysis of total cell protein prenyl groups is that only proteins that become prenylated during the radiolabeling period are detected, and thus the ratio of radiofarnesylated to radio-geranylgeranylated proteins will probably not be the same as the ratio of prenyl groups present in the total pool of cellular prenylated proteins. Also, evidence from our laboratory (see below) has shown that the use of statins together with exogenous radiolabeled mevalonic acid or its lactone leads to a distortion of the ratio of radiolabeled farnesyl versus geranylgeranyl groups attached to total cell protein, presumably owing to an effect of statins on the farnesyl pyrophosphate-to-geranylgeranyl pyrophosphate ratio. For these reasons, prenyl groups present in total cell protein are best analyzed by combined gas chromatography–mass spectrometry of Raney nickel-treated protein from cells grown in the absence of statins.
Only a small fraction of the intracellular radiolabeled mevalonic acid may be incorporated into proteins. Thus it is important to remove radioactive isoprenoids from the protein sample that are not covalently attached to protein. This can be partially accomplished by precipitating cell protein from a cell lysate with trichloroacetic acid and washing the pellet with organic solvent. However, in our hands a significant amount of non-protein-bound radiolabeled material remains, which can interfere with subsequent prenyl group analysis. Complete delipidation of protein is achieved by SDS–polyacrylamide gel electrophoresis (PAGE) followed by extraction of proteins from the gel. As described later, delipidation with organic solvent is adequate for the analysis of prenyl groups by gas chromatography–mass spectrometry.
We describe here a typical procedure that we have used to analyze prenylated proteins in mammalian cells and trypanosomatids. Bloodstream form Trypanosoma brucei (T. brucei) strain 427 (107 mid-log-phase cells, corresponding in protein content to ~105 mammalian cells) is labeled for 24 hr in a humidified atmosphere of 5% CO2 at 37° in 1 ml of culture medium [HMI-9, 10% (v/v) fetal calf serum] containing 1 mCi of RS-[5-3H]mevalonolactone (50 Ci/mmol) and 2 μM saponified simvastatin (obtained as a gift from Merck Research Laboratories, Whitehouse Station, NJ). Cells are harvested in a 1.5-ml Eppendorf tube by centrifugation and washed once with phosphate-buffered saline (PBS) in the usual way. The cell pellet is resuspended in 1 ml of ice-cold lysis buffer [20 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 30 μM Nα-tosyl-l-lysine chloromethyl ketone, 30 μM Nα-tosyl-l-phenylalanine chloromethyl ketone, pepstatin A (10 μg/ml), leupeptin (10 μg/ml), and aprotinin (10 μg/ml)]. Protease inhibitors are added fresh from properly stored stock solutions. The suspension is frozen in a −80° freezer. The frozen sample can be stored at −80° or processed further. The frozen sample is allowed to thaw on ice to lyse the cells (note, freeze–thawing may not be sufficient to lyse other types of cells, and the appropriate cell disruption technique should be used).
To the suspension of disrupted cells (cell debris including membranes should not be removed as the latter contain prenylated proteins) is added 150 μl of 100% (w/v) ice-cold trichloroacetic acid. The sample is briefly vortexed, incubated on ice for 30 min, and microcentrifuged at ~10,000g for 10 min at 4°. The supernatant is decanted, and tissue paper is used to soak up the last bit of liquid from the lip of the inverted tube. To the pellet is added 1 ml of room temperature CHCl3–methanol (2:1, v/v). The sample is vortexed at room temperature and microcentrifuged for 10 min at 4°. After decantation, the pellet is washed once more with organic solvent as above. To the pellet is added 1 ml of ice-cold reagent-grade acetone, and the sample is vortexed and microcentrifuged as described above. The acetone is decanted, and the pellet is allowed to air dry at room temperature. The protein pellet can be stored at −80° or processed further for SDS–PAGE. To the pellet is added 20 μl of water, and the Eppendorf tube is placed in a bath sonicator until most of the pellet is dispersed (several minutes). Laemmli sample loading buffer (20 μl of 2× buffer) supplemented with freshly added 4% (v/v) β-mercaptoethanol is added, and the sample is boiled for 3 min.
After brief centrifugation, a small portion of the supernatant (1 μl) is subjected to scintillation counting to estimate the total amount of radiolabeled protein. Most of the sample (38 μl) is applied to two lanes of a standard Laemmli minigel [12.5% (w/v) running gel, 0.75 mm thick] for SDS–PAGE. Prestained molecular weight markers are also applied to lanes that bracket the lanes with radiolabeled protein. A second gel containing molecular weight markers and 1 μl of radiolabeled protein is run for subsequent fluorography. This gel is prepared for fluorography as described.21 Using the fluorograph and the molecular weight markers as guides, the desired region(s) of the nonfluorographed gel containing the desired radiolabeled proteins is sliced out of the lane with a razor blade (the gel should not be fixed or dried). Slices are placed in Eppendorf tubes. The amount of gel per tube should be no more than a piece that has the width of a gel lane and a length of ~2 cm. To each Eppendorf tube is added 0.6 ml of elution buffer [50 mM ammonium bicarbonate, 0.08% (w/v) SDS, 1% (v/v) β-mercaptoethanol, made fresh] and 20 μg of bovine serum albumin (BSA) as carrier protein.
The gel slice(s) in each tube is crushed into small pieces with a small plastic pestle designed to fit conical Eppendorf tubes (typically several minutes of crushing). Another 0.6 ml of elution buffer is used to rinse the pestle, collecting the liquid into the tube with crushed gel. The sample is boiled for 5 min and rotated overnight at room temperature. The sample is microcentrifuged for a few minutes at room temperature, and most of the supernatant is transferred to a new Eppendorf tube with a Pasteur pipette. To the gel pellet is added 0.3 ml of elution buffer, and after briefly vortexing, the sample is rotated for 2 hr. After centrifugation, the supernatant is combined with the first supernatant. To ensure complete removal of gel pieces, the tube of combined supernatant is microcentrifuged for a few minutes and the supernatant is carefully transferred to a new Eppendorf tube (avoid taking gel slices). The sample can be stored at −80° or further processed. The remaining gel pellet is dissolved in 0.5 ml of 30% (v/v) H2O2 (overnight at 90°), and the solution is subjected to scintillation counting to determine the amount of labeled protein remaining in the gel.
The sample of extracted protein is concentrated to 200 μl in a SpeedVac (Savant Instruments, Holbrook, NY), and a small aliquot is subjected to scintillation counting to determine the protein extraction yield. The final sample volume is important and is estimated by comparing the meniscus height with that of a second Eppendorf tube containing 200 μl of water. We concentrate the volume to <200 μl and then add back the appropriate amount of water. In our experience we are able to extract >90% of the radiolabeled proteins from gel slices. A high yield of gel-extracted radioprenylated proteins is required if accurate quantification of the amount of protein prenyl groups is desired.
To the concentrate is added 50 μl of ice-cold 100% (w/v) trichloroacetic acid. After briefly vortexing, the tube is incubated on ice for 2 hr and microcentrifuged at ~10,000g for 10 min at 4°. The supernatant is decanted, and the last bit of liquid is removed from the lip of the tube with a tissue. Ice-cold reagent-grade acetone (0.5 ml) is added. The tube is inverted twice and microcentrifuged as described above. The supernatant is removed as described above, and the last trace of acetone is removed by placing the open tube in a 37° water bath for several minutes. The sample is now ready for Raney nickel treatment or it can be stored at −80°.
Release of Radiolabeled Prenyl Groups from Proteins with Raney Nickel
To the pellet of delipidated protein (total cell protein or protein eluted from the SDS–polyacrylamide gel; see previous section) is added 0.4 ml of 8 M guanidine hydrochloride, 0.2 M sodium phosphate (pH 7.0). To dissolve the protein pellet, the sample is vortexed and sonicated briefly in a bath-type device. The solution is transferred to a 16 × 100 mm glass tube fitted with a Teflon-lined screw cap. To prevent loss of pentane (see below) due to deformation of the Teflon liner, it is important to use a screw cap that does not have a hole in the center of the cap. To the solution of dissolved protein is added 1 ml of pentane. The tube is briefly vortexed, and most of the pentane is removed and discarded with a pipette. For samples containing large amounts of protein, centrifugation is required after vortexing to separate the solvent layers. Pentane extraction is repeated once more. This pentane extraction is important for removing hydrophobic material that may be present in the protein sample and that is not thioether linked to proteins. Pentane (1 ml) and internal standards [20 μg each of N-acetyl-S-farnesyl-l-cysteine and N-acetyl-S-geranylgeranyl-l-cysteine, both from CalBiochem (La Jolla, CA), added from 1-μg/μl stock solutions in ethanol] are then added to the tube followed by ~50 mg of Raney nickel. Raney nickel is prepared as described.17 Caution is advised because Raney nickel can ignite if allowed to dry in air. A slurry of Raney nickel from an ethanol suspension is transferred with a Pasteur pipette to a piece of weigh paper on a balance. After most of the excess ethanol has evaporated, the moist solid is weighed, and additional Raney nickel is added as needed to give ~50 mg. Most of the Raney nickel is scraped off the paper with a spatula and transferred to the reaction tube. The tube is vortexed briefly to suspend any Raney nickel from the walls of the tube above the liquid phase. A small magnetic stir bar is added (use the proper size stir bar so that rapid mixing can occur), and the tube is tightly capped and held with a clamp and ring stand in an oil bath at 100 ± 5° for 15 hr with sufficient stirring to break the pentane phase into small droplets. The tube is positioned so that the pentane–water interface is at the oil level.
The tube is allowed to cool to room temperature before removing the cap. Most of the pentane is transferred to a new glass vial with a Pasteur pipette. To the aqueous phase is added 0.5 ml of pentane, the tube is vortexed briefly, and the pentane layer is combined with the first pentane extract. The extract can be stored at −20° in a tightly capped vial. To concentrate the pentane solution of released hydrocarbons, a portion is transferred to a small glass tube with a conical bottom (a Pasteur pipette flame sealed at its narrow end with a Bunsen burner works well). The tube is placed in an ice–NaCl bath, and a gentle stream of N2 is focused on the solution to remove much of the pentane, leaving about 100 μl. More pentane solution is transferred to the tube, and the evaporation process is repeated until the entire pentane extract has been concentrated. Finally, the sample is concentrated to a final volume of ~20 μl of pentane, and 100 μl of methanol is immediately added. This concentration procedure ensures that minimal amounts of radiolabeled hydrocarbons are lost (routinely <10%). Routinely, 60–80% of the radioactivity is released into pentane.
Raney nickel-released prenyl groups are analyzed by HPLC on a C18 reversed-phase, analytical column [Vydac 218TP52 (The Separations Group, Hesperia, CA) or similar column]. Prior to HPLC, a small aliquot of sample is subjected to scintillation counting so that yields of HPLC-eluted hydrocarbons can be obtained. The column is developed at a flow rate of 0.5 ml/min with the following solvent program: 0–5 min, 100% water; 5–10 min, 0–60% acetonitrile linear gradient; 10–70 min, 60–100% acetonitrile linear gradient; 70–100 min, 100% acetonitrile. A UV detector set at 210 nm is used to detect the hydrocarbon products, all-trans-2,6,10-Trimethyl-2,6,10-dodecatriene from farnesyl groups elutes with a retention time of 58.5 min, and all-trans-2,6,10,14-tetramethyl-2,6,10,14-hexa-decatetraene from geranylgeranyl groups elutes with a retention time of 73.7 min. Eluent fractions (0.5 ml) are collected with a fraction collector and mixed with 5 ml of scintillation fluid for counting.
Figure 1 shows a typical chromatogram of radiolabeled prenyl groups released from T. brucei protein by Raney nickel treatment. A major peak of radioactivity for the 15-carbon hydrocarbon is seen precisely at the elution position for hydrocarbon standard (UV detection, not shown). Although the unlabeled 20-carbon hydrocarbon elutes as a sharp peak at 73.7 min (UV detection), little if any radiolabeled material is seen in this sample, indicating that little, if any, geranylgeranylated proteins became radiolabeled. Routinely, 75% of the counts per minute applied to the HPLC column are eluted. In contrast to these results, methyl iodide cleavage of prenyl groups from proteins gives rise to multiple prenols derived from each type of protein prenyl group owing to multiple rearrangement pathways for the farnesyl carbocation.22,23 A further disadvantage of the methyl iodide method is that radiolabeled prenols can also come from prenyl pyrophosphates that may be present in large amounts in protein samples. Raney nickel cleavage uniquely generates hydrocarbons rather than alcohols and is selective for carbon–sulfur bond cleavage.
Fig. 1.
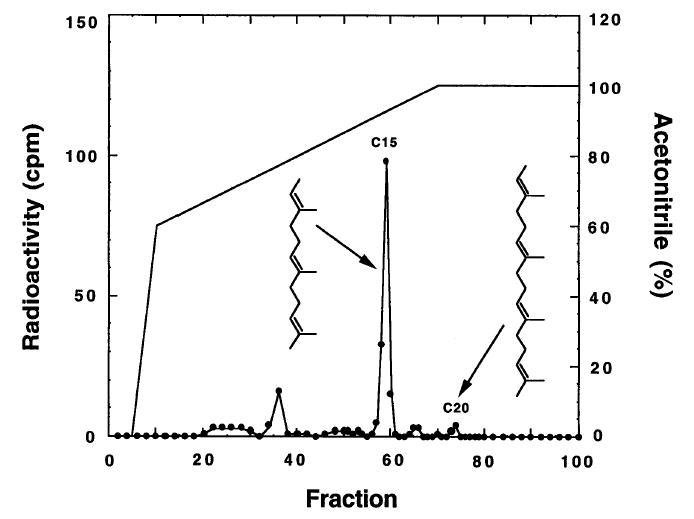
HPLC analysis of prenyl group-derived hydrocarbons released from proteins by Raney nickel cleavage. HPLC conditions are given in text. Arrows show the elution position of authentic hydrocarbon standards (obtained by Raney nickel cleavage of N-acetyl-S-farnesyl-l-cysteine and N-acetyl-S-geranylgeranyl-l-cysteine).
Analysis of Nonradiolabeled Protein Prenyl Groups by Combined Gas Chromatography–Mass Spectrometry
The Raney nickel cleavage procedure when combined with gas chromatography–mass Spectrometry can also be used to analyze prenyl groups without radiolabeling. We have been able to use this method to analyze total cell protein that has been delipidated with organic solvent only (i.e., no need for SDS–PAGE). Delipidated total cell protein (see previous section) is treated with Raney nickel as described in the previous section. The concentrated pentane extract is analyzed by combined gas chromatography–electron impact mass Spectrometry exactly as described in detail.17 In this case, internal standards other than N-acetyl-S-prenyl-l-cysteines must obviously be used. Suitable internal standards are eicosane and phytane.17 Gas chromatogram retention times and mass Spectrometry fragmentation patterns for the prenyl group-derived hydrocarbons have been published.17
Raney nickel-released prenyl groups from ~40 μg of total delipidated cell protein (from 107 T. brucei cells) is more than sufficient for combined gas chromatography–mass Spectrometry including fragmentation pattern analysis of the prenyl group-derived hydrocarbons.17 For example, when 39 μg of total delipidated cell protein is subjected to Raney nickel treatment to obtain the released hydrocarbons in 100 μl of methanol (see previous section), injection of 2 μl of this solution is sufficient for gas chromatography–mass Spectrometry. On the basis of these numbers, it seems reasonable that gas chromatography–mass Spectrometry can be carried out on <1 μg of a single prenylated protein extracted from an SDS–polyacrylamide gel slice. A more precise calculation based on the detection limits of hydrocarbons released from N-acetyl-S-prenyl-l-cysteine internal standards is that 30–1000 ng of prenylated protein is needed depending on the molecular weight of the protein (6000–200,000).
It is important to note that the ratio of C20 to C15 hydrocarbons in the gas chromatography analysis is much higher than the ratio in the radiometric analysis (Fig. 1). These results strongly argue that treatment of cells with statins in the presence of radiolabeled mevalonolactone leads to a distortion of the relative amounts of C15,20-prenyl pyrophosphate in cells, which in turn distorts the relative amounts of the various prenyl groups added to newly synthesized proteins during radiolabeling. Thus, caution is advised, and gas chromatography–mass spectrometry is the method of choice for analyzing the mixture of prenyl groups attached to a collection of proteins.
Analysis of Prenyl Groups and C-Terminal Proteolysis and Methylation in Complex Peptide Mixtures by Electrospray Mass Spectrometry
Preparation of Prenylated Peptides and Peptide Methyl Esters
As is described below, electrospray mass spectrometry is a powerful method for detection of prenylated peptides and peptide methyl esters present in complex peptide mixtures such as tryptic digests of proteins. These studies are facilitated by the availability of appropriate synthetic radiolabeled and nonradiolabeled prenylated peptides and peptide methyl esters. As described in the next section, such peptides are useful for determining mass spectrometry detection limits, for providing HPLC retention times of prenylated tryptic peptides, for ensuring that prenylated peptides remain soluble, and for ensuring that prenylated peptide methyl esters have not undergone hydrolytic demethylation during sample handling.
The synthesis of nonradiolabeled prenylated peptides and prenylated peptide methyl esters by solution-phase prenylation of peptides made by solid-phase synthesis and the purification of these materials have been described in detail.24 The same methods are used to prepare and purify radioprenylated peptides using tritiated farnesyl bromide and geranylgeranyl bromide, which are prepared as described here. There are several methods for converting prenols into their respective prenyl halides, but most are difficult to execute for the preparation of high specific activity tritiated material on the submilligram scale. We have found that bromination with CBr4/triphenyl phosphine (PPh3) works well for this purpose, and is simple to execute.
A solution of [1-3H]farnesol [1 mCi, 22.2 Ci/mmol (American Radiolabeled Chemicals); or prepared by reduction of farnesal with NaB3H4] in ethanol is added to 1 mg of unlabeled farnesol [all-trans (Aldrich, Milwaukee, WI)] in a 3-ml glass screwcap vial, and solvent is evaporated under a stream of dry N2. Toluene (50 μl) is added and subsequently evaporated under N2. This step is repeated twice to ensure that all of the ethanol is removed. Dry CH2Cl2 (350 μl, distilled from CaH2) is added, followed by 2,6-lutidine (0.7 ml, 6 mmol, ≥99%; Eastman Kodak, Rochester, NY), and the vial is left capped on ice for 1 min. Triphenylphosphine (2.6 mg, 9.9 μmol, Aldrich) in 350 μl of dry CH2Cl2 is added, followed by carbon tetrabromide (3 mg, 9 μmol; Aldrich) in 350 μl of dry CH2Cl2. The reaction is incubated on ice for 1 hr in the dark. The solvent is evaporated under a stream of N2, and the residue is taken up in 100 μl of acetonitrile for HPLC purification. The same procedure is used with [1-3H]geranylgeraniol (1 mCi) mixed with 1.3 mg of unlabeled geranylgeraniol (both from American Radiolabeled Chemicals).
The reaction mixture in acetonitrile is injected onto a C18 reversed-phase semipreparative HPLC column (Vydac, 10 μm, 10 × 250 mm). The column is washed at a flow rate of 1.5 ml/min with 30% (v/v) acetonitrile in water (Milli-Q; Millipore, Bedford, MA) for 10 min, followed by a linear gradient from 30 to 100% acetonitrile over 40 min, and then holding at 100% acetonitrile for 30 min. UV detection is performed at 215 nm. The retention times for farnesyl bromide and geranylgeranyl bromide are 60 and 66 min, respectively. The radiochemical purity is verified by thin-layer chromatography on a C18 reversed-phase plate (20% ethyl acetate/hexane) followed by fluorography with EN3HANCE (NEN, Boston, MA). Unlabeled prenol is spotted as a standard (detected with I2 vapor staining). The reaction typically yields approximately 80 μCi of [1-3H]farnesyl bromide or 120 μCi of [1-3H]geranylgeranyl bromide that is >95% radiochemically pure. Unreacted farnesol (51 min) and geranylgeraniol (61 min) are also recovered (0.55 mCi of farnesol and 0.45 mCi of geranylgeraniol), which can be reused.
Figure 2 shows the HPLC trace of four variants of the C-terminal tryptic peptide of bovine rhodopsin kinase (SGMC(S-[1-3H]farnesyl)-COOH, SGMC(S-[1-3H]farnesyl)-COOCH3, SGMC(S-[1-3H]geranylgeranyl)-COOH, and SGMC(S-[1-3H]geranylgeranyl)-COOCH3). The peptides SGMC-COOH and SGMC-COOCH3 are made by solid-phase synthesis24 and prenylated with radiolabeled prenyl bromides (1 Ci/mol) as described.24 Radioprenylated peptides are purified by reversed phase HPLC as shown in Fig. 2.
Fig. 2.
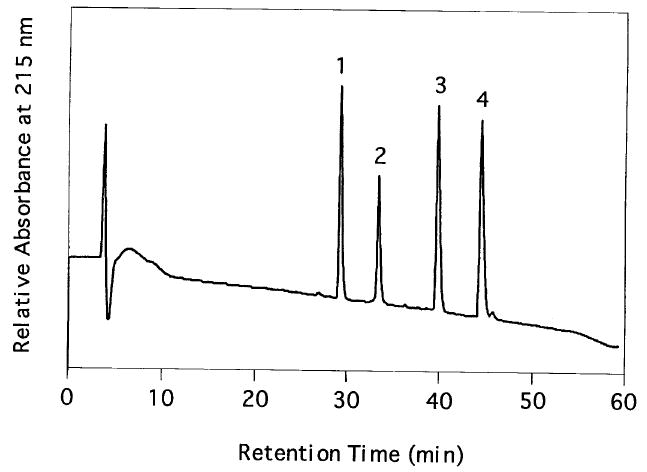
Reversed-phase HPLC analysis of synthetic bovine rhodopsin kinase prenylated peptides: column, Vydac, 4.6 × 250 mm; flow rate, 1.5 ml/min; solvent gradient, 5–30% acetonitrile in water with 0.1% trifluoroacetic acid over 5 min, then 30–85% over 55 min; UV detector at 215 nm. Retention times: peak 1, 29.1 min, SGMC(S-farnesyl)-COOH; peak 2, 33.2 min, SGMC(S-farnesyl-COOCH3; peak 3, 39.9 min, SGMC(S-geranylgeranyl)-COOH; peak 4, 44.8 min, SGMC(S-geranylgeranyl)-COOCH3.
Trypsin Digestion of Bovine Rhodopsin Kinase
Partially purified bovine rhodopsin kinase (purified from whole retina rather than rod outer segments25) in 20 mM Bis–Tris propane, 1 mM dodecyl-β-maltoside (~30% pure as judged by SDS–PAGE) is concentrated to 50 μg/ml in a Centricon-30 ultrafiltration device (Amicon Danvers, MA). For trypsin digestion, 25 μg of protein containing ~7 μg of rhodopsin kinase is mixed with 0.25 μg of sequencing-grade modified trypsin (Promega, Madison, WI) in 1 M urea, 66.7 mM NH4HCO3, 3 mM CaCl2 (pH 8.0) in a total volume of 250 μl. The sample is incubated at 30° for 18 hr. The digest can be stored at −20° prior to analysis by mass spectrometry (see below).
With the radioprenylated tryptic peptide standards in hand, it is possible to gauge the solubility of these peptides in the buffer used for trypsin digestion and also to gauge the stability of the methyl esters under the conditions used for purification of rhodopsin kinase and for trypsinization. Such information is obviously critical for addressing by mass spectrometry (see below) the question of which C-terminal variants of rhodopsin kinase are present in native enzyme purified from retina. The synthetic peptide SGMC(S-[1-3H]farnesyl)-COOCH3 is incubated at 30° for 18 hr in the buffer used for trypsin digestion. Scintillation counting of an aliquot of the solution showed that 83% of the radiolabeled peptide is soluble, and reversed-phase HPLC analysis of the mixture shows that 60% of the methyl ester is recovered from the column with less than 20% of the peptide converted to the free acid.
Combined High-Performance Liquid Chromatography–Electrospray Mass Spectrometry
Analysis of the rhodopsin–kinase tryptic digest is performed by micro-column microelectrospray HPLC–tandem mass spectrometry. The micro-column is constructed from fused silica capillary (365 × 100 μm; J & W Scientific, Folsom, CA) and pulled to a tip of ~2 μm with a laser puller.26 Poros R2 reversed phase material (10 μm; PerSeptive Biosystems, Framingham, MA) is packed against the tip to a length of approximately 15 cm.
The tryptic digest (10 μl of 250 μl) is loaded onto the column at 150 nl/min according to the method described by Yates et al.27,28 An HP 1100 binary HPLC pump (Hewlett-Packard, Wilmington, DE) is used to deliver the gradient to the column. A precolumn split allows a flow rate of 150 nl/min through the column to be achieved. Buffer A consists of 0.5% (v/v) acetic acid and buffer B consisted of acetonitrile–water (80:20, v/v) with 0.5% (v/v) acetic acid (Optima-grade solvents; Fisher, Pittsburgh, PA). The gradient is ramped from 2% (v/v) solvent B to 25% (v/v) solvent B in 2.5 min, followed by 25% (v/v) solvent B to 80% (v/v) solvent B in 27.5 min. Throughout the gradient, a Finnigan LCQ mass spectrometer (Finnigan, San Jose, CA) is programmed for selected ion monitoring (SIM) followed by tandem mass spectrometry (MS/MS). The peptide species monitored are the farnesylated nonmethylated tryptic peptide, m/z 602.4; the geranylgeranyl nonmethylated peptide, m/z 670.6; the farnesylated methylated peptide, m/z 616.3; and the geranylgeranyl methylated peptide, m/z 685.6 (structures of peptides given above). The instrument acquires three microscans of each m/z value in an SIM mode and then acquires tandem mass spectra for the m/z values. MS/MS data provide important verification that the m/z values represent the peptides. The rhodopsin kinase tryptic digest is a complex mixture of peptides. It is therefore possible that a finite amount of ion signal could be produced at the selected m/z values by multiply charged species or fragment ions from peptides other than those of interest. The MS–MS spectrum provides a fingerprint of the prenylated peptide that enables most, if not all, of the peptide sequence to be deduced from the data.
Figure 3A shows the tandem mass spectrum of the synthetic farnesylated and nonmethylated C-terminal tryptic peptide based on the bovine rhodopsin kinase C terminus [SGMC(S-farnesyl)-COOH]. The retention time for this m/z 601.4 ion is 19.23 min. From the tryptic digest of retina-derived bovine rhodopsin kinase, only the farnesylated nonmethylated peptide is observed. The retention time for this m/z 601.4 peptide ion is 19.05 min. Figure 3B shows the tandem mass spectrum for this peptide obtained from native kinase. The spectrum shows fragment ions for the y1–y3 and the b3 ion. A signal is seen for the loss of the farnesyl group and the loss of the farnesyl group including SH is observed at m/z 397.0 and m/z 363.1, respectively. These fragment ions are identical to the signals observed in the tandem mass spectrum of the peptide standard. Studies with synthetic SGMC(S-farnesyl)-COOCH3, and the geranylgeranylated peptide and its methyl ester, show that these compounds are readily detected by mass electrospray spectrometry (not shown). These peptides are also detected when the rhodopsin kinase trypsin digest is spiked with the synthetic standards. A final note is that given the high sensitivity of the method, a blank run should be carried out prior to the analysis of trypsin digests to be sure that there is no carryover of peptide standards from a previous analysis.
Fig. 3.
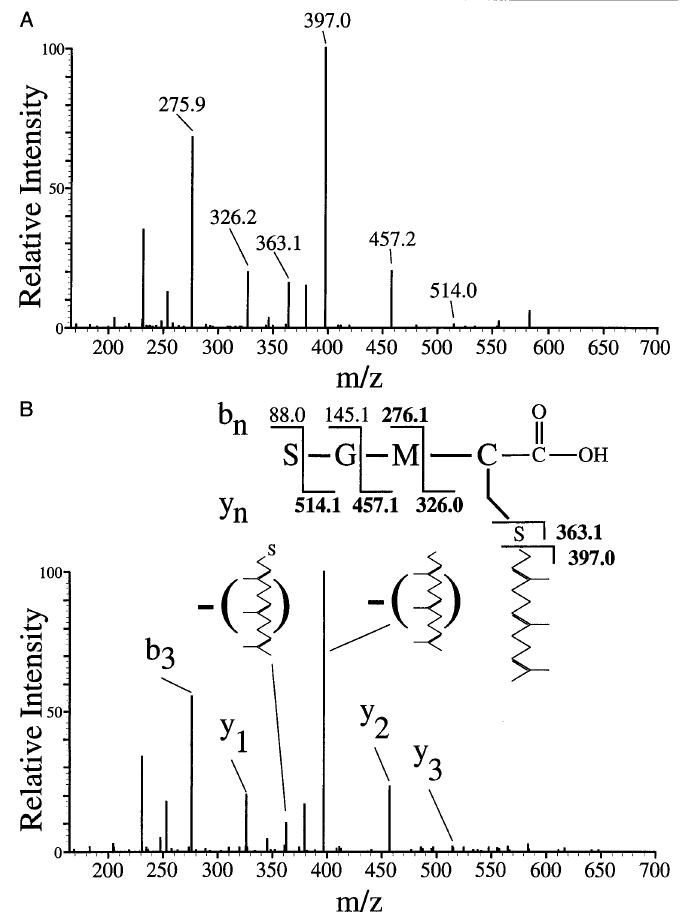
(A) Tandem mass spectrum of the (M + H)+ farnesylated nonmethylated peptide standard ion at m/z 601.4. This peptide eluted at a retention time of 19.23 min. (B) Tandem mass spectrum of the (M + H)+ farnesylated nonmethylated peptide at m/z 601.4 from the tryptic digest of native bovine rhodopsin kinase. This peptide has a retention time of 19.05 min. The amino acid sequence and location of modification are shown above the spectrum. Sequence ions in boldface were found in the spectra.
It is anticipated that the HPLC–MS/MS method for the identification of prenylated and methylated peptides in complex peptide mixtures can be applied to most tryptic digests, whether the source of protein be a partially purified preparation, an SDS–polyacrylamide gel band, or an immunoprecipitate. The method is sensitive; 0.3 μg of rhodopsin kinase is more than sufficient for trypsin digestion and tandem mass spectrometry. The protein to be analyzed does not have to be homogeneous as long as its C-terminal sequence is known. Presumably the method can be used for the analysis of doubly geranylgeranylated peptides, as these peptides elute from reversed-phase HPLC columns in good yield and have been detected by fast atom bombardment mass spectrometry.11 Synthesis of doubly geranylgeranylated peptides has been reported.24
Conclusions
The methods described in this chapter for the structural analysis of prenyl groups released from proteins and of C-terminal lipidated proteolytic fragments derived from native proteins offer many advantages over methods that have been used previously. Raney nickel cleavage yields single cleavage products in high yield. These fragments can be conveniently analyzed by HPLC methods when radiolabeling is carried out or by gas chromatography–mass spectrometry when proof of structure and accurate ratios of the different length prenyl groups are desired. Tandem mass spectrometry, when combined with micro-HPLC, provides a method for characterizing the entire C-terminal structure of the protein of interest (prenylation, proteolysis, and methylation). Because of mass speciation, this method is useful for analyzing complex mixtures such as tryptic digests of native proteins. Finally, all of the methods are sufficiently sensitive to permit the analysis of relatively small amounts of proteins extracted from SDS–polyacrylamide gel bands.
Acknowledgments
Supported by National Institutes of Health Grants CA52874 (M.H.G.), 118223 (J.R.Y.), and EY08061 (K.P.) and by Molecular Biophysics Training Grant T32 GM08268 (M.E.W.).
References
- 1.Glomset JA, Gelb MH, Farnsworth CC. Trends Biochem Sci. 1990;15:139. doi: 10.1016/0968-0004(90)90213-u. [DOI] [PubMed] [Google Scholar]
- 2.Maltese WA. FASEB J. 1990;4:3319. doi: 10.1096/fasebj.4.15.2123808. [DOI] [PubMed] [Google Scholar]
- 3.Clarke S. Annu Rev Biochem. 1992;61:355. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- 4.Zhang FL, Casey PJ. Annu Rev Biochem. 1996;65:241. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 5.Ma YT, Rando RR. Proc Natl Acad Sci USA. 1992;89:6275. doi: 10.1073/pnas.89.14.6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jang GF, Yokoyama K, Gelb MH. Biochemistry. 1993;32:9500. doi: 10.1021/bi00087a031. [DOI] [PubMed] [Google Scholar]
- 7.Boyartchuck VL, Ashby MN, Rine J. Science. 1997;275:1796. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt WK, Tarn A, Fujimura-Kamada K, Michaelis S. Proc Natl Acad Sci USA. 1998;95:11175. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. EMBO J. 1991;10:1699. doi: 10.1002/j.1460-2075.1991.tb07694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romano JD, Schmidt WK, Michaelis S. Mol Biol Cell. 1998;9:2231. doi: 10.1091/mbc.9.8.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farnsworth CC, Kawata M, Yoshida Y, Takai Y, Gelb MH, Glomset JA. Proc Natl Acad Sci USA. 1991;88:6196. doi: 10.1073/pnas.88.14.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokoyama K, Goodwin GW, Ghomashchi F, Glomset J, Gelb MH. Biochem Soc Trans. 1992;20:479. doi: 10.1042/bst0200489. [DOI] [PubMed] [Google Scholar]
- 13.Casey PJ, Seabra MC. J Biol Chem. 1996;271:5289. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 14.Farnsworth CC, Wolda SL, Gelb MH, Glomset JA. J Biol Chem. 1989;264:20422. [PMC free article] [PubMed] [Google Scholar]
- 15.Farnsworth CC, Gelb MH, Glomset JA. Science. 1990;247:320. doi: 10.1126/science.2296721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farnsworth CC, Casey PJ, Howald WN, Glomset JA, Gelb MH. Methods. 1990;1:231. [Google Scholar]
- 17.M. H. Gelb, C. C. Farnsworth, and J. A. Glomset, in “Lipidation of Proteins: A Practical Approach” (A. J. Turner), p. 231. IRL Press, Oxford, 1992.
- 18.Sakagami Y, Isogai A, Suzuki A, Tamura S, Kitada C, Fujino M. Agric Biol Chem. 1979;43:2643. [Google Scholar]
- 19.Kawata M, Farnsworth CC, Yoshida Y, Gelb MH, Glomset JA, Takai Y. Proc Natl Acad Sci USA. 1990;87:8960. doi: 10.1073/pnas.87.22.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamane HK, Farnsworth CC, Xie H, Howald W, Fung BKK, Clarke S, Gelb MH, Glomset JA. Proc Natl Acad Sci USA. 1990;87:5868. doi: 10.1073/pnas.87.15.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGeady P, Kuroda S, Shimizu K, Takai Y, Gelb MH. J Biol Chem. 1995;270:26347. doi: 10.1074/jbc.270.44.26347. [DOI] [PubMed] [Google Scholar]
- 22.Sakagami Y, Isogai A, Suzuki A, Tamura S, Kitada C, Fujino M. Agric Biol Chem. 1978;42:1093. [Google Scholar]
- 23.Ishibashi Y, Sakagami Y, Isogai A, Suzuki A. Biochemistry. 1984;23:1399. [Google Scholar]
- 24.Liu L, Jang GF, Yokoyama K, Ghomashchi F, Farnsworth CC, Glomset JA, Gelb MH. Methods Enzymol. 1994;250:189. doi: 10.1016/0076-6879(95)50072-3. [DOI] [PubMed] [Google Scholar]
- 25.Palczewski K. Methods Neurosci. 1993;15:217. [Google Scholar]
- 26.Gatlin CL, Kleemann GR, Hays LG, Link AJ, Yates JR., III Anal Biochem. 1998;263:93. doi: 10.1006/abio.1998.2809. [DOI] [PubMed] [Google Scholar]
- 27.J. R. Yates III, A. L. McCormack, J. B. Hayden, and M. P. Davey, in “Cell Biology: A Laboratory Handbook” (J. E. Celis), p. 380. Academic Press, San Diego, California, 1994.
- 28.McCormack AL, Eng JK, Yates JR., III Methods. 1994;6:274. [Google Scholar]