

Abstract
Previous studies have shown apolipoprotein E (apoE) recruitment to medial layers of carotid arteries after vascular injury in vivo and apoE activation of inducible nitric oxide synthase (iNOS) in smooth muscle cells in vitro. This investigation explored the relationship between medial apoE recruitment and iNOS activation in protection against neointimal hyperplasia. ApoE was present in both neointimal-resistant C57BL/6 mice and neointimal-susceptible FVB/N mice 24 h after carotid denudation, but iNOS expression was observed only in the neointimal-resistant C57BL/6 mice. However, iNOS was not observed in apoE-defective C57BL/6 mice. In contrast, over-expression of apoE in FVB/N mice activated iNOS expression in the injured vessels resulting in protection against neointimal hyperplasia. ApoE and iNOS were colocalized in the medial layer of neointimal-resistant mouse strains. Endothelial denudation of carotid arteries in the iNOS-deficient NOS2−/− mice did not increase neointimal hyperplasia but significantly increased medial thickness and area. The iNOS-specific inhibitor also abrogated the apoE protective effects on vascular response to injury in apoE over-expressing FVB/N mice. Thus, injury-induced activation of iNOS requires apoE recruitment. Moreover, both apoE and iNOS are necessary for suppression of cell proliferation, and apoE recruitment without iNOS expression resulted in medial hyperplasia without cell migration to the intima.
Supplementary key words: neointimal hyperplasia, smooth muscle cells, endothelial denudation, vascular biology, transgenic mice
Abbreviations Used: Apo, apolipoprotein; LRP-1; LDL receptor related protein-1; iNOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2
Neointimal hyperplasia is a critical pathological process in several vascular occlusive diseases, including atherosclerosis, vein graft-induced arteriosclerosis, and the restenotic response to angioplasty and stent placement (1–4). The key etiology of its pathology is endothelial damage, which exposes the underlying smooth muscle cells in the media to cytokines, growth factors, and other plasma components in circulation resulting in a loss of contractile characteristics and adoption of a synthetic phenotype (5, 6). The phenotypic activation of the underlying smooth muscle cells results in their migration from the media to the intima where their proliferation and increased synthetic potential leads to the formation of the occlusive neointima (7).
Recent studies in mice have shown that one determinant regulating the severity of neointimal formation is the level of apolipoprotein E (apoE) in circulation. Results of these studies showed that transgenic over-expression of apoE is protective and apoE gene ablation exacerbates neointimal hyperplasia caused by mechanically induced endothelial denudation (8). In fact, subphysiological levels of apoE were shown to have beneficial effects in limiting neointimal hyperplasia without correcting for hypercholesterolemia in apoE-null mice (9). The protective role of apoE has been established to involve the recruitment of circulating apoE to the medial layers of the injured vessel (10). It is likely that apoE interaction with the medial smooth muscle cells limits their migration and proliferation in response to growth factors present in the serum (11, 12).
In vitro cell culture studies revealed that apoE inhibits smooth muscle cell migration by binding to the cell surface receptor LRP-1 (13). The interaction between apoE and LRP-1 increases intracellular cyclic AMP level, resulting in activation of protein kinase A pathways that limit smooth muscle cell migration (14). In contrast, LRP-1 is not necessary for apoE inhibition of smooth muscle cell proliferation, and the anti-proliferative response is mediated by apoE binding to heparan sulfate proteoglycans (15). The mechanism by which apoE inhibits growth factor-induced cell proliferation was shown to involve the activation of inducible nitric oxide synthase (iNOS) in one study (12). However, another study suggested that apoE inhibits cell proliferation by activation of cyclooxygenase-2 (COX-2), leading to prostacyclin synthesis and inhibition of cyclin A gene expression (16). The latter study failed to detect iNOS activation by apoE (16). The difference between the two in vitro studies may be related to differences in incubation conditions and/or different mitogens used for the respective experiments.
Given the strong relationship between apoE and the hyperplasic response to vascular injury, this study sought to determine a possible relationship between apoE and iNOS induction in vivo by examining apoE and iNOS expression in our mouse model of injury-induced neointimal hyperplasia. The data showed that apoE present in injured carotid arteries colocalizes with iNOS in strains that are resistant to neointimal hyperplasia, with the expression of iNOS correlating to the presence of apoE. Importantly, the data revealed that failure of iNOS activation results in a medial hyperplasic response without cell migration to the intima and resultant neointimal formation.
Methods
Animals
Wild type C57BL/6J and FVB/N mice, as well as the apoE−/− and the iNOS-deficient NOS2−/− mice were obtained from the Jackson Laboratories (Bar Harbor, MI). Transgenic mice with liver-specific human apoE gene expression were generously provided by Dr. John Taylor (Gladstone Institute, San Francisco, CA) and were backcrossed to the FVB/N background in our institution as described previously (8, 10). The genotype of the apoE transgenic mice was determined based on the amount of human apoE present in the serum, which averaged about 30 mg/dL (10). All animals were maintained on a 12-hour light/12-hour dark cycle and were fed a normal mouse chow diet (Harlan Teklad, Madison WI). Food and water were available ad libitum. All animals used for experimentation were male, between 6 to 8 weeks of age, and weighed between 25 to 30 grams. At the time of experiment, the average plasma cholesterol levels in C57BL/6, FVB/N, and apoE-transgenic FVB/N mice were all within the normal range of 70–95 mg/dL, as reported previously (8). The iNOS-deficient NOS2−/− mice were also normocholesterolemic with an average plasma cholesterol level of 71 ± 9.6 mg/dL, but the apoE−/− mice were hypercholesterolemic with plasma cholesterol levels averaging ~270–280 mg/dL. All animal experimentation protocols were performed under the guidelines of animal welfare prescribed by the University of Cincinnati, in accordance with National Institutes of Health guidelines.
Carotid Artery Injury
Mechanically induced endothelial denudation was performed as described previously (8, 10). Briefly, mice were anesthetized by intraperitoneal injection with a solution of ketamine (80 mg/kg body weight; Phoenix Scientific, Inc., St. Joseph, MO) and xylazine (16 mg/kg; Phoenix Scientific) diluted in 0.9% NaCl. Animals were also given buprenorphine (0.05mg/kg body weight; Abbott Laboratories, Abbott Park, IL) to alleviate any post-operative pain. The mice were then immobilized and the fur covering the neck from sternum to chin was removed with lotion hair remover (Nair; Carter-Wallace, Inc., New York, NY). Surgery was performed under a dissection microscope (Leica GZ6; Leica, Buffalo, NY). The entire length of the left carotid artery was exposed and the artery was ligated immediately proximal from the point of bifurcation with a 7–0 silk suture (Ethicon, Inc., Somerville, NJ). Another 7–0 suture was placed around the common carotid artery immediately distal from the branch point of the external carotid. A transverse arteriotomy was made between the 7–0 sutures and the resin probe was inserted and advanced toward the aorta arch and withdrawn five times. The probe was removed and the proximal 7–0 suture was ligated. Once restoration of blood flow through the carotid branch points was confirmed, the incision was closed with a 6-0 sterile surgical silk (Ethicon, Inc.). The entire procedure was performed within 20 min. Animals were allowed to recover in a cage warmed to 37°C. The identical surgical procedure was applied to each animal to assure reproducibility of the results.
Tissue Preparation and Histological Staining
Animals were anesthetized as described above and perfused with 0.9% NaCl by placement of a 22-gauge butterfly angiocatheter in the left ventricle. The mice were subsequently perfusion-fixed in situ by infusion with 10% neutral-buffered formalin for 10 min at a constant pressure of 100 mm Hg. The entire neck was dissected using a surgical scalpel, using the suture knot and the aorta as reference points. Tissues were placed in formalin for an additional 24 hr, then transferred to a formic acid buffer (Formical-2000; Decal Chemical Corp., Tallman, NY) until decalcification endpoint was reached. Tissues were paraffin-embedded and cross sections were cut at a thickness of 5 μm. Whole-neck sections were used to evaluate both the injured and the uninjured control vessels on the same section. An average of four levels of serial sections separated by 500 μm intervals was analyzed to allow for the representation of hyperplasic response along the entire length of the artery. Both hematoxylin and eosin (H&E) staining as well as Verhoeff Van-Gieson (VVG) staining of elastic lamina was performed on parallel sections. The unstained sections from each level were used for immunohistochemistry.
Immunohistochemistry
Sections were deparaffinized by acetone/ethanol gradient, then equilibrated in phosphate-buffered saline. When relevant, nonspecific binding of anti-mouse antibodies was inhibited with Mouse Ig Blocking Reagent (Vector Laboratories, Inc., Burlingame, CA). General nonspecific binding was inhibited by incubation for 1 hr with 10% normal horse serum (Vector Laboratories, Inc., Burlingame, CA) in phosphate-buffered saline. ApoE was detected using a polyclonal goat antibody that recognizes both mouse and human apoE (Clone M-20; Santa Cruz Biotechnology, Santa Cruz, CA). The presence of iNOS was detected using a monoclonal mouse antibody (Clone 6; BD Biosciences Pharmingen, San Jose, CA). Smooth muscle α-actin was detected using a monoclonal mouse antibody (Clone 1A4; Sigma, St. Louis, MO). All primary antibodies were administered at a 1:1000 dilution in Antibody Diluent (Zymed, San Francisco, CA). All sections were incubated overnight at 4°C with primary antibodies to identify the antigen of choice. The slides were washed three times for 15 min each with phosphate-buffered saline containing 0.01% Triton X-100 and then incubated for 1 hr at 23°C with either donkey anti-mouse ALEXA 594 or donkey anti-goat ALEXA 488 (Molecular Probes, Eugene, OR). All sections were counterstained with 300 nM DAPI (Molecular Probes, Eugene, OR). Sections were mounted using aqueous Gelvatol mounting medium with 0.3% DABCO added.
In Vivo Inhibition of iNOS
Wild type FVB/N and apoE transgenic FVB/N littermates were subjected to endothelial denudation as described above. From the apoE transgenic group, 10 mice were selected to receive 20 mg/kg/day of the specific iNOS pharmacological inhibitor 1400W (Cayman Chemicals, Ann Arbor, MI), and 10 mice were selected to receive an isovolumetric injection of saline. All injections were administered subcutaneously. The first injection was administered just prior to arterial injury, in concert with injection of anesthetics. Daily injections were given for 14 days following injury, at the same time each day. All mice were sacrificed at 24 hr after the last injection and tissues were harvested for morphometric analysis.
Morphometry
Morphometric analyses were performed on elastin-stained (VVG) tissues. For each animal, four whole-neck cross-sections with both injured left and uninjured-control right carotid arteries were measured. Images were digitized and captured using an Olympus digital camera (Olympus America Inc., Melville, NY) connected to a personal computer. Measurements were performed at a magnification of x200 using the ImageJ analysis software (National Institutes of Health, Bethesda, MD). The luminal area, area inside the internal elastic lamina, and the area encircled by external elastic lamina were measured in all four serial sections of each artery. Medial area was calculated as area inside the internal elastic lamina subtracted from the area encircled by external elastic lamina, and intimal area was calculated as luminal area subtracted from the area inside the internal elastic lamina. To calculate the medial thickness for each vessel cross-section, the linear distance between internal elastic lamina and external elastic lamina was measured independently in four places, each at 90° apart and averaged.
COX-2 Western Blot
Both C57BL/6 and FVB/N mice were anesthetized and intracardially perfused with ice-cold saline 24 hr after arterial injury. Both left (injured) and right (uninjured) carotid arteries were immediately dissected separately and snap frozen in liquid nitrogen. Tissues were then placed in lysis buffer composed of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 0.2 mM PMSF. Tissues were then homogenized using a ground glass mortar and pestle, after which solubilized lysate was separated by centrifugation. Total protein content was quantified and 25 μg of total protein was applied to SDS polyacrylamide gel electrophoresis, transferred to PVDF membrane, and immunoblotted with rabbit monoclonal antibodies against COX-2 (Vector Laboratories, Burlingame CA). Immunoreactivity was detected using an infrared fluorophore-conjugated secondary antibody raised against rabbit IgG (Molecular Probes). Detection of secondary antibody was performed using an Odyssey infrared imaging scanner (Li-Cor Biosciences, Lincoln, NE).
Statistical Analysis
All values were expressed as mean ± S.E.M. When only two groups were compared, differences were assessed by using a paired Student’s t test. Multiple comparisons were first tested by using ANOVA. When the ANOVA demonstrated significant differences, individual mean differences were analyzed with the Student-Newman Keuls test. Statistical software SigmaStat (Jandel Co.) was used in statistical analysis. For all analyses, a P < 0.05 was considered significant difference.
Results
The relationship between apoE recruitment and iNOS expression in modulating neointimal hyperplasia was investigated using two different mouse strains differing in their hyperplasic response to endothelial denudation. In these experiments, the carotid arteries of the neointima-resistant C57BL/6 mice and the neointima-susceptible FVB/N mice were injured by mechanical denudation of their endothelium. After a recovery period of 24 hr, mice were sacrificed and whole necks were isolated for tissue sectioning. Fixed sections were immunohistochemically probed with both anti-apoE and anti-iNOS antibodies. Strong positive apoE signal was observed in both medial layers of the denuded carotid arteries in both C57BL/6 and FVB/N mice (Fig. 1 a,b). In contrast, positive iNOS signal was observed only in the medial layers of the neointima-resistant C57BL/6 mice, and iNOS expression was not apparent in the injured arteries of the neointimal-susceptible FVB/N mice (Fig. 1 c, d). Overlaying of the fluorescent signals from both apoE and iNOS immunohistochemical staining of the injured arteries in C57BL/6 mice revealed their colocalization (Fig. 1 e,f). The latter observation is consistent with our previous in vitro studies showing that apoE activates iNOS expression in smooth muscle cells (12). Interestingly, whereas our previous study documented apoE immunoreactivity in the medial layer up to 14 days after vascular injury (10), iNOS expression was no longer observed after 3 days (data not shown). Thus, apoE-dependent activation of smooth muscle iNOS expression appeared to be an early response to endothelial denudation injury. In contrast, no difference in COX-2 levels was observed in extracts from injured or uninjured arteries of C57BL/6 and FVB/N mice (Fig. 2).
Figure 1.
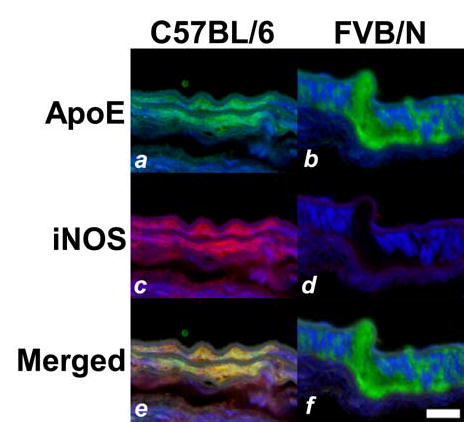
Immunostaining of apoE and iNOS in denuded carotid arteries of inbred mice. Carotid arteries in C57BL/6 (a,c,e) and FVB/N (b,d,f) mice were injured by mechanical denudation. Tissues were harvested after 24 hr and sections were immunostained with fluorescent antibodies against apoE (a,b) and iNOS (c,d). Panels e and f show the overlay of apoE (green) and iNOS (red) signals, with colocalization appearing as yellow signal in the medial smooth muscle cell layers of the C57BL/6 arteries (e) but not in FVB/N arteries (f). Scale bar represents 20 μm.
Figure 2.
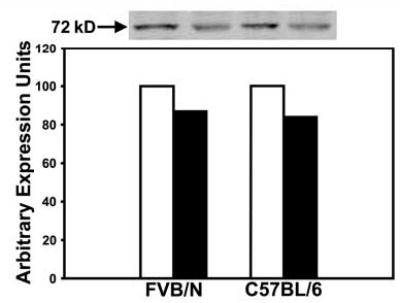
Immunoblot analysis of COX-2 expression in FVB/N and C57BL/6 mice. The uninjured (open bars) and injured (filled bars) carotid arteries were harvested from FVB/N and C57BL/6 mice 24 hr after endothelial denudation. The tissues were homogenized and extracts containing 25 μg of proteins were applied to each lane of a SDS-polyacrylamide gel for electrophoresis. The samples were transferred to PVDF membranes and immunoblotted with antibodies against COX-2. Immunoreactivity was detected using an infrared fluorophore-conjugated secondary antibody against mouse IgG and the image is shown on the top panel. The intensity of the bands was determined with an Odyssey infrared imaging scanner and reported as artibrary unit of expression.
The next series of experiments examined the cause-and-effect relationship between apoE recruitment and the activation of iNOS expression in medial smooth muscle cells in response to arterial injury. In the first set of experiments, the carotid arteries of apoE−/− mice (in C57BL/6 background) were injured by mechanical denudation of their endothelium. The mice were sacrificed after 24 hr and whole necks were isolated for tissue sectioning and immunohistochemical detection of apoE and iNOS as described above. The lack of apoE gene expression in the apoE−/− mice abolished apoE immunoreactivity in the vessel wall as expected (Fig. 3a). The lack of apoE recruitment also resulted in the absence of iNOS expression in the vessel wall of these animals after endothelial denudation (Fig. 3 c,e).
Figure 3.
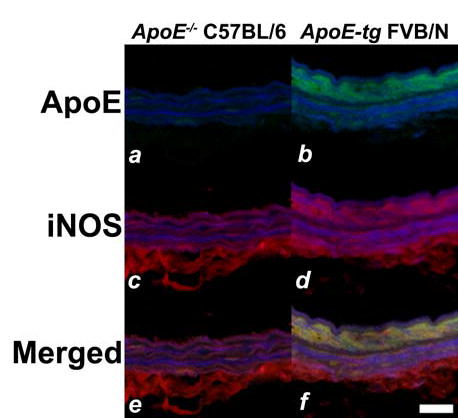
Immunodetection of apoE and iNOS after vascular injury in genetically-modified mice. Immnuofluorescent analysis of injured arteries from C57BL/6 apoE−/− mice (a,c,e) and apoE transgenic FVB/N mice (b,d,f) 24 hr after endothelial denudation. The sections were stained with fluorescent antibodies against apoE (a,b) or iNOS (c,d) to yield green and red signals, respectively. Panels e and f show the overlay of apoE (green) and iNOS (red) signals. Note that colocalization of signals appeared in yellow was detectable only in the medial smooth muscle cell layers of the arteries in FVB/N-apoE transgenic mice (f) but not in arteries of the C57BL/6 apoE-null mice (e). Scale bar represents 20 μm.
The next experiment determined if the suppression of neointimal hyperplasia in FVB/N mice by high circulating levels of apoE that we have reported previously (8) may be due to the activation of smooth muscle iNOS expression in this strain as a consequence of increased recruitment of human apoE to the site of arterial injury (10). We compared apoE recruitment and iNOS expression in injured arteries of FVB/N mice with or without transgenic over-expression of human apoE in the liver. Strong positive apoE immunoreactivity was observed in the abluminal medial layer of the FVB/N human apoE transgenic mice (Fig. 3b). The apoE immunoreactivity in the medial layers of the apoE-transgenic mice, although not a quantitative measurement, reflected the 10-fold increase of circulating apoE concentration in the transgenic mice compared to wild type FVB/N mice (8). The increased circulating apoE in the apoE transgenic mice also resulted in notable iNOS immunoreactivity even in the FVB/N background (Fig. 3d and contrast to Fig. 1d). Overlaying of the fluorescent signals from both apoE and iNOS immunohistochemical staining indicated their co-localization (Fig. 3f). These results, taken together with observations of iNOS expression in C57BL/6 wild type mice but not in C57BL/6 apoE-null mice, documented that apoE recruitment is required for activation of smooth muscle iNOS expression in response to arterial injury.
The results showing apoE recruitment and iNOS expression in C57BL/6 wild type and apoE-transgenic FVB/N mice, both of which are resistant to injury-induced neointimal hyperplasia, and the lack of iNOS expression in the apoE-null C57BL/6 and FVB/N wild type mice, both of which are susceptible to injury-induced neointimal hyperplasia, suggested that both apoE recruitment and its subsequent activation of smooth muscle iNOS expression are required to limit neointimal hyperplasia in response to endothelial denudation. To substantiate this hypothesis, we investigated the effect of iNOS gene ablation on the hyperplasic response to endothelial denudation by performing the same procedure on both C57BL/6 wild type and NOS2−/− mice. After a recovery period of 14 days, mice were sacrificed and whole necks were dissected and fixed for sectioning. Morphometric analysis of the histology data revealed minimal neointima in the C57BL/6 mice, compared to the neointima size of apoE−/− mice 14 days after endothelial injury reported previously (8, 17). The iNOS-defective C57BL/6 mice (NOS2−/−) mice also showed only minimal neointima area after endothelial denudation. Importantly, no significant difference in neointimal area was observed between C57BL/6 mice with or without a functional iNOS gene (Fig. 4a). Interestingly, the medial area and medial thickness were significantly increased (P < 0.001) in the injured arteries of the iNOS-defective NOS2−/− mice in comparison to those observed in the injured arteries of their wild type C57BL/6 counterparts (Fig. 4 b,c).
Figure 4.
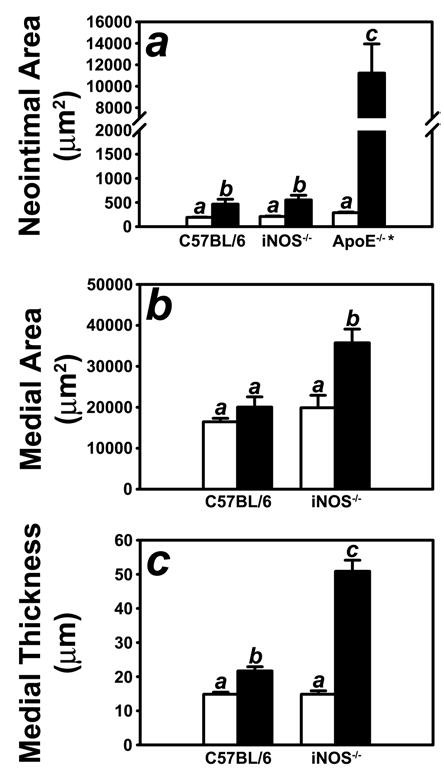
Morphometric quantification of control (open bars) and injured (filled bars) carotid arteries from C57BL/6 wild-type and iNOS-defective NOS2−/− mice. Panel A shows neointimal area of the injury carotid arteries of C57BL/6 wild type and iNOS-deficient mice in comparison with the neointima formed in the injured arteries of apoE-null (apoE(−/−) mice (* reported in ref 8). The Neointimal areas were determined by subtracting the luminal area from the area encircled by the internal elastic lamina. Panel B shows the morphometric data for medial area, which was calculated as the area encircled by external elastic lamina minus the area encircled by the internal elastic lamina. Panel C shows the mophometric data for medial thickness which was calculated as the average linear distance between the internal and external elastic lamina measured in four places at 90° apart. The data represent the mean ± SEM from ten C57BL/6 and ten iNOS(−/−) mice. Bars with different letters in each graph indicate significant difference at P < 0.05.
Histological analysis of the injured arteries in the iNOS-deficient NOS2−/− mice indicated a medial hyperplasia phenotype instead of hyperplasic response of intimal cells after their migration from the media. Verhoeff-Van Geison stained sections showed a widening of the abluminal medial layer compared to that in the contralateral uninjured artery (Fig. 5 a,b). Hematoxylin and eosin staining of parallel sections indicated that the thickening of the medial layer was cellular, most likely due to hyperplasia of the medial smooth muscle cells (Fig. 5 c,d). Immunofluorescent staining of parallel sections with anti-smooth muscle α-actin and anti-apoE antibodies showed that apoE was recruited to the medial layers of the injured arteries (Fig. 5 e,f).
Figure 5.
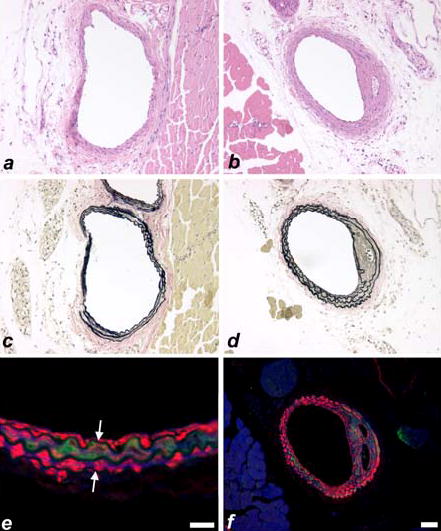
Representative histochemical (a–d) and immunohistochemical (e,f) staining in control (a,c) and injured (b,d,e,f) carotid arteries from C57BL/6 iNOS-null mice. Hematoxylin and eosin staining (a,b) indicates a cellular thickening in the injured iNOS-null carotid artery (b). Verhoeff-Van Geison staining (c,d) indicates a medial thickening in the injured iNOS-null carotid artery. Immunofluorescent detection (e,f) of alpha-smooth muscle actin (red) and apoE (green) indicate the presence of apoE within the medial smooth muscle cell layers. Internal and external elastic laminae are indicated by white arrows (e). Scale bar in panel e represents 20 μm and the scale bar in panel f represents 50 μm.
The requirement of iNOS induction for apoE suppression of neointimal hyperplasia was examined further by determining the effect of iNOS inhibition on arterial response to injury in FVB/N mice with hepatic over-expression of the apoE transgene. Similar to results reported previously showing apoE transgenic mice were protected from injury-induced neointimal hyperplasia (8), apoE transgenic FVB/N mice receiving daily injection of saline displayed significantly less neointimal hyperplasia compared to nontransgenic FVB/N mice (Fig. 6). In contrast, apoE transgenic FVB/N mice receiving daily injections of the iNOS inhibitor 1400W (18) developed a neointimal lesion indistinguishable in size from the neointima observed in nontransgenic FVB/N mice (Fig. 6a). In addition, apoE transgenic mice treated with 1400W developed significantly larger medial walls, as evidenced by measurements of medial area and thickness (Fig. 6 b,c). Thus, these results indicated that iNOS inhibition abrogated the apoE protection on vascular response to injury.
Figure 6.
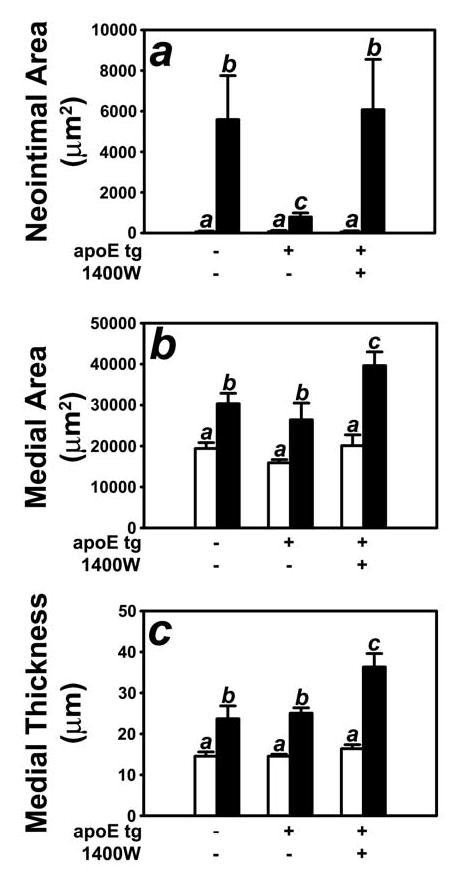
Morphometric quantification of control (open bars) and injured (filled bars) carotid arteries from FVB/N wild type and apoE transgenic mice given daily injections of saline or the iNOS inhibitor 1400W. Panel A shows neointimal area of the injury carotid arteries of apoE transgenic mice with or without 1400W. Neointimal area was determined by subtracting the luminal area from the area encircled by the internal elastic lamina. Panel B shows the morphometric data for medial area, which was calculated as the area encircled by external elastic lumina minus the area encircled by the internal elastic lamina. Panel C shows the morphometric data for medial thickness which was calculated as the average linear distance between the internal and external elastic lamina measured in four places at 90° apart. The data represent 10 mice from each group. Bars with different letters in each graph indicate significant difference at P < 0.05.
Discussion
Vascular response to injury is a complex phenomenon, with contributions by various molecules and mechanisms at varying time points (3). The role of apoE in modulating vascular response to injury has been clearly established, with a mechanism related to its inhibition of smooth muscle cell migration and proliferation. Our in vitro data showed that apoE inhibition of cell migration and its inhibition of cell proliferation are controlled by different mechanisms (12, 15). In particular, apoE inhibition of smooth muscle cell proliferation, but not its inhibition of cell migration, has been shown to be mediated by expression of iNOS in smooth muscle cells (12).
Nitric oxide, the product of iNOS, has been shown to have a number of anti-hyperplasic properties, including the inhibition of smooth muscle cell migration and proliferation (19, 20), inhibition of platelet aggregation (21), and promotion of endothelial regeneration (22). Despite these findings, however, the contribution of nitric oxide deficiency to vascular disease has been controversial. Kuhlencort et al reported that iNOS deficiency in mice resulted in lowering of lipid peroxides and decreased lesion size in an atherosclerosis model (23), whereas Ihrig et al reported that iNOS deficiency resulted in elevated cholesterol levels and an increased lesion incidence (24). In the context of injury-induced hyperplasia, Chyu et al reported that iNOS ablation resulted in a decrease of neointimal hyperplasia (25), but Koglin et al concluded from their study in a vascular allograft model that iNOS deficiency resulted in an increased neointimal hyperplasia (26). The discrepancies between these findings remain unresolved.
This investigation builds upon our previously published data and confirms the role of iNOS in apoE-dependent inhibition of smooth muscle cell proliferation. Previous research in our laboratory has shown that the C57BL/6 wild type mice are resistant to endothelial denudation-induced neointimal hyperplasia, whereas the FVB/N mice are susceptible (27). Comparison of apoE recruitment and iNOS expression in injured carotid arteries from both strains showed that, whereas apoE was recruited to the media in both hyperplasia-resistant and -susceptible strains after endothelial denudation, iNOS expression was only detectable in the resistant strains. In addition, double-labeling using fluorescent antibodies indicated that the recruited apoE colocalized with iNOS expression in the medial layers of the injured carotid arteries of the resistant strains. Further studies with genetically-modified mouse models showed that iNOS was not expressed in the absence of apoE, and that over-expression of apoE in a hyperplasia-susceptible strain resulted in the activation of iNOS expression and a resistant phenotype. This correlation strongly indicated apoE-dependent induction of iNOS as an early response event that is necessary for its inhibition of neointimal formation.
In this investigation, we also took advantage of the iNOS-null mouse model, which is available on a hyperplasia-resistant genetic background (28), and compared vascular response to injury in C57BL/6 mice with or without iNOS gene expression. Our results showed that, although neointimal hyperplasia was not different between the two strains, significant increase in both medial area and medial thickening was observed in mice lacking a functional iNOS gene. Given the established importance of iNOS in apoE-dependent inhibition of smooth muscle cell proliferation (12), we interpret the data to indicate a phenotype of medial hyperplasia, as opposed to neointimal hyperplasia. These results further underscore the importance of iNOS in mediating apoE inhibition of smooth muscle cell proliferation but not its inhibition of cell migration. However, these results are in conflict with the data published by Chyu et al, who showed that deficiency of iNOS resulted in a decrease of neointimal hyperplasia with no concurrent change in medial morphometry (25). The difference between the two studies may be due to differences in time points for the analysis of vascular pathology, but more importantly differences in the procedure used to induce vascular injury. The Chyu study induced vascular injury with a cuff model by applying a periadventitial collar around the carotid artery. This model involves significant inflammatory cell participation in the injury response. In contrast, our studies induced vascular injury by mechanical denudation of the endothelium, in which lymphocyte infiltration appeared to be an early protective event against neointimal hyperplasia (17). Thus, the different methodology used may have different impact on inflammatory cell functions, with the resulting differences in the role of iNOS in smooth muscle cell activation and/or endothelial repair responses.
The implication of iNOS as a necessary mediator of apoE-dependent inhibition of vascular cell proliferation in the C57BL/6 strain does not fully address the potential mechanism by which the FVB/N strain of mice are more susceptible to endothelial denudation-induced neointimal hyperplasia, requiring elevated levels of apoE to protect against this pathological phenotype (8). The expression level of COX-2 cannot explain the difference between C57BL/6 and FVB/N mice in vascular response to injury as both strains displayed similar levels of COX-2 protein in the arterial wall before and after endothelial denudation (Fig. 2). In the current study, we showed the expression of iNOS in injured vessels of apoE transgenic FVB/N mice but not in injured vessels of wild type FVB/N mice. Importantly, we used the iNOS specific inhibitor 1400W (18) and showed that inhibition of iNOS activity also obliterated the protective effects of the apoE transgenic expression against neointimal hyperplasia in the FVB/N mice. Thus, the dysregulation of iNOS may be a critical determinant for the increase in injury-induced neointimal hyperplasia in wild type FVB/N mice compared to that observed in C57BL/6 mice.
The current study clearly illustrated that the in vivo activation of iNOS expression in smooth muscle cells is necessary for apoE inhibition of endothelial denudation-induced neointimal hyperplasia. Although the mechanism of nitric oxide inhibition of smooth muscle cell proliferation remains to be identified, previous work from our laboratory showed that apoE inhibition of cell proliferation also requires inhibition of mitogen activated kinase (11), a property that has been attributed to nitric oxide (29, 30). Additional studies elucidating the intermediate signaling pathway(s) between apoE binding to heparan sulfate proteoglycans, which is required for apoE inhibition of cell proliferation (15), and expression of iNOS in smooth muscle cells would be beneficial for a complete understanding of the mechanism by which apoE is involved in regulation of cell proliferation.
Acknowledgments
The authors wish to thank Pravina Desai for her histological expertise. This research was supported by National Institutes of Health Grant HL-61332 (to D.Y.H.) and a Predoctoral Fellowship (Grant 0415267B) from the Ohio Valley Affiliates of the American Heart Association (to Z.W.Q.M).
References
- 1.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Fan Y-S, Chow LH, Van den Diepstraten C, van der Veer E, Sims SM, Pickering JG. Innate diversity of adult human arterial smooth muscle cells: Cloning of distinct subtypes from the internal thoracic artery. Circ Res. 2001;89:517–525. doi: 10.1161/hh1801.097165. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RS, Henry TD. Pathophysiology of coronary artery restenosis. Rev Cardiovasc Med. 2002;3:S4–9. [PubMed] [Google Scholar]
- 4.Kearney M, Pieczek A, Haley L, Losordo DW, Andres V, Schainfeld R, Rosenfield K, Isner JM. Histopathology of instent restenosis in patients with peripheral artery disease. Circulation. 1997;95:1998–2002. doi: 10.1161/01.cir.95.8.1998. [DOI] [PubMed] [Google Scholar]
- 5.Wilensky RL, March KL, Gradus-Pizlo I, Sandusky G, Fineberg N, Hathaway DR. Vascular injury, repair, and restenosis after percutaneous transluminal angioplasty in the atherosclerotic rabbit. Circulation. 1995;92:2995–3005. doi: 10.1161/01.cir.92.10.2995. [DOI] [PubMed] [Google Scholar]
- 6.Reidy MA. Factors controlling smooth muscle cell proliferation. Arch Pathol Lab Med. 1992;116:1276–1280. [PubMed] [Google Scholar]
- 7.Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;100:S87–S89. [PubMed] [Google Scholar]
- 8.Zhu B, Kuhel DG, Witte DP, Hui DY. Apolipoprotein E inhibits neointimal hyperplasia after arterial injury in mice. Am J Pathol. 2000;157:1839–1848. doi: 10.1016/S0002-9440(10)64823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wientgen H, Thorngate FE, Omerhodzic S, Rolnitzky L, Fallo JT, Williams DL, Fisher EA. Subphysiologic apolipoprotein E (ApoE) plasma levels inhibit neointimal formation after arterial injury in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:1460–1465. doi: 10.1161/01.ATV.0000134297.61979.3c. [DOI] [PubMed] [Google Scholar]
- 10.Moore ZWQ, Zhu B, Kuhel DG, Hui DY. Vascular apolipoprotein E expression and recruitment from circulation to modulate smooth muscle cell response to endothelial denudation. Am J Pathol. 2004;164:2109–2116. doi: 10.1016/S0002-9440(10)63769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishigami M, Swertfeger DK, Granholm NA, Hui DY. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J Biol Chem. 1998;273:20156–20161. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- 12.Ishigami M, Swertfeger DK, Hui MS, Granholm NA, Hui DY. Apolipoprotein E inhibition of vascular smooth muscle cell proliferation but not the inhibition of migration is mediated through activation of inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2000;20:1020–1026. doi: 10.1161/01.atv.20.4.1020. [DOI] [PubMed] [Google Scholar]
- 13.Swertfeger DK, Bu G, Hui DY. Low density lipoprotein receptor-related protein mediates apolipoprotein E inhibition of smooth muscle cell migration. J Biol Chem. 2002;277:4141–4146. doi: 10.1074/jbc.M109124200. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Y, Hui DY. Apolipoprotein E binding to low density lipoprotein receptor-related protein-1 inhibits cell migration via activation of cAMP-dependent protein kinase A. J Biol Chem. 2003;278:36257–36263. doi: 10.1074/jbc.M303171200. [DOI] [PubMed] [Google Scholar]
- 15.Swertfeger DK, Hui DY. Apolipoprotein E receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J Biol Chem. 2001;276:25043–25048. doi: 10.1074/jbc.M102357200. [DOI] [PubMed] [Google Scholar]
- 16.Kothapalli D, Fuki I, Ali K, Stewart SA, Zhao L, Yahil R, Kwiatkowski D, Hawthorne EA, FitzGerald GA, Phillips MC, Lund-Katz S, Pure E, Rader DJ, Assoian RK. Antimitogenic effects of HDL and APOE mediated by Cox-2-dependent IP activation. J Clin Invest. 2004;113:609–618. doi: 10.1172/JCI19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu B, Reardon CA, Getz GS, Hui DY. Both apolipoprotein E and immune deficiency exacerbate neointimal hyperplasia after vascular injury in mice. Arterioscler Thromb Vasc Biol. 2002;22:450–455. doi: 10.1161/hq0302.105377. [DOI] [PubMed] [Google Scholar]
- 18.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJR, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric oxide synthease in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- 20.Lee JS, Adrie C, Jacob HJ, Roberts JD, Jr, Zapol WM, Bloch KD. Chronic inhalation of nitric oxide inhibits neointimal formation after balloon-induced arterial injury. Circ Res 1996. 1996;78:337–342. doi: 10.1161/01.res.78.2.337. [DOI] [PubMed] [Google Scholar]
- 21.Yan Z-Q, Yokota T, Zhang W, Hansson GK. Expression of inducible nitric oxide synthase inhibits platelet adhesion and restores blood flow in the injured artery. Circ Res. 1996;79:38–44. doi: 10.1161/01.res.79.1.38. [DOI] [PubMed] [Google Scholar]
- 22.Ziche M, Morbidelli L, Masini E, Smerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099–3104. doi: 10.1161/01.cir.103.25.3099. [DOI] [PubMed] [Google Scholar]
- 24.Ihrig M, Dangler CA, Fox JG. Mice lacking inducible nitric oxide synthase develop spontaneous hypercholesterolemia and aortic atheromas. Atherosclerosis. 2001;156:103–107. doi: 10.1016/s0021-9150(00)00636-5. [DOI] [PubMed] [Google Scholar]
- 25.Chyu KY, Dimayuga P, Zhu J, Nilsson J, Kaul S, Shah PK, Cercek B. Decreased neointimal thickening after arterial wall injury in inducible nitric oxide synthase knockout mice. Circ Res. 1999;85:1192–1198. doi: 10.1161/01.res.85.12.1192. [DOI] [PubMed] [Google Scholar]
- 26.Koglin J, Glysing-Jensen T, Mudgett JS, Russell ME. Exacerbated transplant arteriosclerosis in inducible nitric oxide-deficient mice. Circulation. 1998;97:2059–2065. doi: 10.1161/01.cir.97.20.2059. [DOI] [PubMed] [Google Scholar]
- 27.Kuhel DG, Zhu B, Witte DP, Hui DY. Distinction in genetic determinants for injury-induced neointimal hyperplasia and diet-induced atherosclerosis in inbred mice. Arterioscler Thromb Vasc Biol. 2002;22:955–960. doi: 10.1161/01.atv.0000017994.77066.75. [DOI] [PubMed] [Google Scholar]
- 28.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarkar R, Gordon D, Stanley JC, Webb RC. Cell cycle effects of nitric oxide on vascular smooth muscle cells. Am J Physiol. 1997;272:H1810–H1818. doi: 10.1152/ajpheart.1997.272.4.H1810. [DOI] [PubMed] [Google Scholar]
- 30.Yu SM, Hung LM, Lin CC. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway. Circulation. 1997;95:1269–1277. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]