

Abstract
Although the anticancer effects of selenium have been demonstrated in clinical, preclinical, and laboratory studies, the underlying mechanism(s) remain unclear. Our previous study demonstrated that sodium selenite induced LNCaP human prostate cancer cell apoptosis in association with production of reactive oxygen species, alteration of cell redox state, and mitochondrial damage. In the present study, we demonstrated that selenite-induced apoptosis was superoxide-mediated and p53-dependent via mitochondrial pathways. In addition, we also demonstrated that superoxide production by selenite was p53-dependent. Our study showed that wild-type p53 expressing LNCaP cells were more sensitive to selenite-induced apoptosis than p53-null PC3 cells. Selenite treatment resulted in high levels of superoxide production in LNCaP cells, but only low levels in PC3 cells. LNCaP cells also showed sequential increases in levels of phosphorylated p53 (Serine15), total p53, Bax, and p21Waf1 proteins following selenite treatment. The effects of selenite were suppressed by pre-treatment with a synthetic superoxide dismutase mimic or by knockdown of p53 via RNA interference. LNCaP cells treated with selenite also showed p53 translocation to mitochondria, cytochrome c release into the cytosol, and activation of caspase 9. On the other hand, restoration of wild-type p53 expression in PC3 cells increased cellular sensitivity to selenite and resulted in increased superoxide production, caspase 9 activation, and apoptosis following selenite treatment. These results suggest that selenite induces apoptosis by producing superoxide to activate p53 and to induce p53 mitochondrial translocation. Activation of p53 in turn synergistically enhances superoxide production and apoptosis induced by selenite.
Keywords: apoptosis, mitochondria, p53 tumor suppressor, prostate cancer, redox regulation, selenite, superoxide
The abbreviations used are: CuZnSOD, copper and zinc-containing superoxide dismutase; H2AX, phosphorylated histone H2AX (serine 139); GSH, reduced glutathione; GSSG, glutathione disulfide; GPx, glutathione peroxidase; MnSOD, manganese-containing superoxide dismutase; MnTMPyP, manganese (III) tetrakis (N-methyl-2-pyridyl) porphyrin; MOI, multiplicity of infectivity; MTT, 3-[4,5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2 tetrazolium bromide; RLU, relative light unit; ROS, reactive oxygen species; Sel, selenite; siRNA, small interfering RNA; SOD, superoxide dismutase
INTRODUCTION
Selenium, an essential nutritional trace element, is a key element in maintenance of the activity of some antioxidant enzymes and redox-regulatory proteins (1, 2). A number of epidemiological studies have shown an inverse association between selenium levels and cancer risks in humans (3–6). Studies found that individuals with higher selenium levels in toenails or serum had significant reduction in prostate cancer incidence and the risk of advanced prostate cancer (7, 8). The most compelling findings were from a double-blind, placebo-controlled, randomized cancer prevention trial conducted by Clark et al. (9). In this study, human subjects given a daily dose of 200 μg of selenium supplementation showed a 63% decrease in prostate cancer incidence compared with the placebo group. The results of this study led to a current larger phase III, double-blind, placebo-controlled clinical trial, Selenium and Vitamin E Chemoprevention Trial (SELECT) (10).
At present, the anticancer mechanism(s) of selenium are still unclear. Several mechanisms have been postulated: 1) maintenance of glutathione peroxidase (GPx) activity to protect against oxidative damage to DNA, proteins, and membrane lipids; 2) prevention and/or detoxification of carcinogenic intermediate metabolites of chemical carcinogens; 3) stimulation of the immune system; 4) effects of selenium intermediate metabolites on the cell cycle and apoptosis; 5) modulation of thioredoxin reductase activity and cell reduction/oxidation (redox) state, which in turn regulate cell signal transduction, transcription factor activation and DNA repair; and 6) inhibition of angiogenesis (1,2,11,12). Among these potential mechanisms of selenium action, cell cycle regulation and apoptosis have received the most attention and have been postulated to be critical for cancer chemoprevention by selenium compounds (13). Although apoptotic effects of selenium are dramatic and reproducible in experimental studies, the exact pathways have yet to be elucidated. There is an increasing interest in cell redox modulation by selenium because studies have shown that selenium treatment altered the intracellular redox state, resulting in alteration in the activity of transcription factors, cell cycle regulatory proteins, signal transduction molecules, and cell apoptosis (14–20).
Our previous study showed that selenite induced apoptosis and growth inhibition of human prostate cancer cells in association with production of reactive oxygen species (ROS), alterations in levels of intracellular reduced glutathione (GSH) and glutathione disulfide (GSSG), and mitochondrial damage (15). Selenite treatment also up-regulated the antioxidant enzymes manganese superoxide dismutase (MnSOD), copper-zinc superoxide dismutase (CuZnSOD), GPx, and the cell cycle arrest gene p21Waf1. These effects of selenite were suppressed by a synthetic superoxide dismutase (SOD) mimic. A number of studies from other laboratories have confirmed that selenite-induced apoptosis was mediated by ROS production (21–25). We also demonstrated that overexpression of MnSOD prevented cell death from selenite treatment (26). In addition, studies from other laboratories have demonstrated that selenium compounds modulated p53 (14, 27–29). Taken together, these results suggest that superoxide and p53 may be involved in apoptosis induced by selenite.
The aims of the present study were to investigate the role of p53 and superoxide and apoptotic pathways in selenite-induced apoptosis in human prostate cancer cells. We compared cellular sensitivity to selenite between wild-type (wt) p53-containing LNCaP and p53-null PC3 human prostate cancer cell lines and determined that downregulation or reexpression of wt-p53 altered the cellular effects of selenite. We used an SOD mimic MnTMPyP to determine that activation of p53 by selenite treatment was mediated by superoxide production via posttranscriptional modification. Finally, we determined that superoxide production by selenite was also p53-dependent and apoptosis induced by selenite was via mitochondrial pathways.
MATERIAL AND METHODS
Chemicals and antibodies
Sodium selenite and anti-β-actin antibody were purchased from Sigma Chemical Co. (St. Louis, MO). Lucigenin (bis-N-methylacridinium nitrate) was purchased from Molecular Probes (Eugene, OR). Manganese (III) tetrakis (N-methyl-2-pyridyl) porphyrin (MnTMPyP) was purchased from Alexis Biochemicals (San Diego, CA). p53 and Bax siRNAs and anti-p53 (ser15) antibody were purchased from Cell Signaling Technology (Beverly, MA). siRNA Duplex control (non-silence) and RNAiFect™ Transfection Reagent were purchased from QIAGEN (Valencia, CA). Apoptotic DNA-Ladder kit was purchased from Roche Diagnostic Co. (Indianapolis, IN). Caspase-Glo™ 9 Assay kit was purchased from Promega Co. (Madison, WI). SuperSignal® West Pico Stable Peroxide and Luminol/Enhancer Solutions, M-per® Mammalian Protein Extraction Reagent, and Mitochondria Isolation Kit were purchased from Pierce Biotechnology Inc. (Rockford, IL). Anti-p21Waf1 (C-19) antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-Bax and anti-phosphohistone H2AX (Ser139) antibodies were purchased from Upstate USA Inc. (Charlottesville, VA).
Cell culture
LNCaP (ATCC CRL-1740) and PC3 (ATCC CRL-1435) cells were obtained from the American Type Culture Collection (Manassas, VA) and routinely maintained in 100-mm tissue culture dishes (Corning) in RPMI 1640 supplemented with 5% heat-inactivated fetal bovine serum and 1% antibiotic-antimycotic (Life Technologies, Inc., Rockville, MD) at 37°C in a humidified atmosphere of 95% air and 5% CO2. For biochemical analyses, cells were collected by rinsing in PBS three times, scraping with a rubber policeman in 10-ml PBS, and then centrifuging at 2000 rpm for 5 min. After removing the PBS, the cell pellets were stored at −80°C until use.
Superoxide measurement
Lucigenin-dependent chemiluminescence (CL) in cells was measured by a modified method as described previously (30). The stock solution of lucigenin (10 mM) was prepared in PBS and stored at −20°C in the dark. Lucigenin (100 μM) was added to 1×105 cells in 100 μL PBS, and incubated with or without 5 μM MnTMPyP for 30 min before selenite treatment. The reaction was initiated by the addition of lucigenin and Se to the cells and the CL level was monitored as relative light unit (RLU) using a Lumat LB9501 luminometer (Berthold Tech USA, Oak Ridge, TN) for a total period of 15 min at 30 sec intervals.
Flow cytometric analysis
Cell samples were prepared and analyzed as described previously (15). After selenite treatment and trypsinization, 1 × 106 cells were washed with PBS/EDTA/BSA buffer (PBS, 1 mm EDTA, and 0.1% BSA) and fixed in 100 μl of PBS/EDTA/BSA buffer plus 900 μl of 70% ethanol for 30 min at −20°C. After washing with phosphate-citric acid buffer (0.192 m Na2HPO4 and 4 mm citric acid, pH 7.8), the cells were stained in 500 μl of propidium iodide staining solution (33 μg/ml propidium iodide, 200 μg/ml DNase-free RNase A, and 0.2% Triton X-100) overnight at 4°C. Both cell cycle distribution and apoptotic cells were simultaneously measured in a Becton Dickinson FACScan flow cytometer (San Jose, CA) using 488-nm laser excitation.
Apoptotic DNA ladder analysis
DNA isolation and gel electrophoresis were performed according to the manufacturer’s instructions. Briefly, after selenite exposure, cells were scraped in PBS and harvested by centrifugation at 500 × g for 5 min at room temperature and then lysed in 400 μL lysis buffer for 10 min at room temperature. Following an addition of 100 μL isopropanol, the lysate was centrifuged through a filter and washed with the washing buffer. Genomic DNA was eluted with 100 μL elution buffer. DNA samples were loaded onto a 1.5% agarose gel containing 0.1 mg/ml ethidium bromide and electrophoresed. The gel was photographed with Kodak Image Station 2000R using UV illumination and digitized with Kodak 1D 3.6 software (Eastman Kodak Company, Rochester, NY).
MTT assay
Cells were seeded at 1 × 105 cells/well in 24-well plates overnight before treatment with different agents and then allowed to grow for an additional 5 days. MTT solution (10 μl; 5 mg/ml in PBS) was added to each well of the plate and incubated for 3 hr at 37°C. MTT lysis buffer (100 μl of 10% SDS, 45% dimethyl formamide, adjusted to pH 4.5 by glacial acetic acid) was then added to dissolve the formazan. The optical density was measured at 570 nm using a Beckman Coulter DU-640 Spectrophotometer (Beckman Coulter, Inc. Fullerton, CA). The percentage of viable cells was calculated as the relative ratio of optical density to the control.
Western blot analysis
Cell pellets were lysed with M-PER mammalian protein extraction reagent, and protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA). Cell lysates (20–50 μg) were electrophoresed in 12.5 % SDS polyacrylamide gels and then transferred onto nitrocellulose membranes. After blotting in 5% nonfat dry milk in Tween 20 Tris-buffered saline (TTBS), the membranes were incubated with primary antibodies at 1:1,000–2,000 dilutions in TTBS overnight at 4°C, and then secondary antibodies conjugated with horseradish peroxidase at 1:10,000 dilution in TTBS for 1 hr at room temperature. Protein bands were visualized on X-ray film using an enhanced chemiluminescence system (Pierce Biotechnology, Rockford, IL).
Small interfering RNA (siRNA) transfection
Cells were seeded at 1 × 105 cells/well in 6-well plates, and allowed to grow to 60% confluence. Cells were transfected with 50 nM of p53 or Bax siRNA with 2 μL RNAiFect™ Transfection reagent in 1 ml serum-free medium for 12 hr, and then 1 ml fresh medium with 10% FBS was added to each well for 24 hr before selenite treatment. Cells were also transfected with non-silencing, negative control siRNA which has no known homology to mammalian genes and allows assessing the possibility of non-specific gene silencing effects.
Adenoviral p53 transduction
PC3 cells were seeded at 4 × 105 in 60 mm tissue culture dishes for western blot analysis and at 1 × 105/well in 24-well plates for viability assay. Approximately 20 hr later, cells were infected with the indicated multiplicity of infection (MOI) of recombinant Ad5 CMV wt p53-GFP adenoviral constructs (Ad-p53) or with media alone (mock) in serum-free medium. After 12 hr, an equal volume of fresh medium with 10% FBS was added to each dish or well for 24 hr before selenite treatment. Cells were also transduced with adenoviral empty constructs (Ad empty) as a control.
Activity assay of caspase-9
Cells were seeded at 3 × 104 cells/well in a 96-well plate with 100 μL medium. Approximately 16 hr later, cells were treated with 2.5 μM selenite for 18 hr to induce apoptosis. Caspase-Glo™ 9 Reagent (100 μl) was directly added into each well to a final volume of 200 μl/well. Chemiluminescence was measured using a Tropix TR717™ Microplate Luminometer (Applied Biosystems, Bedford, MA).
Mitochondria fractionation
Cells were seeded at 6 × 105 cells in 100 mm tissue culture dishes, and allowed to grow to 60% confluence. Cells were treated with or without 2.5 μM selenite for 18 hr to induce apoptosis, and then mitochondria and cytosol fractions were separated from cells according to the manufacturer’s instructions (Pierce Biotechnology Inc., Rockford, IL).
Laser scanning confocal microscopy of p53 mitochondrial translocation
2000 cells were seeded in 8-well Lab-Tek Chamber Slides (Nalge Nunc International, Rochester, NY) in 400-μL culture medium. Twenty-four hr after plating, cells were treated with 2.5 μM selenite for 18 hr and then incubated with 200 nmol/L MitoTracker Red CM-H2XRos (Molecular Probes, Eugene, OR) in culture medium for 30 minutes. After washing 3 times with PBS, the cells were fixed in 4% formaldehyde for 30 minutes, permeabilized with 0.5% Triton X-100 for 10 minutes at room temperature and then incubated with a monoclonal anti-p53 antibody at a 1:100 dilution (Ab-6, EMD Biosciences, San Diego, CA) followed by incubation with anti-mouse IgG-FITC (BD Biosciences, San Diego, CA) at a 1:200 dilution for 1 hour at 37°C. After rinsing with PBS, the slides were mounted with a 90% glycerol medium. Images were immediately observed and captured using a BioRad MRC 1024 laser scanning confocal microscope at magnification 60X (Bio-Rad Laboratories, Hercules, CA).
Statistical analysis
All data were presented as means ± standard deviation (SD) from at least three sets of independent experiments. ANOVA analysis with Tukey’s multiple comparisons was used to determine the significance of statistical differences between data at the level of p <0.05 using SPSS computer statistics software (SPSS, Inc., Chicago, IL).
RESULTS
Superoxide-mediated apoptosis in selenite-treated LNCaP cells
LNCaP cells were treated with different doses of selenite for different time periods and cell viability was assessed by MTT assay. Selenite treatment decreased cell viability in a dose-dependent manner (Fig. 1A). Significant cell viability decreases occurred in cells treated with 1.5 μM and higher doses of selenite. Time-dependent decreases in viability which started at 24 hr were demonstrated in cells treated with 2.5 μM selenite over 120 hr (data not shown). Gel electrophoresis showed DNA laddering (fragmentation) in cells treated with 2.5 μM selenite, but not in cells treated with 0.5 μM selenite (Fig. 1B). Flow cytometry analysis demonstrated time-dependent increases in sub-G1 cell populations in cells treated with 2.5 μM selenite (Fig. 1C). These data demonstrated that cells underwent apoptosis following selenite treatment in dose- and time-dependent manners. To assess possible involvement of superoxide in selenite -induced apoptosis, cells were pre-treated with a synthetic SOD mimic, MnTMPyP. As shown in Fig. 1D, pre-treatment with MnTMPyP significantly reduced selenite -induced apoptosis in LNCaP cells. Using a lucigenin-dependent chemiluminescence assay for assessment of superoxide levels, chemiluminescence increased in cells treated with 2.5 μM selenite in 3 min and reached a peak value in 6 min. Chemiluminescence produced by selenite treatment was suppressed by MnTMPyP pre-treatment (Fig. 1E). There was only minimal chemiluminescence detected in a mixture of only the culture medium and lucigenin (data not shown). These results demonstrated that selenite treatment increased superoxide production to trigger apoptosis in LNCaP cells.
Figure 1.
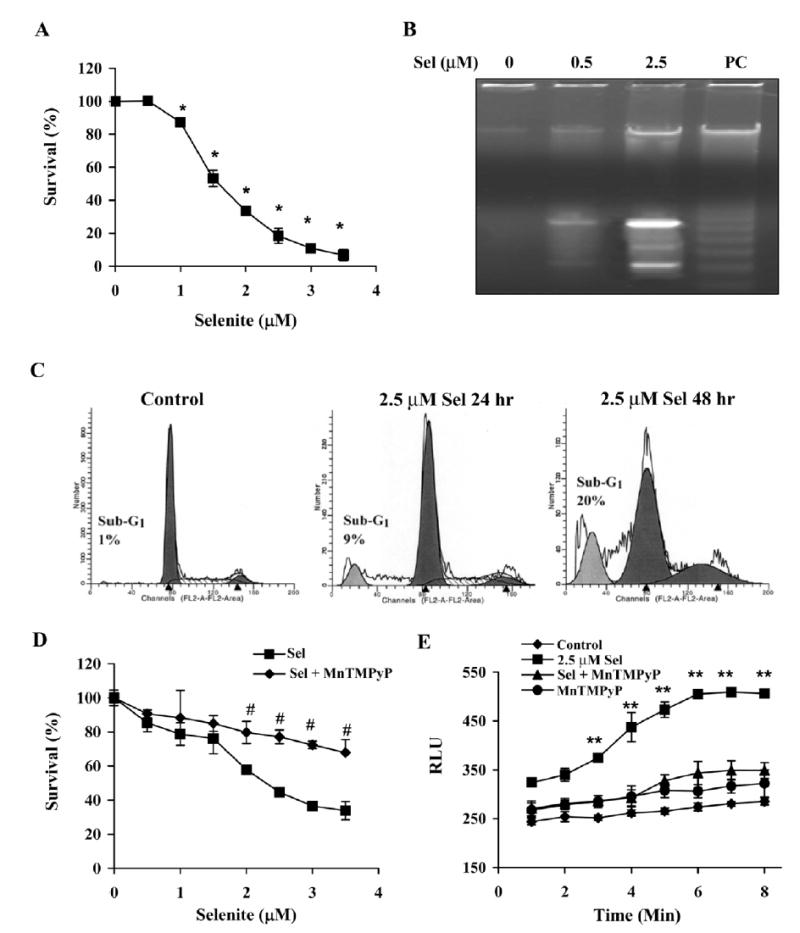
Selenite causes apoptosis with superoxide accumulation in wt-p53 expressing LNCaP cells. A, Dose-dependent effect of selenite on cell viability as demonstrated by the MTT assay. Cells were treated with 0-3.5 μM selenite for 5 days. B, Agarose gel electrophoretic detection of DNA fragmentation as a marker of cell apoptosis induced by selenite. Cells were treated with 0–2.5 μM selenite for 24 hr. C, Flow cytometry analyzed cell apoptosis by measuring the sub-G1 cell population. Cells were treated with 2.5 μM selenite for 24 and 48 hr. D, The SOD mimic MnTMPyP protected against cytotoxicity of selenite. Cells were treated with 0–3.5 μM selenite with or without 5 μM MnTMPyP for 5 days. E, Chemiluminescence assay showing production of superoxide radicals in cells treated with selenite. Cells were treated with 2.5 μM selenite, 5 μM MnTMPyP or selenite plus MnTMPyP. The treatment agents were added into the cell suspensions in test tubes and chemiluminescence was immediately measured. Data are presented as means ± SD of three independent experiments. * p<0.05 compared with 0 μM; # p<0.05 compared with the corresponding concentration of selenite without MnTMPyP; ** p<0.05 compared with control (cells without treatment), MnTMPyP, Se + MnTMPyP, and Se at 1 min.
Activation of p53 in LNCaP cells by selenite
Our previous study demonstrated that p21Waf1 was upregulated in selenite -treated LNCaP cells (15), suggesting possible involvement of p53 in selenite-induced apoptosis. To determine whether p53 is activated by selenite treatment, western blot analysis was used to detect levels of total p53 and phosphorylated p53 and its target genes p21Waf1 and Bax. As shown in Fig. 2A, selenite treatment resulted in elevations of total and phosphorylated p53 at Serine 15 (p-p53 Ser 15) in LNCaP cells in a dose-dependent manner. Significant elevations of p53 occurred in cells treated with selenite at doses of 2.0 μM and higher. The effect of selenite treatment on p53 was also time-dependent (Fig. 2B). An elevation of total p53 was detected at 1 hr following 2.5 μM selenite treatment, reached a peak value at 6 hr, and was persistent up to 36 hr, though there was a slight decrease at 24 and 36 hr. An increase in p-p53 Ser15 was detected at 3 hr following selenite treatment, reached a peak value at 6 hr, and was maintained at a steady state up to 36 hr. Protein levels of both p21Waf1 and Bax were elevated in dose- and time-dependent manners, corresponding to the elevation of p53 observed following selenite treatment (Fig. 2A and B). The effects of selenite on these proteins were suppressed by MnTMPyP pre-treatment (Fig. 2C). It has been previously reported that selenite treatment caused DNA damage (31–33). A DNA damage marker, phosphorylated histone H2AX on serine 139 (H2AX), was analyzed by western blotting (34). As shown in Fig. 2A and B, there were no significant increases in this phosphorylated protein in cells treated with different doses of selenite or 2.5 μM selenite for different times. This result suggests that DNA damage is not likely to be the mechanism of cell killing at the concentrations of selenite used in this study.
Figure 2.
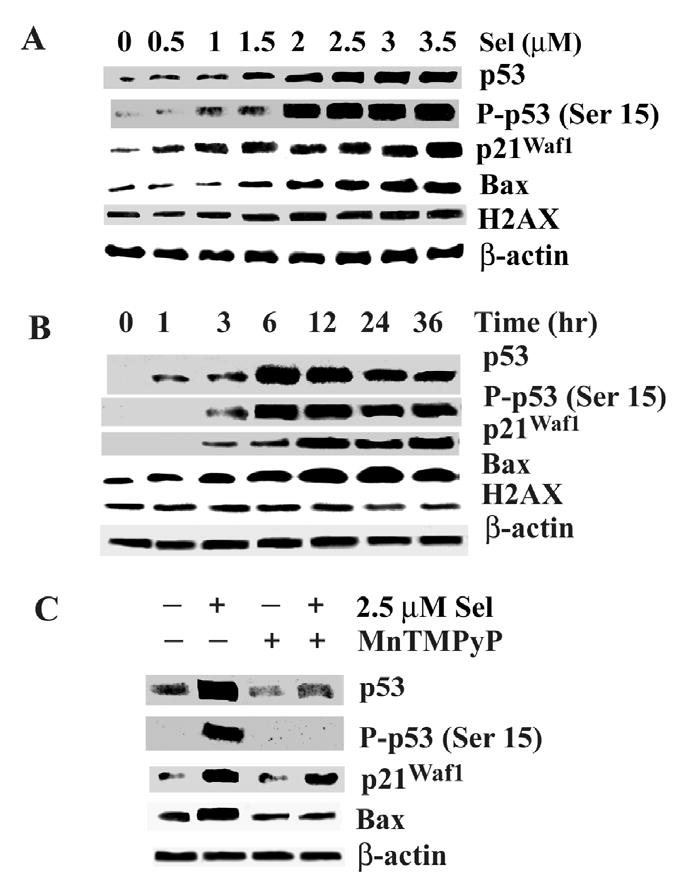
Western blot analysis of effects of selenite on expression of p53, p21Waf1, and Bax and phosphorylation of p53 and histone (H2AX) in LNCaP cells. A, Dose-dependent effect of selenite. Cells were treated with 0–3.5 μM selenite for 18 hr. B, Time-dependent effect of selenite. Cells were treated with 2.5 μM selenite for 0–36 hr. C, The SOD mimic MnTMPyP suppressed selenite effects on p53, p21Waf1, and Bax. Cells were treated with 2.5 μM selenite, 5 μM MnTMPyP, or combination of selenite and MnTMPyP for 18 hr. Protein loading: 40 μg for p53, p-p53 Ser15, p21Waf1, Bax, and H2AX and 20 μg for β-actin.
Role of p53 and Bax in selenite-induced apoptosis in LNCaP cells
We next determined the role of p53 and Bax in selenite -induced apoptosis using RNA interference to reduce mRNA of these two genes. As shown in Fig. 3A, cells transfected with p53 siRNA had decreased sensitivity to selenite, while selenite sensitivity did not change in cells transfected with the control siRNA. The western blot analysis showed that levels of total p53, p-p53 Ser 15, p21Waf1, and Bax increased in cells treated with selenite and these effects of selenite were suppressed by p53 siRNA transfection, but not by the negative control siRNA, indicating that the effects of p53 siRNA were specific and selective (Fig. 3B). However, p53 siRNA treatment only partially suppressed Bax expression, suggesting upregulation of Bax by selenite treatment may also be p53-independent. This conclusion is supported by the results of p53-null PC3 cells reported in the next figure, in which selenite treatment upregulated Bax in the absence of p53 expression. Although Bax siRNA treatment also reduced cellular sensitivity to selenite, the magnitude was much lower than that of p53 siRNA (Fig. 3C). The western blot analysis showed that Bax siRNA transfection decreased levels of Bax protein, but did not affect elevation of p53 and p21Waf1 induced by selenite treatment, indicating that Bax is a downstream gene. The results demonstrated that siRNA selectively downregulated p53 and Bax, respectively. Downregulation of p53 decreased the cellular sensitivity to selenite. Regulation of Bax was not completely p53-dependent and selenite-induced apoptosis was not completely mediated by Bax.
Figure 3.
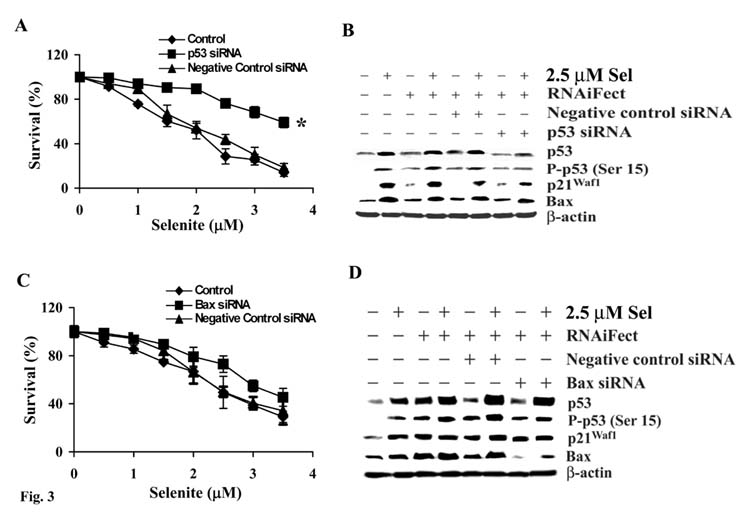
Downregulation of p53, but not Bax, by RNA interference causes resistance to selenite-mediated cytotoxicity in LNCaP cells. A, MTT assay of viability of LNCaP cells with p53 siRNA transfection and selenite treatment. B, Western blot analysis showing suppressive effects of p53 siRNA transfection on expression of p53, p21Waf1, and Bax in cells with and without selenite treatment. C, MTT assay of cellular response to selenite following downregulation of Bax by siRNA transfection. D, Western blot analysis of effects of Bax siRNA transfection on p53, p21Waf1, and Bax in cells with and without selenite treatment. Cells were transfected with 50 nM of p53 or Bax siRNA, respectively, for 36 hr and then treated with selenite for 5 days for cell viability analysis or with 2.5 μM selenite for 18 hr for Western blot analysis. Protein loading: 40 μg for p53, p-p53 Ser15, p21Waf1, Bax, and H2AX and 20 μg for β-actin. The data were obtained from three independent experiments and the results shown are the mean ± SD. *p<0.05 compared with RNAiFect and negative control siRNA.
Effect of wt-p53 on cellular response to selenite in p53-null PC3 cells
To verify that cellular sensitivity to selenite is dependent on p53, we next tested the sensitivity of p53-null PC3 cells to selenite and whether restoration of wt-p53 expression altered sensitivity of PC3 cells to selenite. Fig. 4A and B show dose- and time-dependent effects of selenite in PC3 cells. PC3 cells were twofold more resistant to selenite than LNCaP cells (Fig. 1A and B). To reduce cell viability by 50% required 1.5 μM selenite for LNCaP cells (Fig. 1A) and 3 μM selenite for PC3 cells (Fig. 4A). Time course studies showed that 50% of LNCaP cells were killed by 2.5 μM selenite at 48 hr (Fig. 1B), while only 20% of PC3 cells were killed at 48 hr and less than 40% of cells were killed at 120 hr (Fig. 4B). Western blot analyses showed that PC3 cells had no detectable p53 and p21Waf1 and had very low levels of Bax (Fig. 4C). Following transduction of Ad-p53, PC3 re-expressed p53 and p21Waf1 in a dose-dependent manner. At 0.5 MOI of Ad-p53 transduction, selenite treatment induced phosphorylation of p53 Ser15, whereas p53 Ser15 was also phosphorylated in cells transduced with 1 or higher MOI Ad-p53 without selenite treatment. Selenite treatment alone or transduction of control adenoviral constructs alone significantly increased levels of Bax. Levels of Bax were further increased in cells transduced with 2 or 4 MOI of Ad-p53. These data demonstrated that expression of p21Waf1 was p53-dependent, while Bax was not completely p53-dependent. These results were consistent with the results of LNCaP cells (Fig. 3B and D). Fig. 4D shows that re-expression of wt-p53 enhanced sensitivity of PC3 cells to selenite in a dose-dependent manner. Transduction of control adenoviral constructs alone did not alter cellular response to selenite
Figure 4.
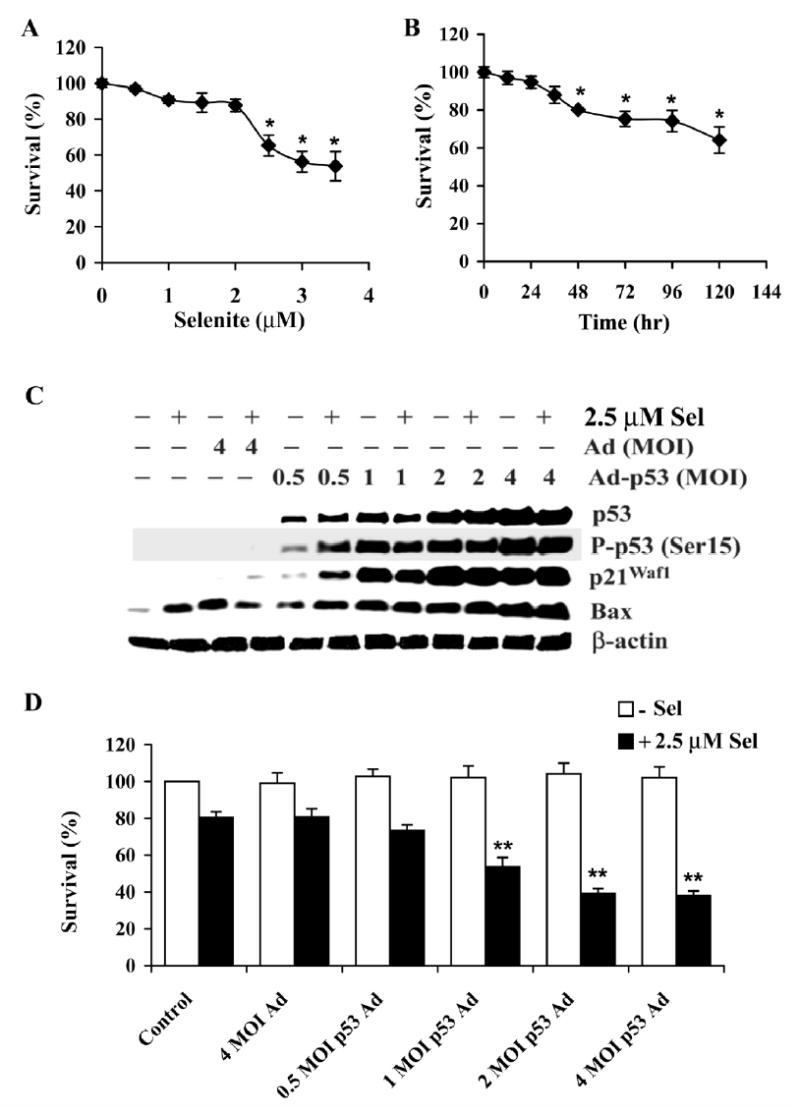
Forced p53 expression sensitizes PC3 cells to selenite-mediated cytotoxicity. A, MTT assay of a dose-dependent effect of selenite on cell viability. Cells were treated with selenite for 5 days. B, MTT assay of a time-dependent effect of selenite on cell viability. Cells were treated with 2.5 μM selenite. C, Western blot analysis of levels of p53, p-p53 Ser 15, p21Waf1, and Bax in cells following Ad-p53 transfection and selenite treatment. D, MTT assay of effect of p53 on viability of cells with or without selenite treatment. Cells were transduced with Ad-p53 constructs for 36 hr and then treated with 2.5 μM selenite for 18 hr for western blot analysis and for 5 days for cell viability analysis. The data were obtained from three independent experiments and the results shown are the mean± SD. *p<0.05 compared with 0 μM selenite or 0 hr. ** p<0.05 compared with control and 4 MOI Ad.
Dependence of selenite-induced superoxide production on p53 status in LNCaP and PC3 cells
Since selenite-induced cell apoptosis was more dependent on p53 than Bax, we next analyzed other mechanisms by which selenite induced p53-dependent apoptosis. We first reduced levels of p53 in LNCaP cells by siRNA transfection. As shown in Fig. 5A, transfection of p53 siRNA suppressed selenite-induced elevation of superoxide, though p53 siRNA alone increased levels of superoxide. The latter was mostly likely due to the combined effects of RNAiFect and siRNA, suggesting that the transfection reagent may cause oxidative stress in cells. Similar effects on protein levels of p53 and p21Waf1 were also seen in RNA interference studies (Fig. 3B and D). In contrast, re-expression of wt-p53 in PC3 cells significantly increased superoxide production following selenite treatment (Fig. 5B). There were no significant changes in superoxide production in cells treated with 0.5 μM selenite (data not shown). These results demonstrated that superoxide production and apoptosis by selenite treatment were enhanced by p53. Superoxide may act as a downstream mediator of apoptosis as well as a mediator of p53 phosphorylation and activation.
Figure 5.
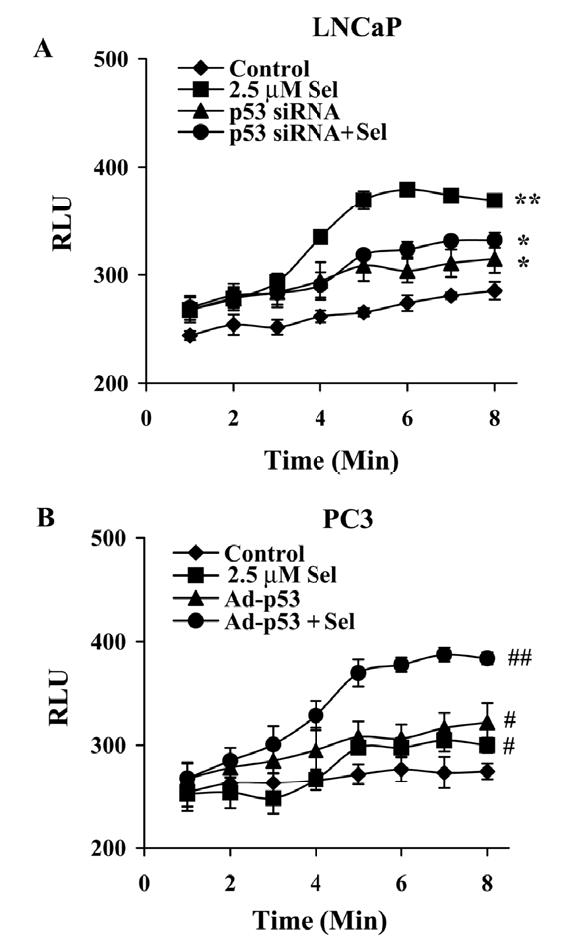
p53 modulates selenite-induced production of superoxide radical in LNCaP and PC3 cells. A, Chemiluminescence assay of suppression of superoxide production by p53 siRNA transfection in LNCaP cells treated with selenite. B, Enhancement of superoxide production by Ad-p53 transduction in PC3 cells treated with selenite. Cells were transfected with 50 nM p53 siRNA or transduced with 4 MOI Ad-p53 for 36 hr and then treated with 2.5 μM selenite in suspension. Superoxide production was immediately measured using a luminometer. The data were obtained from three independent experiments and the results shown are the mean± SD. *p<0.05 control (cells without treatment) vs. p53 siRNA and p53 siRNA + Se. ** p<0.05 2.5 μM Se vs. control, p53 siRNA, and p53 siRNA + Se. #p<0.05 control (cells without treatment) vs. 2.5 μM Se and Ad-p53. ##p<0.05 Ad-p53 + Se vs. control, 2.5 μM Se, and Ad-p53.
p53-dependent, superoxide-mediated mitochondrial pathways of apoptosis induced by selenite treatment
To explore involvement of mitochondrial pathways in cell apoptosis, mitochondria were isolated from the cytosol of LNCaP cells. Mitochondrial translocation of p53 and cytochrome c release from the mitochondria into the cytosol were determined by the western blot analysis. As shown in Fig. 6A, protein levels of p53, p-p53 Ser 15, p21Waf1, and Bax increased in LNCaP cells following treatment with 2.5 μM selenite for 18 hr. In addition, a substantial amount of p53 translocated to mitochondria. The levels of Bax in the mitochondria also increased, but the magnitude was much less than p53. There was also a substantial amount of cytochrome c release into the cytosol. To verify mitochondrial translocation of p53, we performed indirect immunofluorescence staining for p53 using a monoclonal antibody and labeled mitochondria with a fluorescent marker, MitoTracker, to analyze co-localization between p53 and the MitoTracker in LNCaP cells. Fig. 6B shows that mitochondria in the control and 2.5 μM selenite-treated LNCaP cells were labeled in red by Mitotracker and an elevation of p53 in LNCaP cells at 12 hr following 2.5 μM selenite treatment was demonstrated by the immunofluorescent stain using a monoclonal anti-p53 antibody and FITC-conjugated anti-mouse IgG as shown in green. Fig. 6B (overlay) shows increased orange fluorescence in LNCaP cells treated with selenite, indicating a co-localization of MitoTracker (red fluorescence) and p53 (green fluorescence) and also indicating mitochondrial translocation of p53. The results confirmed that the level of p53 increased and the translocation of p53 to mitochondria occurred in intact LNCaP cells following selenite treatment.
Figure 6.
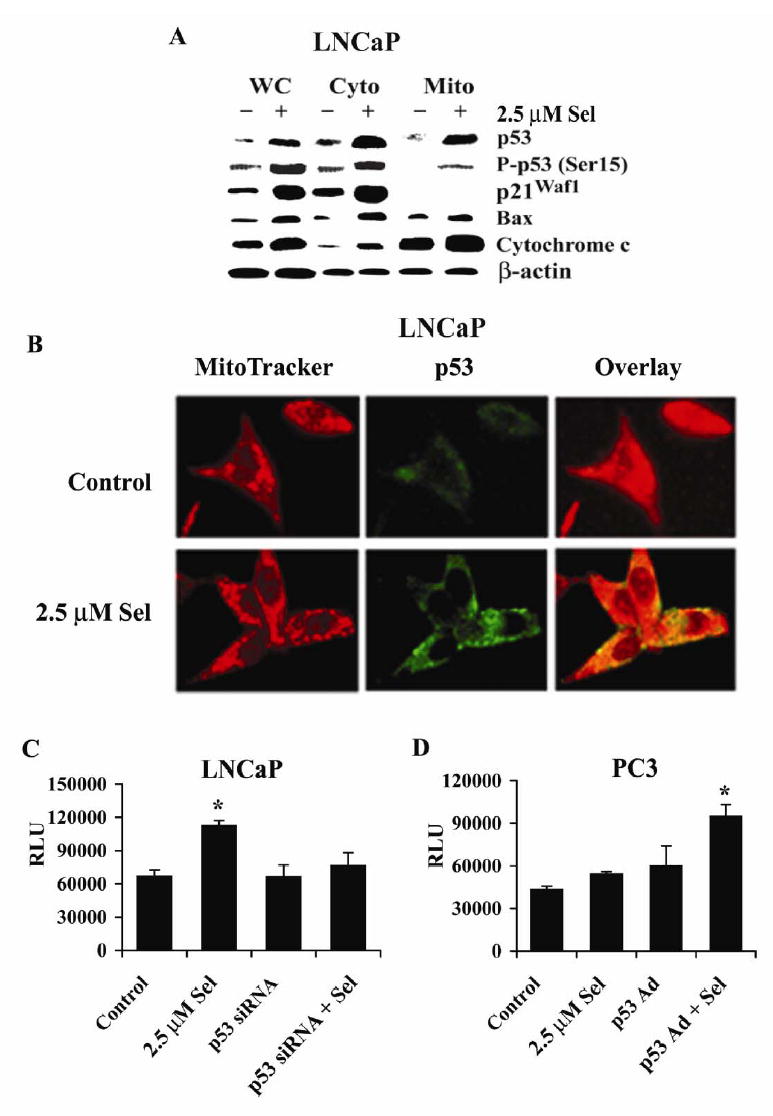
Selenite causes p53 mitochondrial translocation, cytochrome c release from mitochondria, and activation of caspase 9 in LNCaP and PC3 cells. A, Western blot analysis of selenite-induced p53 accumulation in mitochondria and release of cytochrome c into cytosol in LNCaP cells. WC, whole cell lysate; Cyto, cytosol extract; and Mito, mitochondrial extract. Cells were treated with 2.5 μM selenite for 18 hr. p21Waf1 was used as a control for the purity of mitochondrial extracts and also for p53 transcriptional activity. β-actin was used as a control for equal sample loading. Protein loading: 40 μg for p53, p-p53 Ser15, p21Waf1, Bax, and cytochrome c, and 20 μg for β-actin. B, Laser scanning confocal microscopic photographs showing co-localization of p53 and MitoTracker in mitochondria of LNCaP cells. Red, MitoTracker as a mitochondrial maker; green, p53 labeled with a p53 antibody and a FITC-conjugated secondary antibody; overlay, merging of MitoTracker (red) and p53 (green). C, Chemiluminescence assay of activation of caspase 9 by selenite treatment and the suppressive effect of p53 siRNA transfection in LNCaP cells. D, Chemiluminescence assay of activation of caspase 9 by selenite treatment and the enhancing effect of Ad-p53 transduction in PC3 cells. Cells were transfected with 5 nM p53 siRNAs or transduced with 4 MOI p53 Ad for 36 hr and then treated with 2.5 μM selenite for 18 hr. Chemiluminescence was measured using a luminometer. The data were obtained from three independent experiments and the results shown are the mean± SD. *p<0.05 compared with control (cells without treatment), p53 siRNA, and p53 siRNA+Se or control (LNCaP), Se, and Ad-p53 (PC3).
We next analyzed caspase 9 activity with a chemiluminescence assay. As shown in Fig. 6C, caspase 9 activity significantly increased in LNCaP cells treated with 2.5 μM selenite for 18 hr. Activation of caspase 9 by selenite treatment was suppressed by transfection of p53 siRNA. In contrast to LNCaP cells, PC3 cells showed no significant increase in caspase 9 activity following selenite treatment (Fig. 6D). After restoration of wt-p53 expression by andenoviral transduction, selenite treatment significantly increased caspase 9 activity in PC3 cells (Fig. 6D). A mixture of culture media and chemical reagents without cells produced only minimal background chemiluminescence (data not shown). These data demonstrate that apoptosis induced by selenite was p53-dependent via mitochondrial pathways.
DISCUSSION
Studies have demonstrated that ROS, particularly superoxide, are produced by several selenium compounds when they interact with thiols (21). The prooxidant effects of these selenium compounds have been proposed to be one of the mechanism(s) by which selenium exerts the anticancer effect (21). Selenite is one of these selenium compounds. Metabolism of selenite to elemental selenium is involved in oxidation of GSH and superoxide production in biologic systems (1, 21, 35). Proposed mechanisms are:
| (A) |
| (B) |
| (C) |
Balances in equations A, B, and C are incomplete. As shown in equation A, selenite nonenzymatically reacts with GSH to form selenodiglutathione (GSSeSG) and glutathione disulfide (GSSG). GSSeSG reacts with NADPH or GSH to produce hydrogen selenide (H2Se) in equation B. Hydrogen selenide is oxidized by O2 to produce elemental selenium (Se0) and superoxide radical (O2·−) in equation C. It has been proposed that the intermediate metabolite selenotrisulfide from interaction of selenite with GSH may also produce superoxide and other ROS (21). One study reported that selenocystamine (RSeSeR) can interact with GSH to form the reduced diselenide (RSe−) that interacts with O2 to produce superoxide (35), suggesting that the selenopersulfide anion (GSSe−) formed from selenite may react with O2 to produce superoxide in a similar pathway (21). Therefore, selenite redox catalysis can result in oxidation of GSH and superoxide production with resultant oxidative shift in the redox state of cells. Superoxide is most likely to play a major role in selenite-induced prooxidant effect in cells. Several studies have demonstrated that some selenium compounds can produce superoxide and induce cell apoptosis (21–25). Selenite-induced cell death can be inhibited by treatment with a SOD mimic or by overexpression of MnSOD (15, 26). Studies also found that selenium compounds with superoxide production generally have better anticancer activity (21), suggesting that the subtoxic yet prooxidative effect of selenium compounds may be the anticancer effect of selenium by induction of cell cycle arrest and apoptosis (13, 14).
Our study showed that selenite treatment resulted in immediate elevation of superoxide, subsequent p53 activation, and apoptosis of LNCaP cells. These effects of selenite were inhibited by the superoxide scavenger MnTMPyP or by MnSOD overexpression as demonstrated by this and previous studies (15, 26). These results suggest that superoxide acts as a mediator for p53 activation and apoptosis induced by selenite treatment. Activation of p53 was via posttranslational modification evidenced by p53 phosphorylation and upregulation of p21Waf1 and Bax by the western blot analysis. One study reported that apoptosis induced by selenite was mediated by DNA damage with an involvement of the ataxia-telangiectasia mutated (ATM) protein and induction of H2AX phosphorylation (31). Our study shows that selenite treatment did not induce H2AX phosphorylation and pretreatment with caffeine, an ATM inhibitor, did not alter the effects of selenite (data not shown), suggesting that DNA damage was not the underlying mechanism of selenite-induced p53 activation and cell apoptosis in our study. These experimental differences are most likely due to the different doses of selenite used between studies (31). The doses of selenite used by others were 100 and 500 μM, which were extremely toxic levels of selenite to all cells and therefore induced DNA damage. In our study, we used only 2.5 and 3 μM selenite which were 30- to 160-fold lower, but higher than the requirement for maintenance of the levels of GPx in tissue culture (15). The doses of selenite used in our studies most likely induced a prooxidative effect resulting in an oxidative shift in the cell redox state and growth inhibition or apoptosis as described instead of DNA damage. Our results suggest that selenite treatment alters the cell redox state by production of superoxide and activates p53 triggering apoptosis.
The tumor suppressor p53 protein plays an important role in DNA repair, cell cycle arrest, and apoptosis (35–38). Induction of apoptosis is considered to be central to the tumor suppressive function of p53. Through transcription-dependent pathways, p53 functions as a transactivator to up-regulate downstream proapoptotic genes, such as Bax, and/or functions as a repressor to down-regulate antiapoptotic genes, such as Bcl-2, promoting apoptosis. Through transcription-independent pathways, p53 can translocate to mitochondria in response to DNA damage or other stressors, resulting in apoptosis via interaction with anti-apoptotic Bcl-2 and Bcl-XL proteins that alter the mitochondrial membrane potential and induce cytochrome c release into the cytosol with resultant caspase activation (39). p53-dependent apoptosis has also been demonstrated to be mediated by ROS (40, 41). Apoptosis triggered by p53 has been reported to be dependent on an increase in ROS and the release of apoptotic factors from mitochondrial damage (42). These studies suggest that ROS are downstream mediators in p53-dependent apoptosis in transcription-dependent or independent pathways. ROS are known to play an important role in apoptosis (43, 44). When cells are exposed to oxidative stress, such as hypoxia or genomic damage, p53 is expressed at high levels by post-translational modifications, including phosphorylation, acetylation and glycosylation (45, 46). These modifications occur rapidly and lead to the activation of p53, resulting in either G1 or G2/M cell cycle arrest or apoptosis. Therefore, ROS can function as p53 activators or p53 downstream effectors.
Studies have shown that selenite-induced cell apoptosis was associated with superoxide production and was suppressed by antioxidants, especially by MnSOD or SOD mimics (15, 23, 25, 26). Recent studies have demonstrated that selenite treatment upregulated p53 and induced p53 phosphorylation on Ser 15 to induce caspase-mediated apoptosis (27–29). However, the link between superoxide production and p53 regulation in selenite-induced apoptosis has not been elucidated. Our present study showed that superoxide levels dramatically increased in p53-expressing LNCaP cells following treatment with selenite. Meanwhile, protein levels of total p53 and p-p53 Ser 15, and its target genes p21Waf1 and Bax were also up-regulated by selenite treatment. These effects of selenite were suppressed by pre-treatment with the SOD mimic MnTMPyP. These results demonstrated that treatment with selenite produced superoxide and subsequently activated p53 via phosphorylation, suggesting that superoxide may be a mediator for p53 phosphorylation/activation in anticancer effects of selenite. Transfection of p53 siRNA downregulated p53, p21Waf1, and Bax, and decreased superoxide production by selenite in LNCaP cells. Conversely, PC3 cells transduced with Ad-p53 reexpressed wt-p53 and p21Waf1 elevated Bax protein, and produced high levels of superoxide following selenite treatment. Cell viability demonstrated that LNCaP cells transfected with p53 siRNA decreased sensitivity to selenite, while PC3 cells with restoration of p53 expression increased sensitivity to selenite. Our study demonstrated that superoxide production and apoptosis by selenite were p53-dependent and p53 synergistically enhanced superoxide production by selenite. These results suggest that p53 may function to regulate superoxide production from selenite treatment and superoxide may also act as a downstream effector for apoptosis. Therefore, p53 levels would affect selenite-induced apoptosis in cancer cells.
Results of p53 mitochondrial translocation, increased Bax protein, cytochrome c release, and caspase-9 activation suggest that Se-induced apoptosis is via p53-dependent mitochondrial pathways. Bax is a mitochondrial proapoptotic protein and is regulated by p53 (36–38, 47). Our results showed that Bax was upregulated in LNCaP cells corresponding to p53 upregulation and phosphorylation following selenite treatment or after restoration of p53 expression in PC3 cells. These findings are consistent with the results of previous studies demonstrating that Bax is a p53 target gene. In addition, our study demonstrated that Bax expression was also p53-independent. p53-null PC3 cells expressed low levels of Bax and Bax was upregulated by selenite treatment in the absence of p53 in PC3 cells. Bax siRNA transfection only slightly decreased selenite-induced apoptosis in LNCaP cells, though levels of Bax protein were downregulated. In contrast, p53 siRNA transfection and SOD mimic treatment were more effective in reducing apoptosis by selenite treatment. These results suggest that p53 and superoxide play an important role in selenite-induced apoptosis, while Bax plays a minor role.
In summary, our study demonstrates that induction of apoptosis by selenite is superoxide-mediated and p53-dependent via mitochondrial pathways. Activation of p53 by selenite treatment is mediated by superoxide production. On the other hand, p53 can synergistically enhance superoxide production and cell apoptosis by selenite. The mitochondrial translocation of p53 may alter mitochondrial functions with resultant increased levels of superoxide triggering apoptosis. Therefore, cells with wt-p53 expression are more sensitive to selenite than p53-null cells. These results suggest that superoxide production and p53 may enhance selenite-induced apoptosis, and are crucial for apoptotic chemoprevention of prostate cancer by selenite. Our results provide a link between superoxide and p53 in selenite-induced apoptosis in cancer cells. Our results also suggest that levels of intracellular antioxidants, particularly SOD may influence the chemopreventive efficacy of selenite.
Footnotes
Grant support: The University of Wisconsin-Madison, the American Cancer Society, and Grant CA1114281 (to W. Z.) from the National Cancer Institute. This study was also supported by the use of resources and facilities of the William S. Middleton Veterans Administration Hospital, Madison, WI.
References
- 1.Combs GF, Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79:179–92. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 2.Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–66. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- 3.Shamberger RJ, Tytko SA, Willis CE. Antioxidants and cancer. Part VI. Selenium and age-adjusted human cancer mortality. Arch Environ Health. 1976;31:231–5. doi: 10.1080/00039896.1976.10667225. [DOI] [PubMed] [Google Scholar]
- 4.Schrauzer GN, White DA, Schneider CJ. Cancer mortality correlation studies. III. Statistical association with dietary selenium intakes. Bioinorg Chem. 1977;7:35–56. doi: 10.1016/s0006-3061(00)80126-x. [DOI] [PubMed] [Google Scholar]
- 5.Clark LC, Cantor KP, Allaway WH. Selenium in forage crops and cancer mortality in US counties. Arch Environ Health. 1991;46:37–42. doi: 10.1080/00039896.1991.9937427. [DOI] [PubMed] [Google Scholar]
- 6.Brooks JD, Metter EJ, Chow DW, et al. Plasma selenium level before diagnosis and the risk of prostate cancer development. J Urol. 2001;166:2034–8. [PubMed] [Google Scholar]
- 7.Yoshizawa K, Willett WC, Morris SJ, et al. Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst. 1998;90:1219–24. doi: 10.1093/jnci/90.16.1219. [DOI] [PubMed] [Google Scholar]
- 8.Li HJ, Stampfer MJ, Giovannucci EL, et al. A prospective study of plasma selenium levels and prostate cancer risk. J Natl Cancer Inst. 2004;96:696–703. doi: 10.1093/jnci/djh125. [DOI] [PubMed] [Google Scholar]
- 9.Clark LC, Combs GF, Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group J Am Med Assoc. 1996;276:1957–63. [PubMed] [Google Scholar]
- 10.Klein EA, Thompson IM, Lippman SM, et al. SELECT: the next prostate cancer prevention trial. J Urol. 2001;166:1311–5. doi: 10.1016/s0022-5347(05)65759-x. [DOI] [PubMed] [Google Scholar]
- 11.Combs GF. Status of selenium in prostate cancer prevention. Br J Cancer. 2004;91:195–9. doi: 10.1038/sj.bjc.6601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whanger PD. Selenium and its relationship to cancer: an update. Br J Nutr. 2004;91:11–28. doi: 10.1079/bjn20031015. [DOI] [PubMed] [Google Scholar]
- 13.Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4:13–28. doi: 10.2174/1568009043481614. [DOI] [PubMed] [Google Scholar]
- 14.Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci USA. 2002;99:14548–53. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong W, Oberley TD. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001;61:7071–8. [PubMed] [Google Scholar]
- 16.Park HS, Park E, Kim MS, et al. Selenite inhibits the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) through a thiol redox mechanism. J Biol Chem. 2000;275:2527–31. doi: 10.1074/jbc.275.4.2527. [DOI] [PubMed] [Google Scholar]
- 17.Powis G, Gasdaska JR, Gasdaska PY, et al. Selenium and the thioredoxin redoxsystem: effects on cell growth and death. Oncol Res. 1997;9:303–12. [PubMed] [Google Scholar]
- 18.Kim IY, Stadtman TC. Inhibition of NF-kappaB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc Natl Acad Sci USA. 1997;94:12904–7. doi: 10.1073/pnas.94.24.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gopalakrishna R, Chen ZH, Gundimeda U. Selenocompounds induce a redox modulation of protein kinase C in the cell, compartmentally independent from cytosolic glutathione: its role in inhibition of tumor promotion. Arch Biochem Biophys. 1997;348:37–48. doi: 10.1006/abbi.1997.0335. [DOI] [PubMed] [Google Scholar]
- 20.McKenzie RC, Arthur JR, Beckett GJ. Selenium and the regulation of cell signaling, growth, and survival: molecular and mechanistic aspects. Antioxid Redox Signal. 2002;4:339–51. doi: 10.1089/152308602753666398. [DOI] [PubMed] [Google Scholar]
- 21.Spallholz JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med. 1994;17:45–64. doi: 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 22.Spallholz JE, Shriver BJ, Reid TW. Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: implications for the anticarcinogenic activity of L-selenomethionine and L-Se-methylselenocysteine. Nutr Cancer. 2001;40:34–41. doi: 10.1207/S15327914NC401_8. [DOI] [PubMed] [Google Scholar]
- 23.Shen H-M, Yang C-F, Ong C-N. Sodium selenite-induced oxidative stress and apoptosis in human hepatoma HepG2 cells. Int J Cancer. 1999;81:820–8. doi: 10.1002/(sici)1097-0215(19990531)81:5<820::aid-ijc25>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 24.Jung U, Zheng X, Yoon SO, Chung AS. Se-methylselenocysteine induces apoptosis mediated by reactive oxygen species in HL-60 cells. Free Radic Biol Med. 2001;31:479–89. doi: 10.1016/s0891-5849(01)00604-9. [DOI] [PubMed] [Google Scholar]
- 25.Kim TS, Yun BY, Kim IY. Induction of the mitochondrial permeability transition by selenium compounds mediated by oxidation of the protein thiol groups and generation of the superoxide. Biochem Pharmacol. 2003;66:2301–11. doi: 10.1016/j.bcp.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Zhong W, Yan T, Webber MM, et al. Alteration of cellular phenotype and responses to oxidative stress by manganese superoxide dismutase and a superoxide dismutase mimic in RWPE-2 human prostate adenocarcinoma cells. Antioxid Redox Signal. 2004;6:513–22. doi: 10.1089/152308604773934279. [DOI] [PubMed] [Google Scholar]
- 27.Lanfear J, Fleming J, Wu L, et al. The selenium metabolite selenodiglutathione induces p53 and apoptosis: relevance to the chemopreventive effects of selenium? Carcinogenesis. 1994;15:1387–92. doi: 10.1093/carcin/15.7.1387. [DOI] [PubMed] [Google Scholar]
- 28.Wei Y, Cao X, Qu Y, et al. SeO(2) induces apoptosis with down-regulation of Bcl-2 and up-regulation of p53 expression in both immortal human hepatic cell line and hepatoma cell line. Mutat Res. 2001;490:113–21. doi: 10.1016/s1383-5718(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 29.Jiang C, Hu H, Malewicz B, et al. Selenite-induced p53 Ser-15 phosphorylation and caspase-mediated apoptosis in LNCaP human prostate cancer cells. Mol Cancer Ther. 2004;3:877–84. [PubMed] [Google Scholar]
- 30.Li Y, Zhu H, Kuppusamy P, et al. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem. 1998;273:2015–23. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- 31.Zhou N, Xiao H, Li TK, et al. DNA damage-mediated apoptosis induced by selenium compounds. J Biol Chem. 2003;278:29532–7. doi: 10.1074/jbc.M301877200. [DOI] [PubMed] [Google Scholar]
- 32.Garberg P, Stahl A, Warholm M, et al. Studies of the role of DNA fragmentation in selenium toxicity. Biochem Pharmacol. 1988;37:3401–6. doi: 10.1016/0006-2952(88)90688-0. [DOI] [PubMed] [Google Scholar]
- 33.Jiang C, Wang Z, Ganther H, et al. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001;61:3062–70. [PubMed] [Google Scholar]
- 34.Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 35.Chaudiere J, Courtin O, Leclaire J. Glutathione oxidase activity of selenocystamine: a mechanistic study. Arch Biochem Biophys. 1992;296:328–36. doi: 10.1016/0003-9861(92)90580-p. [DOI] [PubMed] [Google Scholar]
- 36.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 37.Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–81. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Burns TF, El-Deiry WS. The p53 pathway and apoptosis. J Cell Physiol. 1999;181:231–9. doi: 10.1002/(SICI)1097-4652(199911)181:2<231::AID-JCP5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–12. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 40.Johnson TM, Yu ZX, Ferrans VJ, et al. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA. 1996;93:11848–52. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 42.Macip S, Igarashi M, Berggren P, et al. Influence of Induced Reactive Oxygen Species in p53-Mediated Cell Fate Decisions. Mol Cell Biol. 2003;23:8576–85. doi: 10.1128/MCB.23.23.8576-8585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 44.Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 1999;18:6027–36. doi: 10.1093/emboj/18.21.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandel NS, Vander Heiden MG, Thompson CB, et al. Redox regulation of p53 during hypoxia. Oncogene. 2000;19:3840–8. doi: 10.1038/sj.onc.1203727. [DOI] [PubMed] [Google Scholar]
- 46.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nature Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 47.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]