


Abstract
Clerocidin (CL), a diterpenoid natural product, alkylates DNA through its epoxide moiety and exhibits both anticancer and antibacterial activities. We have examined CL action in the presence of topoisomerase IV from Streptococcus pneumoniae. CL promoted irreversible enzyme-mediated DNA cleavage leading to single- and double-stranded DNA breaks at specific sites. Reaction required the diterpenoid function: no cleavage was seen using a naphthalene-substituted analogue. Moreover, drug-induced DNA breakage was not observed using a mutant topoisomerase IV (ParC Y118F) unable to form a cleavage complex with DNA. Sequence analysis of 102 single-stranded DNA breaks and 79 double-stranded breaks revealed an overwhelming preference for G at the −1 position, i.e. immediately 5′ of the enzyme DNA scission site. This specificity contrasts with that of topoisomerase IV cleavage with antibacterial quinolones. Indeed, CL stimulated DNA breakage by a quinolone-resistant topoisomerase IV (ParC S79F). Overall, the results indicate that topoisomerase IV facilitates selective irreversible CL attack at guanine and that its cleavage complex differs markedly from that of mammalian topoisomerase II which promotes both irreversible and reversible CL attack at guanine and cytosine, respectively. The unique ability to form exclusively irreversible DNA breaks suggests topoisomerase IV may be a key intracellular target of CL in bacteria.
INTRODUCTION
Clerocidin (CL) (Figure 1) is a natural drug (1), which has been shown to inhibit bacterial DNA gyrase and eukaryotic DNA topoisomerase II (2,3). CL differs from other known topoisomerase poisons since it can induce the formation of a cleavage complex, which is partially stable to heat and salt treatment (3). In particular, the sequence specificity of CL-stimulated eukaryotic topoisomerase II-mediated DNA damage showed that when the base immediately preceding the site of enzyme attack, i.e. position −1, was a guanine (G), the cleavage was irreversible, whereas in the case of a cytosine (C), the cleavage was reversible (4,5).
Figure 1.

Chemical structures of CL and its analogue NA1, both shown in the cyclic hemiacetal form.
CL can also induce irreversible damage per se on single-stranded DNA regions (6). The C12–C15 moiety of the drug is responsible for the cleavage activity; in particular, spontaneous DNA fragmentation derives from depurination at the G level caused by the electrophilic attack of the epoxide ring of CL on the N7 group of exposed Gs (6). The hemi-acetalic form of the α-ketoaldehyde function at C14–C15 enhances epoxide reactivity by applying additional strain to the oxirane ring (7). On the other hand, the diterpenoid portion of CL is dispensable for drug activity per se; in fact the NA1 analogue of CL (Figure 1), which preserves the C12–C15 ring but has the diterpenoid function substituted with naphthalene retains intrinsic DNA cleavage activity (7).
The modes of CL activity towards the DNA in the absence or presence of a mammalian topoisomerase II are distinctive. Direct CL-stimulated DNA damage requires longer reaction times (6–24 h versus 30 min for the enzyme) and, unlike the topoisomerase II-mediated reaction (4,5), the results are irreversible at both G and C nucleotides (7,8). Therefore, topoisomerases substantially modulate CL activity towards DNA.
Studies of CL potentiation have focussed almost entirely on mammalian topoisomerase II. However, CL also exhibits antibacterial activity against Escherichia coli and Gram-positive pathogens, such as Staphylococcus aureus (2,3). Moreover, it retains potency against Gram-positive (but not E.coli) isolates resistant to quinolones, another class of antibacterial topoisomerase poison. These properties raise the issue of whether the bacterial type II enzymes topoisomerase IV and gyrase are targeted by CL and potentiate its action. Although topoisomerase IV and gyrase share important common features with eukaryotic topoisomerase II, such as homology and acting through a double-strand DNA break, nevertheless their detailed mechanisms of action and quaternary organization appear to be individually different. Thus, eukaryotic topoisomerase II controls the global topology of DNA (9–11) whereas DNA gyrase unwinds replicating DNA by introducing negative supercoils, while topoisomerase IV decatenates interlocked daughter chromosomes (12–14). The mechanism of topoisomerase II-catalyzed reactions and the interaction of these enzymes with the DNA are hence inevitably different: for instance, eukaryotic topoisomerase II binding to DNA protects 25–28 bp (15), similar to topoisomerase IV (34 bp) (16), and gyrase wraps about 150 bp of nucleic acid (17). Given that CL reactivity with the DNA rests on the accessibility of nucleic acid reactive functions (6), distinct DNA processing by bacterial topoisomerases could result in a different CL poisoning mode to that observed for mammalian topoisomerase II.
Early data have reported on CL activity against (E.coli) gyrase, the quinolone target in Gram-negative species (2,3), but surprisingly no information is available for CL action towards topoisomerase IV, the primary target of many quinolones in Gram-positive bacteria, such as S.aureus and Streptococcus pneumoniae (18,19). To gain insight on CL mechanism potentially relevant to its antibacterial properties, we have investigated its interaction with S.pneumoniae topoisomerase IV, one of the best characterized bacterial type II enzymes. We report that CL was able to poison S.pneumoniae topoisomerase IV with unique sequence specificity and irreversibility different from those seen for eukaryotic topoisomerase II. In contrast to what is observed for drug activity per se, both the electrophilic C12–C15 ring and the diterpenoid moiety were required for CL enzyme-mediated biological activity. By using a mutant topoisomerase IV (ParC Y118F) we found that the enzyme must be covalently bound to the DNA to promote irreversible CL damage. Finally, CL was active against a quinolone-resistant topoisomerase IV but with differences in DNA cleavage specifity to the wild-type protein, which provide new insights on drug-induced poisoning of topoisomerase IV.
MATERIALS AND METHODS
Chemicals and reagents
CL was a kind gift of Leo Pharmaceutical Products (Ballerup, Denmark). NA1 was generously provided by Prof. S. Kobayashi (University of Tokyo, Japan). CL and NA1 were dissolved in absolute ethanol and the concentration determined by measuring absorbance in ethanol at 230 and 282 nm on a UV/VIS Spectrometer Lambda 12 (Perkin Elmer, MA). Molar extinction coefficients were experimentally determined to be 11 818 M−1 cm−1 for CL and 9493 M−1 cm−1 for NA1. Working drug solutions were obtained by diluting fresh stocks in the appropriate buffer. In each assay, the final ethanol concentration was 2%. Ciprofloxacin (CPF) was provided by GlaxoSmithKline (Verona, Italy). Stock solutions were made in mQ-grade water and diluted to the working concentration in the desired buffer. Buffer components were purchased from Sigma–Aldrich (St. Louis, MO) and electrophoretic reagents, dNTPs and Taq polymerase were from Amersham Biosciences Europe (Freiburg, Germany). [γ-32P]ATP was from Perkin Elmer (MA); T4 polynucleotide kinase and EcoRI were purchased from Invitrogen (Paisley, UK).
Topoisomerases IV from S.pneumoniae
E.coli BL21(λDE3)(pLysS) was from our laboratory collection. Conditions for growth of bacterial strains were as described previously (18). S.pneumoniae ParC and ParE subunits were expressed as His-tagged proteins in E.coli BL21(λDE3)(pLysS) from plasmids pXP13 and pXP14, which contain the respective parC and parE genes cloned from strain 7785, as described previously (20) except that cells were induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and protein expression was carried on for 12 h at 16°C. This modification allowed the recovery of soluble protein in greater yield. The proteins were purified to >95% homogeneity by nickel chelate column chromatography and exhibited specific activities of >2 × 105 U/mg when they were assayed with an excess of the complementing subunit (20). ParC S79F protein, whose purity and activity were comparable to those of their wild-type counterpart, was obtained similarly (21). ParC Y118F was prepared by expression from parC plasmid pXP13 or pEL1 (22) whose parC codon 118 had been altered using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer's instructions. The mutagenic primers used for PCR were MUTFOR (GATCCTCCTGCGGCTATGCGTTTTACTGAGGCACGTTTGTCT) and MUTREV (AGACAAACGTGCCTCAGTAAAACGCATAGCCGCAGGAGGATC). MUTFOR corresponds to nucleotide positions 330–372 in parC, and carries a T instead of A at position 352. Therefore the codon (underlined) specifies phenylalanine instead of tyrosine at position 118. After 1 h incubation at 37°C with DpnI, which digests the parental DNA template, the solution containing the circular mutated plasmid was used to transform E.coli XL1-Blue. Colonies of interest were analysed and sequenced; the correct plasmid was transformed into BL21(λDE3)pLysS. The expression and purification of ParC Y118F was performed as described previously (20). Recombinant proteins were examined by SDS–PAGE, and shown to be more than 95% homogeneous. Protein concentration was determined by Bradford assay and SDS–PAGE.
DNAs
Plasmid pBR322 and SV40 DNA were purchased from MBI Fermentas (MD) and Invitrogen (Paisley, UK), respectively. Kinetoplast DNA from Crithidia fasciculata was obtained from Topogen, Inc., (Ohio). Primers were purchased from Eurogentec (Liege, Belgium) and were named according to the nucleotide position of their 5′ end in the SV40 sequence. Primer sequences are as follows: pr658: GAGGCTCCTGGTGGTG; pr883: CTTTGTGATCCCAGTCAC; pr1640: GAGGCTCCTGGTGGTG; pr1402: TGAAGCTGTCTACTCCAG; pr2026: TGCTCAAACTGTAACCCC; pr2261: GCCCAACACCCTGCTC; pr3368: CTCTGGACTCCCCTCCA; pr3586: CTCTGGACTCCCCTCCA; pr4457: GAGAGTCAGCAGTAGCC; pr4697: CCTTACTTCTGTGGTGTG; pr2533: GATCCAGACATGATAAG; pr2757: AGCCATACCACATTTGTA. These primers were used in PCR to amplify 239 bp fragments using SV40 DNA as template.
5′ end-labeling of primers and PCR
For primer labeling, 10 pmol of primer solution were incubated with 2 µl (10 µCi/µl) of [γ-32P]ATP and 10 U of T4 polynucleotide kinase in 50 mM Tris–HCl (pH 7.5), 7 mM MgCl2 and 10 mM DTT, at 37°C for 30 min. After incubation, DNA was ethanol precipitated and the labelled primers were used to amplify 239 bp fragments of SV40 by PCR. Each PCR was prepared by mixing 200 µM dNTPs, the pellet of the ethanol precipitated labelled primer, 10 pmol of the cold primer, 50 ng of template SV40 DNA and 5 U of Taq polymerase in PCR buffer [10 mM Tris–HCl (pH 9.0), 50 mM KCl and 1.5 mM MgCl2] to a final volume of 100 µl. PCR cycles were 94°C for 30 s, 55°C for 30 s and 72°C for 30 s (30 cycles). DNA fragments were then purified with a QIAquick PCR purification kit (Qiagen, CA). The resulting labelled fragments encompass ∼27% of the total sequence of SV40.
Topoisomerase catalytic and cleavage assays
For decatenation assays, standard reaction mixtures (20 µl) contained 40 mM Tris–HCl (pH 7.5), 6 mM MgCl2, 10 mM DTT, 200 mM potassium glutamate, 1 mM ATP, 50 µg/ml of BSA, 180 ng of kDNA and reconstituted topoisomerase IV (0.03 µg ParC and 0.10 µg ParE), in the presence of different drug concentrations. Reaction mixtures were incubated at 37°C for 1 h, and then loading buffer (50% glycerol, 0.2% xylene cyanol) was added. The treated samples were then loaded on a 1% agarose gel in TBE [89 mM Tris, 89 mM boric acid and 2 mM EDTA (pH 8.0)]. Gels were run at 70 V for 120 min and stained with ethidium bromide; DNA bands were visualized with a GelDoc 1000 ultraviolet (UV) transilluminator (Bio-Rad, Milan, Italy) and their intensity quantified by Bio-Rad ImageQuant software.
For DNA cleavage assays, reconstituted topoisomerase IV (0.45 µg ParC and 1.7 µg ParE) was incubated with 400 ng of supercoiled pBR322 in 25 µl reaction mixtures [40 mM Tris–HCl (pH 7.5), 6 mM MgCl2, 10 mM DTT, 200 mM potassium glutamate, 50 µg/ml BSA], in the presence of different drug concentrations. After a 30 min incubation at 37°C, 1 µl of 10% SDS and 2 µl of a 20 mg/ml stock of proteinase K were added, and incubation continued for 30 min at 45°C. Loading buffer (3 µl) was added to the samples, which were then analysed by electrophoresis in a 1% agarose gel in TBE containing ethidium bromide at 50 µg/ml. The linearized standard was obtained by incubating pBR322 with EcoRI in 10 mM Tris–HCl, 3.8 mM MgCl2 and 20 mM NaCl (pH 8.0), at 37°C for 1 h. Cleavage assays were also performed using labelled PCR products as DNA substrates. In this case, after the incubation with SDS and proteinase K, samples were ethanol precipitated, then resuspended in formamide gel loading buffer [95% formamide, 200 mM EDTA (pH 8.0), 0.1% (w/v) xylene cyanol and 0.1% (w/v) bromophenol blue] and heated at 90°C for 5 min. Cleavage products were resolved in 8% polyacrylamide, 7 M urea gels alongside a G+A ladder obtained according to the Maxam and Gilbert protocol (23). Gels were then transferred to Whatman 3 MM filter paper, dried under vacuum at 80°C and bands were visualized by phosphorimaging analysis (Molecular Dynamics, Amersham Biosciences Europe). Quantification was performed by ImageQuant software (Molecular Dynamics).
Statistical tests
The χ2 one-sample test was used to determine the deviation from the random distribution of bases at each position of the aligned sequences (24). The expected occurrence of each nucleotide was calculated considering the oligonucleotide frequencies on the whole SV40 DNA (A = T = 0.296; C = G = 0.204). Also, nucleotide preference was calculated by computing the standard deviation (SD) of the expected frequency p: SD = (p[1 − p]/n)1/2, where p = 0.296 for A or T and p = 0.204 for G or C, and n is the number of sites analysed in the dataset. A total of 99.9% confidence interval was then calculated [p ± 3.3 SD]. Base frequencies outside confidence intervals were considered significant. The probability (P) of deviation from expectation was calculated as described previously (24,25). Briefly, the expected number of sites having a given base at any particular position is pn. Let m be the observed number of sites, which have the given base at that position. If m > pn, the probability P of the chance occurrence of m or more instances was calculated as
If m < pn, the chance occurrence of m or fewer instances was calculated as follows:
Negative values of the logarithm of P, −log(P), are reported for each base at each position around the cleavage site.
RESULTS
CL stimulates S.pneumoniae topoisomerase IV-mediated DNA cleavage
CL activity against S.pneumoniae topoisomerase IV was examined for both the catalytic and cleavage activities of the enzyme. In the enzymatic assay, the ability of the enzyme to decatenate kDNA was exploited. In the absence of the drug, topoisomerase IV converted all the input kDNA to free minicircles, (lane 2, Figure 2A). CL partially inhibited enzyme activity at 100 µM, but was essentially inactive at lower doses (EC50 >200 µM; lanes 8–12, Figure 2A). CPF, a known topoisomerase IV inhibitor, was used as a positive control (lanes 3–7, Figure 2A). This fluoroquinolone impaired enzyme activity above 100 µM, as shown by the presence of unlinked input kDNA and of intermediate forms of catenated DNA (EC50 80 µM; lanes 6 and 7, Figure 2A). These data for CPF are consistent with previous observations (20,26).
Figure 2.

CL poisons topoisomerase IV. (A) Inhibition of the decatenation activity of the enzyme. Kinetoplast DNA (kDNA) was incubated with topoisomerase IV and increasing amounts (12.5, 25, 50, 100, 200 µM) of CPF, (lanes 3–7) or CL (lanes 8–12). Reaction mixtures were loaded and run on a 1% agarose gel. DNA bands were visualized with ethidium bromide staining. Lane 2 is a control for topoisomerase enzymatic activity in the absence of the drugs and lane 1 is a control for the input kDNA. M denotes released minicircle DNA. Pk indicates partially catenated DNA intermediates. (B and C) Stimulation of enzyme-mediated DNA breaks. (B) Supercoiled plasmid pBR322 was incubated with increasing amounts of CPF (5, 10, 20 µM, lanes 3–5) or CL (12.5, 25, 50, 100, 200 µM, lanes 6–10) and loaded on a 1% agarose gel. DNA bands were visualized with ethidium bromide staining. Lane 2 is a control for the cleavage activity of topoisomerase IV without drugs. Lane 1 shows the input supercoiled plasmid DNA and lane 11 is a control for linear plasmid DNA. Band identities are shown on the left. N, L and SC indicate nicked, linear and supercoiled plasmid DNA, respectively. For any given DNA sample, the percentage of nicked, linear and supercoiled DNA bands were determined by dividing the relevant band intensity by the total DNA intensity loaded into that particular track. Quantification data are shown below the gel figure. (C) A 239 bp SV40 DNA fragment, labelled with 32P at one 5′ end, was incubated with topoisomerase IV and increasing amounts (60 nM–256 µM) of CL (lanes 3–15) or CPF (lanes 16–28). Reaction mixtures were loaded and run on a denaturing urea-8% polyacrylamide gel. DNA fragments were visualized by phosphorimaging. Lane 1 and 2 are controls for non-treated DNA and DNA incubated with the enzyme without drugs. Lane 29 and 30 are respective G+A and G sequencing products obtained according to the Maxam and Gilbert protocol (23). Drug concentrations present in cleavage reactions are shown at the bottom of the gel.
For cleavage experiments, the supercoiled form of plasmid pBR322 was incubated with topoisomerase IV in the absence or presence of increasing drug concentrations. DNA breakage was induced by addition of SDS, and following proteinase K digestion, the DNA was examined by agarose gel electrophoresis (Figure 2B). In the absence of the drug, the enzyme converted ∼15% of the plasmid into the nicked form (lane 2, Figure 2B). When CL was added, the drug-induced detectable single-strand DNA breaks (producing nicked form plasmid) in a dose-dependent fashion, starting from a concentration of 25 µM, (lanes 6–10, Figure 2B). At higher concentrations, CL also induced double-strand DNA cleavage generating dose-dependent increases in the linear DNA (lanes 8–10, Figure 2B). For any given DNA sample, the percentage of nicked, linear and supercoiled DNA bands were determined by dividing the relevant band intensity by the total DNA intensity loaded into that particular track. At 200 µM concentration, CL generated 55 and 16% of the nicked and linear form, respectively, while reduced the supercoiled DNA to 34%. CPF produced both single- and double-strand cleavage at DNA concentrations as low as 5 µM (lanes 3–5, Figure 2B). At 20 µM quinolone concentration, the nicked, linear and supercoiled form were 50, 18 and 32%, respectively (20,26).
To accurately estimate CL efficiency in stabilizing topoisomerase IV-mediated cleavage, we studied the intensity patterns of DNA breaks stimulated by CL or CPF on a 5′ end 32P-labelled SV40 DNA segment amplified by PCR. After SDS and proteinase K treatment, reaction mixtures were loaded on denaturing urea–polyacrylamide gels to separate DNA fragments. Drug concentrations were gradually increased from 60 nM to 250 µM (Figure 2C). Under these conditions, cleavage detected was the sum of single-strand and double-strand breaks: the minimal concentrations for CL- and CPF-mediated DNA fragmentation were 16 and 1 µM, respectively (lanes 11 and 20, Figure 2C). The EC50 was 69 and 13 µM for CL and CPF, respectively.
CL topoisomerase IV-mediated cleavage is selective for G bases
From the cleavage assay reported in Figure 2C, it is clear that CL induced a specific DNA fragmentation pattern that was different from that of CPF. In order to analyse DNA sequence requirements for CL action, six 239 bp-long SV40 segments were 32P-labelled on the forward or reverse strand independently, and amplified by PCR to obtain 27% coverage of the total SV40 sequence. All DNAs were next incubated with topoisomerase IV in the presence of CL and the cleavage patterns were analysed as described above. We were able to collect a large set of CL-mediated cleavage sites, 260 in total. Sequences of cleavage sites were aligned at the point of the phosphodiester bond break in the 5′→3′ orientation, and bases immediately 5′ and 3′ to the cut were numbered −1 and +1, respectively. Positions −1 and +5 are equivalent to topoisomerase IV-mediated DNA double-strand breaks since they are adjacent to each site of cleavage. From a total of 260 strand cuts that were sequenced, 102 (39%) lacked a 4 bp staggered break on the complementary strand.
The deviation of base distribution from the global SV40 DNA base frequencies was first evaluated at each position (± 20 from the cleavage site) by χ2 analyses (Figure 3A). A core region of non-random base composition was found between positions −5 and +10. In particular, the most strikingly biased positions were −1 and +5. Positions −4, −5 and +8, +9 were also slightly preferred, while the centre of the core region (positions from +1 to +4) and positions −2, −3 and +6, +7 showed no preferential base selection. Next, probabilities of the observed base frequency deviations from expectation (G = C = 20.4%; A = T = 29.6%) at each position were assessed for the non-complementary sites (102 in total) and complementary sites (79 on each strand) independently (total 260 sites). When considering the complementary set, 47 and 54 out of 79 sites on the forward and reverse strand, respectively, at positions −1 were guanines, which corresponds to −logP values of 19.3 and 13.4 (Figure 3B). Likewise, 54 and 49 cytosines out of 79 bases (forward and reverse strands, respectively) were found at position +5, which corresponds to −logP values of 19.3 and 15.0 (Figure 3B). On the other hand, T and A at positions −1 and +5 were notably disfavoured (−logP −9.28 and −3.97, respectively). In the non-complementary set, G remained largely preferred at position −1 (−logP 35.9), while T and A were again disfavoured (−logP −10.25 and −5.75, respectively); conversely no base preference or disfavour were detected at position +5 (Figure 3C). It should be noted that −logP values >3 correspond to <0.1% possibility that the preference arose by chance. Therefore, the observed preferences are very strong indeed.
Figure 3.

Sequence analysis of CL-stimulated DNA cleavage by topoisomerase IV. (A) χ2 values of the base distribution at positions ranging from −10 to +10 of the sites of CL stimulation of DNA cleavage. The number −1 indicates the base 5′ of the cleaved phosphodiester bond identified in sequencing gels. (B and C) Probabilities of the observed base frequency deviations from expectation at each position on the forward strand of complementary sites (B) and non-complementary sites (C) (data for the reverse strand are reported in the text). In the ordinate, P is the probability of observing that deviation or more, either as an excess (above base line) or deficit (below base line) relative to the expected frequency of each base. The expected frequencies were calculated using the overall base frequency of SV40 DNA (G = C = 20.4%; A = T = 29.6%).
Comparing sequence specificity of topoisomerase IV-mediated cleavage stimulated by CL or by our control quinolone drug, CPF, we observed that about 25% of CL-stimulated sites were shared with CPF sites and, vice versa, 16% of CPF-stimulated sites were shared with CL. Of all sites exhibiting a G at position −1 stimulated by CPF, 42% were also cleaved by CL.
CL-induced topoisomerase IV-mediated DNA damage is irreversible
All known anti-topoisomerase II drugs show cleavage reversibility upon salt addition or incubation at high temperature (27,28). In contrast, CL was reported to possess the unique capability of poisoning human and murine topoisomerase II in a partially salt- and temperature-independent manner (4,5). To determine if this characteristic was maintained against bacterial topoisomerase IV, CL was incubated with a 5′ end 32P-labelled SV40 DNA fragment in the presence of topoisomerase IV for 20 min at 37°C; then, either 0.6 M NaCl was added and incubation continued for 10 min, or the incubation temperature was increased to 65°C for 10 min. As shown in Figure 4, none of the CL-stimulated cleavage sites were reversed by either treatment (compare lanes 7 and 8 with 6, Figure 4), whereas the totality of CPF-induced sites displayed reversion (compare lanes 4 and 5 with 3, Figure 4). In other experiments (data not shown) we found that EDTA was similarly unable to reverse CL-stimulated DNA cleavage by topoisomerase IV.
Figure 4.
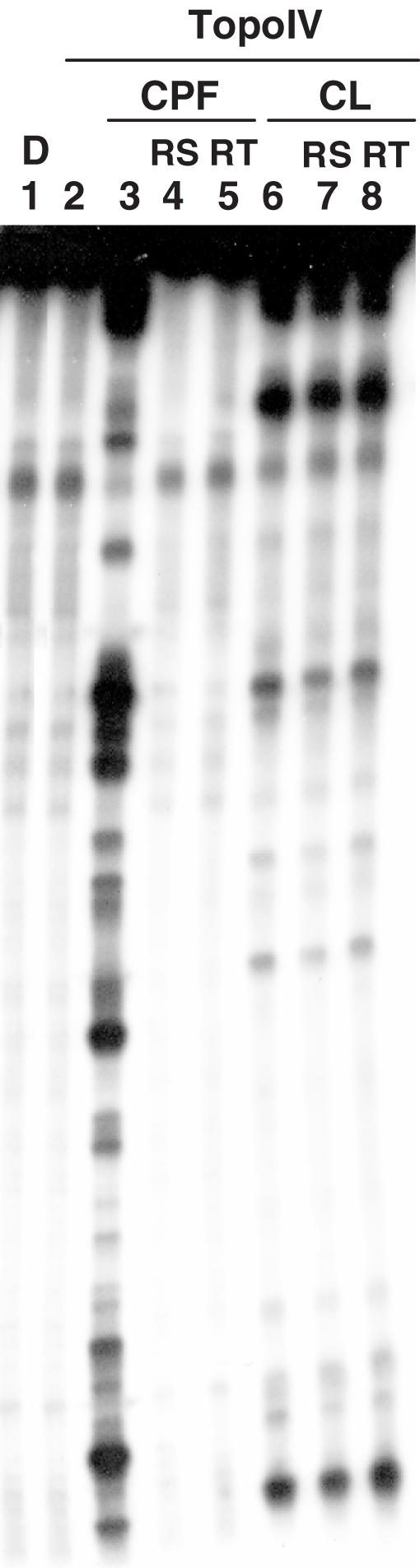
Irreversibility of CL-stimulated topoisomerase IV-mediated DNA cleavage sites. A 5′ end 32P-labelled 239 bp-long SV40 DNA segment was incubated with topoisomerase IV and 25 µM of CPF (lanes 3–5) or 100 µM of CL (lanes 6–8) at 37°C for 20 min. Samples in lanes 4 and 7, and 5 and 8 were further treated with 0.6 M NaCl for 20 min (indicated as RS) or 65°C for 20 min (indicated as RT), respectively. Stopped reaction mixtures were loaded and run on a 8% denaturing polyacrylamide gel. DNA fragments were visualized by phosphorimaging. Lane 1 and 2 are controls for untreated DNA (D) and topoisomerase IV-processed DNA, respectively.
CL is inactive when topoisomerase IV is unable to cleave DNA
In order to understand whether enzyme-mediated DNA cleavage was a prerequisite for irreversible CL-mediated DNA damage, CL activity was measured against a mutant topoisomerase IV reconstituted with Y118F ParC. This enzyme is able to bind the DNA, but lacks the active site tyrosine residue necessary for the covalent nucleophilic attack and phosphodiester bond breakage involved in DNA breakage-reunion. Consequently, the enzyme is inactive in topoisomerase and cleavage assays. The ability of topoisomerase IV Y118F to bind double-stranded DNA was assayed by band shift analysis: enzymes containing the wild-type and mutant ParC subunits were able to bind the nucleic acid with similar efficiency (E. Leo and L. M. Fisher, unpublished data). Experiments with CL showed that the drug did not stimulate DNA cleavage by the mutant enzyme (data not shown).
Quinolone-resistant topoisomerase IV reconstituted with S79F ParC alters CL activity
To investigate enzymatic residues potentially involved in CL poisoning, we assayed CL against a mutant topoisomerase IV carrying an S79F substitution in ParC that confers resistance to quinolones (29). As shown in Figure 5, DNA fragmentation in the presence of CL indicated that the drug was active on the mutant enzyme (lanes 10 and 11, Figure 5). Interestingly, though the overall cleavage pattern resembled that of the wild-type protein, it was apparent that five out of nine cleavage sites were absent or markedly reduced in intensity when using the mutant topoisomerase IV (compare lanes 8 and 9 with 10 and 11, see sites labelled with an asterisk, Figure 5). As expected, the control drug CPF induced DNA fragmentation with the wild-type topoisomerase IV but not with the mutant enzyme (compare lanes 6 and 7 with 4 and 5, Figure 5). The same assay was repeated with two additives non-complementary SV40-based PCR amplified fragments: a total of 11 CL-induced sites were found decreased in intensity with the mutant topoisomerase IV compared with wild-type enzyme. Of these, eight had −1 G; hence the base frequency at the cleavage site remained the same as that observed with the wild-type enzyme (see Figure 3). However, the region between positions +2 and +5 showed a moderately enhanced frequency of G/C nucleotides; in particular the preferred base at position +2 was G for five sites, whereas C was preferred at positions +3 (4 Cs and 3 Gs), +4 (4 Cs and 1 G) and +5 (5 Cs and 3 Gs). It appears that CL cleavage by the mutant enzyme is sensitive to the base composition of the DNA that lies within the 4 bp staggered break induced by topoisomerase IV.
Figure 5.
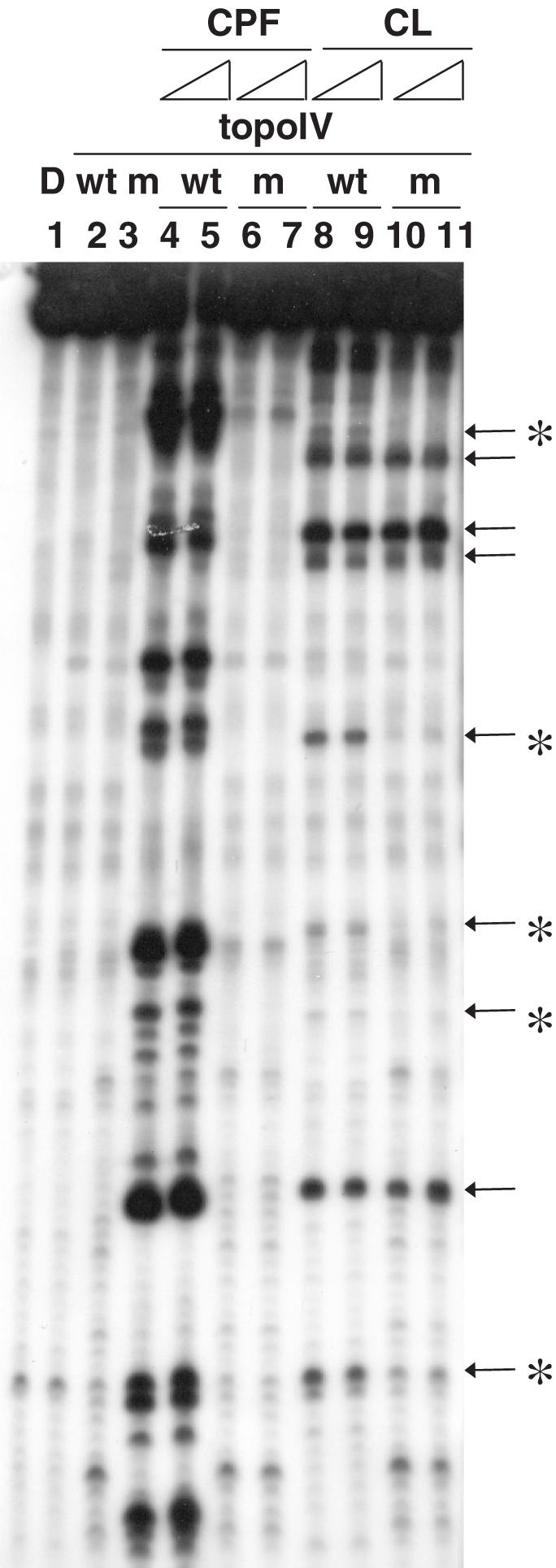
CL-stimulated DNA cleavage in the presence of wild-type topoisomerase IV or mutant quinolone-resistant enzyme reconstituted with ParC S79F. A 5′ end 32P-labelled 239 bp-long SV40 DNA segment was incubated with wild-type (lanes 2, 4–5, 8–9) or S79F mutant topoisomerase IV (lanes 3, 6–7, 10–11) and 25–50 µM of CPF (lanes 4–7) or CL (lanes 8–11) at 37°C for 30 min. Reaction mixtures were stopped and run on a 8% denaturing polyacrylamide gel. DNA fragments were visualized by phosphorimaging. Lane 1 is a control for the untreated input DNA. Lanes 2–3 are controls for wild-type and S79F mutant topoisomerase IV-processed DNA, respectively. Arrows on the right of the gel indicate cleavage bands and asterisks specify cleavage sites that are differently induced by CL-stimulated wild-type and S79F mutant enzymes.
The diterpenoid portion of CL is necessary for topoisomerase IV poisoning
Recent work has shown that the diterpenoid portion of CL (Figure 1) is dispensable for DNA damage in the absence of a topoisomerase (7). To test whether this holds true for topoisomerase IV-mediated cleavage, DNA fragmentation was measured in the presence of NA1, an analogue of CL in which the diterpenic function is substituted with a naphthalene moiety (Figure 1). Freshly-made CL and NA1 solutions were incubated either with 32P-labelled PCR amplified DNA and topoisomerase IV for 30 min, or with supercoiled pBR322 plasmid for 24 h (Figure 6). Surprisingly, NA1 did not induce any cleavage (lanes 8–12, Figure 6) in the presence of the enzyme, unlike CL which was able to induce strand breaks resulting in marked plasmid nicking (lanes 3–7, Figure 6) (52, 17 and 31% of the nicked, linear and supercoiled form, respectively, at 200 µM CL concentration), as described above. Concurrently, in the absence of the enzyme, NA1 was confirmed to be essentially as effective as CL in promoting plasmid DNA cleavage after 24 h incubation at 37°C, while at shorter reaction times (30 min) both drugs generated little cleavage (data not shown) (5,7). To rule out the possibility of enzymatic inhibition by NA1, the drug was tested in a topoisomerase IV decatenation assay, which showed that it was unable to inhibit enzymatic activity at concentrations up to 400 µM (data not shown). Thus, the diterpenoid moiety of CL is important for reaction with topoisomerase IV.
Figure 6.

The CL analogue NA1 does not promote topoisomerase IV-mediated DNA breakage. Supercoiled plasmid pBR322 was incubated with increasing amounts (12.5, 25, 50, 100, 200 µM) of CL (lanes 3–7) or NA1 (lanes 8–12) and after SDS-proteinase K treatment, DNA products were separated on a 1% agarose gel and visualized by ethidium bromide staining. Lanes 1 and 2 are controls for the input supercoiled plasmid DNA and for the cleavage activity of topoisomerase IV without drugs, respectively. Band identities are shown on the left. N, L and SC stand for nicked, linear and supercoiled plasmid DNA, respectively. For any given DNA sample, the percentage of nicked, linear and supercoiled DNA bands were determined by dividing the relevant band intensity by the total DNA intensity loaded into that particular track. Quantification data are shown below the gel figure.
DISCUSSION
We have established that CL, an antimicrobial DNA alkylating agent, mediates single- and double-stranded DNA cleavage by S.pneumoniae topoisomerase IV. By characterizing the enzyme-, drug- and sequence-determinants, we could show that the reaction has a number of unique features, notably its irreversibility, the overwhelming preference for attack at guanine bases and differences in sequence specificity and selectivity with CPF, an antibacterial topoisomerase IV poison. These and other data indicate that CL reacts in a unique manner with the cleavage complex of topoisomerase IV and suggest that the enzyme may contribute to the antibacterial action of the drug.
Initial studies revealed that whereas CL is a poor inhibitor of DNA decatenation by topoisomerase IV requiring high drug concentrations (EC50 >100 µM) (Figure 2A), stimulation of topoisomerase IV-mediated DNA cleavage was effective at lower CL levels and increased in a dose-dependent fashion. When CL and topoisomerase IV were incubated with supercoiled plasmid DNA and nucleic acid breaks were visualized by ethidium bromide, drug effects were manifest at 25 µM (Figure 2B). When the drug and the enzyme were instead used against a 32P-labelled 239 bp linear DNA fragment, DNA damage was apparent at lower CL concentrations (16 µM) due to the higher sensitivity of the detection method (Figure 2C). In both experiments, CL was about five times less potent than the control antibacterial fluoroquinolone CPF (calc. EC50 69 µM and 13 µM for CL and CPF, respectively). In this regard, CL activity is similar to that described with murine topoisomerase II: in fact eukaryotic enzyme-mediated CL damage at the DNA level was assessed at 25 µM and was 25–2.5 times less potent than the control anti-topoisomerase II drugs (5).
CL-promoted DNA cleavage by topoisomerase IV occurred at specific sites (Figure 2C) and could not be reversed by prior incubation with high salt or high temperature, in contrast to CPF-promoted cleavage that occurred at a different spectrum of sites and was completely reversible (Figure 4). Within the 260 CL-stimulated strand scission sites that we identified and sequenced, we distinguished two subsets: complementary sites arising from double-strand breaks and non-complementary sites resulting from single-stranded DNA breakage. Statistical analysis of base frequency deviations from expectation showed in both cases an overwhelming preference for G at the cleavage site (position −1) (Figure 3). The complementary set, where sites were analysed independently for the forward and reverse sequences, attested also to a comparable preference for C at position +5, indicating that the vast majority of complementary sites bear a −1 G at the cleavage site on both complementary strands. On the other hand, the non-complementary set of sites lacked this symmetry and exhibited −1 G only on the cut strand. All CL-stimulated cleavage sites were found fully irreversible upon heat or salt treatment.
These results obtained with the bacterial topoisomerase IV are significantly different from those reported for murine topoisomerase II. In fact, in the case of topoisomerase II, the base immediately preceding the site of enzyme attack was either G or C; in addition, the cleavage was irreversible at G sites, but reversible at Cs which comprise one-third of the sites (4,5). A mechanism for the biased behaviour toward G and C in the presence of the eukaryotic enzyme has been proposed. According to the model, the N7 of G is exposed to efficient alkylation by the drug, which blocks enzymatic re-ligation and thereby stabilizes DNA damage (6). In contrast, the NH2 and N3 reactive sites of C, being involved in base pairing, remain effectively protected from CL alkylation. Hence, the C preference is promoted by non-covalent interaction with the drug and, in this model, CL behaves like a classical topoisomerase poison (8).
We note that CL stimulation of topoisomerase IV-mediated cleavage is in general asymmetric. If G is present at the −1 position of both strand-breakage sites, CL induces a double-strand break. If G is present at the −1 position of one of the two strand-breakage sites, CL induces a single-strand break. If G is absent at the −1 position, CL does not affect the strand-breakage and reunion activity of topoisomerase IV. This feature is consistent with our present observation of weak inhibition of topoisomerase IV enzymatic activity in a decatenation assay (Figure 2A). Thus, these results revealed the molecular basis of topoisomerase poisoning by CL and suggested that, as is the case with human topoisomerase II (30), topoisomerase IV could ligate the two scissile bonds of a cleavage site in a non-concerted manner.
It is known that, in the absence of enzyme and at longer reaction times, the electrophilic attack of the CL epoxide on N7 of Gs on locally unpaired regions (i.e. bulges) can induce strand breaks per se (6). Therefore, we assessed whether topoisomerase-mediated DNA cleavage was a condition for CL-mediated nucleic acid damage in the presence of the enzyme. For this purpose, we used a mutant topoisomerase IV reconstituted with ParC in which the catalytic Tyr 118 had been replaced with Phe (Y118F). The mutant topoisomerase IV was able to bind DNA non-covalently and possibly distort it, but was inactive in DNA breakage-reunion. In fact, CL failed to damage the DNA in the presence of the mutant enzyme, indicating that covalent topoisomerase–DNA interactions are required for enzyme-related CL activity. This result supports the idea that CL stimulates cleavage at G nucleotides that are already ‘processed’ by the enzyme i.e. positioned at the new 3′ end of the transient enzyme-induced strand break. Heightened reactivity of the −1G in the cleavage complex coupled with proper alignment of CL via enzyme binding to the deterpenic portion (Figure 6) presumably account for the much shorter DNA reaction times observed for CL in the presence of enzyme.
The sequence specificity of S.pneumoniae topoisomerase IV-mediated cleavage induced by CL is dramatically different from that of quinolones. In the case of the diterpenoid drug, G is always preferred at the cleavage site (−1 position), while a wider consensus DNA sequence (−2A, +1G, +4C, +6T) is driven by quinolones, with no specific base requirement at position −1 (22). Were CL to force the enzyme to cut the DNA at the G level, then one would expect cleavage at every G in DNA. On the contrary, only a subset of Gs is cleaved. We propose that CL cleaves only those G nucleotides that are first processed by topoisomerase IV. The fact that no more than 42% of −1G sites stimulated by CPF are shared with CL indicates that the cleavage sites preferred by the two families of drugs are only partially overlapping.
It is intriguing that the ParC S79F mutation that confers enzyme resistance to quinolones, also affected CL poisoning activity (Figure 5). In fact, even though incubation of CL with the S79F mutant enzyme still induced strand scission and G remained the base largely preferred at the cleavage site, nonetheless, some sites exhibited a dramatic reduction in cleavage intensity compared to the response with wild-type enzyme. Analysis of base frequency at positions −20 to +20 for sites which were less efficiently cleaved by the mutant topoisomerase IV revealed that, again, G was preferred at position −1. Thus, base preference at the cleavage site itself did not discriminate the differential effects found for the wild-type and mutant enzymes. However, in sites whose cleavage was reduced using the quinolone-resistant enzyme, positions from +2 to +5 were particularly rich in G/C nucleotides. Base pairs in this region must be separated during the topoisomerase enzymatic cycle in order to permit strand passage, and many topoisomerase poisons, such as quinolones, are accommodated between the two split DNA strands (22). The mutated ParC residue at position 79 is close in the primary sequence to the active site tyrosine (Y118) and confers high-level resistance to quinolones. We speculate that the S79 ParC residue is somehow involved in promoting or stabilizing DNA strand separation between the two staggered cuts. Although the mutant topoisomerase IV remains active, it possibly separates complementary DNA strands less efficiently. This would be particularly evident when the nucleic acid sequence is rich in strongly hydrogen-bonded G/C base pairs. As a consequence, for sites rich in GC base pairs at positions +2 to +5, CL would fail to reach its G target (and induce irreversible cleavage) due to insufficient distortion of the adjacent double helical DNA region. It should be noted that topoisomerase IV reconstituted with ParC S79F is comparably active to wild-type enzyme in catalysis (29). Therefore, based on our results, DNA unpairing within the staggered break would not be the limiting step in the topoisomerase IV catalytic cycle.
Finally, the ability of CL to poison the S79F mutant topoisomerase IV may help explain data found on drug activity in earlier work. CL was reported to be inactive versus several quinolone-resistant Gram-negative bacterial strains, whereas it remained active against quinolone-resistant Gram-positive S.aureus (2). Since topoisomerase IV is the major quinolone target in Gram-positive bacteria, we propose that susceptibility of the quinolone-resistant S.aureus to CL arises from the continued sensitivity of the mutated topoisomerase IV to the diterpenoid drug. On the other hand, gyrase is mainly poisoned in Gram-negative bacteria and indeed mutations in the gyrA gene produced cross-resistance to quinolones and CL in resistant strains (2). Further progress on these issues will require studies of CL targeting in bacteria.
Acknowledgments
This work was carried out with the financial support of the Italian Ministry for University and Research (MIUR), Rome, Italy. Work in the Fisher group was supported by Project Grant C16747 from the Biotechnology and Biological Sciences Research Council of the United Kingdom. Further funding to support this work and to pay the Open Access publication charges for this article was provided by the European Community STREP Grant No. 503466.
Conflict of interest statement. None declared.
REFERENCES
- 1.Andersen N., Lork H., Rassmussen P. The relative and absolute configuration of clerocidin and its cometabolites. Tetrahedron Lett. 1984;25:469–472. [Google Scholar]
- 2.McCullough J.E., Muller M.T., Howells A.J., Maxwell A., O'Sullivan J, Summerill R.S., Parker W.L., Wells J.S., Bonner D.P., Fernandes P.B. Clerocidin, a terpenoid antibiotic, inhibits bacterial DNA gyrase. J. Antibiot. Tokyo. 1993;46:526–530. doi: 10.7164/antibiotics.46.526. [DOI] [PubMed] [Google Scholar]
- 3.Kawada S., Yamashita Y., Fujii N., Nakano H. Induction of a heat-stable topoisomerase II-DNA cleavable complex by nonintercalative terpenoides, terpentecin and clerocidin. Cancer Res. 1991;51:2922–2925. [PubMed] [Google Scholar]
- 4.Borgnetto M.E., Tinelli S., Carminati L., Capranico G. Genomic sites of topoisomerase II activity determined by comparing DNA breakage enhanced by three distinct poisons. J. Mol. Biol. 1999;285:545–554. doi: 10.1006/jmbi.1998.2330. [DOI] [PubMed] [Google Scholar]
- 5.Binaschi M., Zagotto G., Palumbo M., Zunino F., Farinosi R., Capranico G. Irreversible and reversible topoisomerase II DNA cleavage stimulated by clerocidin: sequence specificity and structural drug determinants. Cancer Res. 1997;57:1710–1716. [PubMed] [Google Scholar]
- 6.Gatto B., Richter S., Moro S., Capranico G., Palumbo M. The topoisomerase II poison clerocidin alkylates non-paired guanines of DNA: implications for irreversible stimulation of DNA cleavage. Nucleic Acids Res. 2001;29:4224–4230. doi: 10.1093/nar/29.20.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richter S., Gatto B., Fabris D., Takao K., Kobayashi S., Palumbo M. Clerocidin alkylates DNA through its epoxide function: evidence for a fine tuned mechanism of action. Nucleic Acids Res. 2003;31:5149–5156. doi: 10.1093/nar/gkg696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richter S.N., Menegazzo I., Fabris D., Palumbo M. Concerted bis-alkylating reactivity of clerocidin towards unpaired cytosine residues in DNA. Nucleic Acids Res. 2004;32:5658–5667. doi: 10.1093/nar/gkh898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rybenkov V.V., Ullsperger C., Vologodskii A.V., Cozzarelli N.R. Simplification of DNA topology below equilibrium values by type II topoisomerases. Science. 1997;277:690–693. doi: 10.1126/science.277.5326.690. [DOI] [PubMed] [Google Scholar]
- 10.Morais Cabral J.H., Jackson A.P., Smith C.V., Shikotra N., Maxwell A., Liddington R.C. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature. 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 11.Berger J.M., Gamblin S.J., Harrison S.C., Wang J.C. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 12.Levine C., Hiasa H., Marians K.J. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys Acta. 1998;1400:29–43. doi: 10.1016/s0167-4781(98)00126-2. [DOI] [PubMed] [Google Scholar]
- 13.Bates A.D., Maxwell A. DNA topology: topoisomerases keep it simple. Curr. Biol. 1997;7:R778–R781. doi: 10.1016/s0960-9822(06)00403-9. [DOI] [PubMed] [Google Scholar]
- 14.Ullsperger C., Cozzarelli N.R. Contrasting enzymatic activities of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 1996;271:31549–31555. doi: 10.1074/jbc.271.49.31549. [DOI] [PubMed] [Google Scholar]
- 15.Lee M.P., Sander M., Hsieh T. Nuclease protection by Drosophila DNA topoisomerase II. Enzyme/DNA contacts at the strong topoisomerase II cleavage sites. J. Biol. Chem. 1989;264:21779–21787. [PubMed] [Google Scholar]
- 16.Peng H., Marians K.J. The interaction of Escherichia coli topoisomerase IV with DNA. J. Biol. Chem. 1995;270:25286–25290. doi: 10.1074/jbc.270.42.25286. [DOI] [PubMed] [Google Scholar]
- 17.Reece R.J., Maxwell A. DNA gyrase: structure and function. Crit. Rev. Biochem. Mol. Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 18.Pan X.S., Fisher L.M. DNA gyrase and topoisomerase IV are dual targets of clinafloxacin action in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 1998;42:2810–2816. doi: 10.1128/aac.42.11.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan X.S., Fisher L.M. Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 1997;41:471–474. doi: 10.1128/aac.41.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan X.S., Fisher L.M. Streptococcus pneumoniae DNA gyrase and topoisomerase IV: overexpression, purification, and differential inhibition by fluoroquinolones. Antimicrob. Agents Chemother. 1999;43:1129–1136. doi: 10.1128/aac.43.5.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan X.S., Yague G., Fisher L.M. Quinolone resistance mutations in Streptococcus pneumoniae GyrA and ParC proteins: mechanistic insights into quinolone action from enzymatic analysis, intracellular levels, and phenotypes of wild-type and mutant proteins. Antimicrob. Agents Chemother. 2001;45:3140–3147. doi: 10.1128/AAC.45.11.3140-3147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leo E., Gould K.A., Pan X.S., Capranico G., Sanderson M.R., Palumbo M., Fisher L.M. Novel symmetric and asymmetric DNA scission determinants for Streptococcus pneumoniae topoisomerase IV and gyrase are clustered at the DNA breakage site. J. Biol. Chem. 2005;280:14252–14263. doi: 10.1074/jbc.M500156200. [DOI] [PubMed] [Google Scholar]
- 23.Maxam A.M., Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Meth. Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 24.Capranico G., Kohn K.W., Pommier Y. Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin. Nucleic Acids Res. 1990;18:6611–6619. doi: 10.1093/nar/18.22.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaxel C., Capranico G., Kerrigan D., Kohn K.W., Pommier Y. Effect of local DNA sequence on topoisomerase I cleavage in the presence or absence of camptothecin. J. Biol. Chem. 1991;266:20418–20423. [PubMed] [Google Scholar]
- 26.Anderson V.E., Zaniewski R.P., Kaczmarek F.S., Gootz T.D., Osheroff N. Action of quinolones against Staphylococcus aureus topoisomerase IV: basis for DNA cleavage enhancement. Biochemistry. 2000;39:2726–2732. doi: 10.1021/bi992302n. [DOI] [PubMed] [Google Scholar]
- 27.Strumberg D., Nitiss J.L., Dong J., Kohn K.W., Pommier Y. Molecular analysis of yeast and human type II topoisomerases. Enzyme-DNA and drug interactions. J. Biol. Chem. 1999;274:28246–28255. doi: 10.1074/jbc.274.40.28246. [DOI] [PubMed] [Google Scholar]
- 28.Hsiang Y.H., Liu L.F. Evidence for the reversibility of cellular DNA lesion induced by mammalian topoisomerase II poisons. J. Biol. Chem. 1989;264:9713–9715. [PubMed] [Google Scholar]
- 29.Yague G., Morris J.E., Pan X.S., Gould K.A., Fisher L.M. Cleavable-complex formation by wild-type and quinolone-resistant Streptococcus pneumoniae type II topoisomerases mediated by gemifloxacin and other fluoroquinolones. Antimicrob. Agents Chemother. 2002;46:413–419. doi: 10.1128/AAC.46.2.413-419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromberg K.D., Velez-Cruz R., Burgin A.B., Osheroff N. DNA ligation catalyzed by human topoisomerase II alpha. Biochemistry. 2004;43:13416–13423. doi: 10.1021/bi049420h. [DOI] [PubMed] [Google Scholar]