

Abstract
The DNA-binding specificities of transcription factors can be used to computationally predict cis-regulatory modules (CRMs) that regulate gene expression1. However, the absence of specificity data for the majority of transcription factors limits the wide-spread implementation of this approach. We have developed a bacterial one-hybrid system that provides a simple and rapid method to determine the DNA-binding specificity of a transcription factor. Using this technology, we successfully determined the DNA-binding specificity of seven previously characterized transcription factors and one novel transcription factor, the Drosophila factor Odd-skipped. Regulatory targets of Odd-skipped were successfully predicted using this information, demonstrating that the data produced by the bacterial one-hybrid system is relevant to in vivo function.
Several methods exist for determining the DNA-binding specificity of a transcription factor. SELEX is the most commonly employed method to define DNA-binding specificity2. More recently, microarrays of short oligonucleotides3 or intergenic sequences4 have been used to characterize transcription factors. Genome-wide chromatin immunoprecipitation (ChIP-chip)5 and DNA immunoprecipitation with microarray detection (DIP-chip)6 have also been used in conjunction with computational analysis to identify statistically overrepresented sequence motifs to extract DNA-binding specificity from the genomic segments that are bound by a transcription factor. These methods, while powerful, have drawbacks: the in vitro technologies require the purification of the transcription factor in its active form; SELEX requires multiple rounds of selection to complete; and microarray-based techniques require the facilities and expertise to analyze the arrays and resulting data.
We have developed a bacterial one-hybrid (B1H) system that provides a simple method for defining the DNA-binding specificity of a transcription factor. The selection procedure is rapid, because only a single round of selection is required to generate a set transcription factor binding sites, and it is readily accessible, because only basic molecular biology expertise is required to employ the technology. Conceptually, this system is similar to yeast one-hybrid systems that can determine the DNA-binding specificity of a transcription factor7 and detect protein-DNA interactions8. However, a bacterial selection system provides advantages over the corresponding system in yeast9. In particular, the higher transformation efficiency of bacteria allows libraries containing more than 100-fold greater complexity to be searched.
The B1H system is derived from a previously described bacterial two-hybrid system9,10. This system contains three components: the transcription factor expression vector, a library of randomized binding sites in the reporter vector, and the bacterial selection strain (Fig. 1). Each DNA-binding domain is expressed as a fusion to the alpha subunit of RNA polymerase. The reporter vector contains restriction sites for introducing a library of randomized oligonucleotides upstream of the promoter of two reporters, the yeast HIS3 and URA3 genes. If a DNA-binding domain (bait) recognizes a target site (prey) in the reporter vector, it will recruit RNA polymerase to the promoter and activate transcription of the reporter genes.
Figure 1.

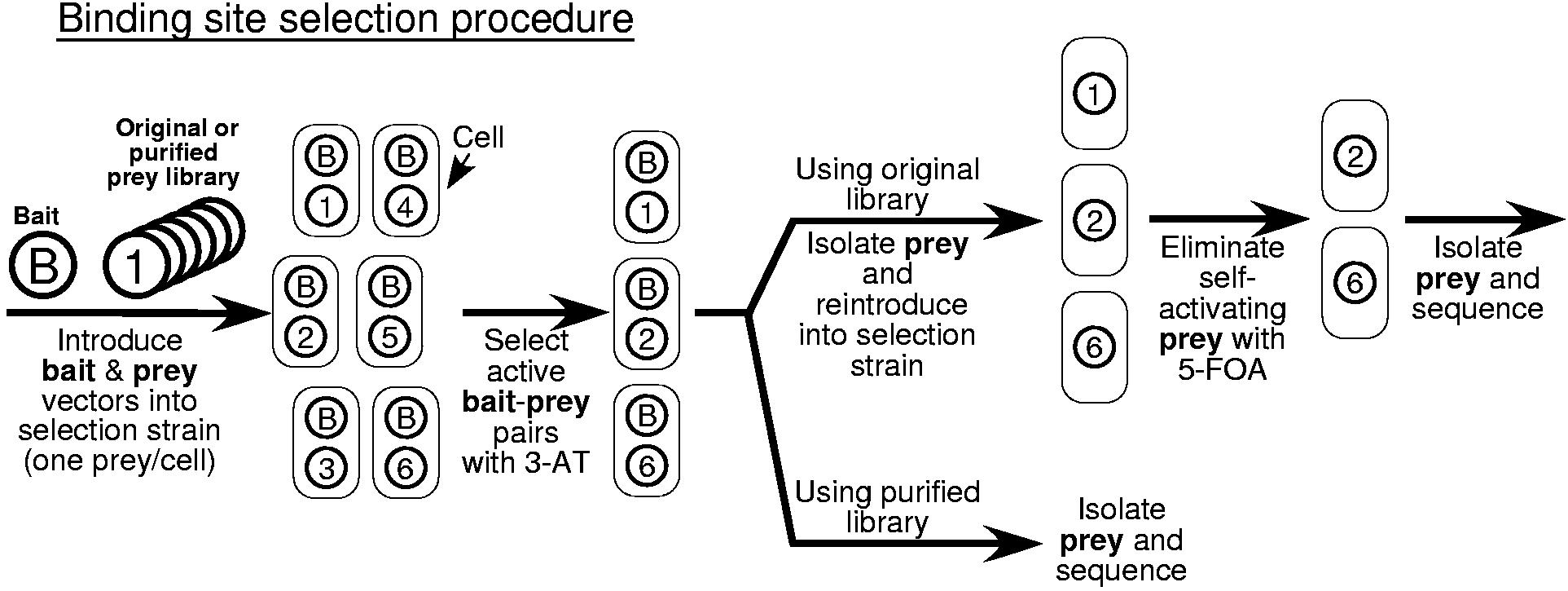

Overview of the bacterial one-hybrid system. a) Schematic representation of the HIS3/URA3 cistron in the pH3U3 prey vector. If the DNA-binding domain (DBD) of the bait recognizes a sequence in the randomized region, the fusion to the alpha subunit will recruit RNA polymerase9,10 to the weak lac promoter and activate transcription of HIS3 and URA3. b) Schematic outline of the B1H selection procedure. The prey library and bait are introduced into the bacterial selection strain. These cells are plated on NM selective media containing the desired concentration of 3-AT to select for bait-prey combinations that activate the reporter. Subsequent steps depend on the prey library (either the original or purified version) that was used for the selection. If the original library is used then prey from the colonies that grow under the selective conditions are isolated and reintroduced into the selection strain in the absence of the bait. These cells are challenged to survive stringent counterselection (5-FOA) to remove any self-activating clones that represent false positives. DNA is isolated from individual colonies that grow under these conditions and the randomized region of each prey is sequenced. If the purified library was used for the selection, then DNA can be isolated and sequenced from colonies on the positive selection plate since the majority of self-activating prey have already been eliminated. The unique DNA sequences recovered from the selection are analyzed by MEME11 to identify any overrepresented sequence motifs, which should represent the recognition sequence of the bait. c) A purified library can be constructed to simplify the selection of bait-prey combinations that activate the reporter genes. The original prey library is introduced into the selection strain and cells are challenged to survive stringent counterselection conditions (5-FOA) to remove any self-activating clones that represent false positives. Prey vectors from the surviving colonies are isolated as a pool to generate the purified prey library for use with any desired bait.
The HIS3 and URA3 reporter genes allow positive and negative selections to be performed in a bacterial strain where the bacterial homologs are deleted. Growth of cells on minimal medium containing 3-amino-triazole (3-AT), a competitive inhibitor of HIS3, provides selection for an active promoter. Growth of cells on medium containing 5-fluoro-orotic acid (5-FOA), which is converted into a toxic compound by the uracil biosynthesis pathway, provides selection against an active promoter. Reporter vectors harboring a binding site for the bait can be isolated by selecting for increased levels of HIS3 expression. Reporter vectors containing DNA sequences that activate the promoter independent of the bait (self-activation) can be eliminated by selection against URA3 expression. Thus, recognition sequences for the bait can be isolated from the library of prey by a combination of positive selection in the presence of the bait and negative selection in the absence of the bait.
Two Cys2His2 zinc finger proteins, Zif268 and PLAG1, were used to test the B1H system. The DNA-binding domains from these proteins were introduced into the bait vector and binding sites were isolated from a prey library containing ∼2×107 unique clones by positive selection with the bait followed by negative selection without the bait (Fig. 1b). The results obtained with Zif268 are representative of selections using other baits in the B1H system. Approximately 4×107 cells containing the Zif268 bait and the prey library were screened on a single plate containing 5 mM 3-AT; approximately 800 colonies appeared after 2 days incubation at 37 °C. Prey plasmids were recovered from these colonies as a pool and reintroduced into the selection strain. Approximately 260 of the 60,000 cells from this population produced colonies in the presence of 5-FOA. Prey from seventeen colonies were sequenced, of which fifteen were unique. The MEME algorithm11 identified an overrepresented sequence motif within the 18 bp randomized region of these clones that strongly resembles the previously defined DNA-binding specificity of Zif268 determined by in vitro SELEX12 (Fig. 2a). The binding site motif determined for PLAG1 using the B1H system is also consistent with its previously described DNA-binding specificity determined by in vitro SELEX13 (Fig. 2b). 14% of the prey isolated in this selection did not contain the computationally identified recognition motif and they were not included when producing the PLAG1 binding motif.
Figure 2.

Binding site motifs for seven proteins determined using the B1H system. The binding site motif determined using each bait in the B1H system is displayed as a Sequence logo28. The maximum possible information content at each position is 2 bits. The Zif268, PLAG-1 and Runt/Bgb binding motifs were generated using the two-step selection method, whereas motifs for the other factors were generated using the purified library in a single selection step. The previously reported consensus sequence for each factor as determined by SELEX is displayed or listed below each Logo. a) Comparison of the binding site motifs produced for Zif268 by the B1H system and from a previously reported in vitro SELEX experiment12. b) PLAG1 can tolerate a 0 to 4 base pair gap between the two recognition motifs based on analysis of the raw sequences by Bioprospector27. A one base pair gap (position 8) is shown in its motif, but a 2 base pair gap was most prevalent in the isolated sequences (7 of 18). The PLAG1 motif is consistent with the previously described PLAG1 consensus sequence (GG(g/a)GGCCNNNNNN(g/a)GG(g/t)) determined by SELEX13. The most 5' base identified in the SELEX analysis could not be conclusively defined in the B1H data because of overlap of the majority of sequences with the edge of the constant region abutting the library. c) The ZnFp53 motif is consistent with the previously described consensus sequence ((g/a)GACACGT) determined by SELEX for a nearly identical clone12. d) The Dorsal motif is consistent with the previously described consensus sequence (GGG(a/t)(a/t)(t/a)(t/c/a)C(c/t)) determined by SELEX16. e) The LAG-1 motif is consistent with the previously described consensus sequence (A(c/g)CGTGGGAA(a/c)) for the mouse homolog of LAG-1 (RBP-Jκ) determined by SELEX18. f) The Paired motif contains at its core a sequence similar to the consensus sequence ((t/c)CGTCACG(g/c)TT(g/c)) determined by SELEX for the paired domain in the absence of the homeodomain19. SELEX selections reported for both DNA-binding domains of Paired resulted a complex mixture of recognition sequences19, a subset contained a core homeodomain binding site abutting the 5' end of the paired domain binding site (AATTAGTCACGC; where the homeodomain element is underlined), which is similar to the 5' end of our motif. g) The Runt/Bgb motif is consistent with the previously described consensus sequence ((t/c)G(t/c)GGT(t/c)) for CBFα/β determined by SELEX20. Raw sequences of the prey from each selection are listed in Supplementary Table 2.
Eliminating the self-activating prey present in the initial library by negative selection would improve the efficiency of the selection system. The resulting “purified” prey library could be used to determine the specificity of any bait in a single selection step. A similar pre-selection procedure has been described for eliminating self-activating baits in the yeast two-hybrid system14. A purified prey library was generated by challenging cells containing the original prey library to grow on media containing 5-FOA and then isolating prey vectors from the surviving cells (∼107 clones) as a pool (Fig. 1c). The counterselection decreased the proportion of constitutively active prey in the purified library by approximately two orders of magnitude (Supplementary Fig. 1).
The purified library was used to determine the DNA-binding specificity of two Cys2His2 zinc finger proteins using a single positive selection step. In a pilot screen using the Zif268 bait, ten of ten sequenced clones contained Zif268 binding sites and eight of these sequences were unique (Supplementary Table 2). Next, the one-step selection procedure was applied to ZnFp53, an artificial zinc finger protein that was previously selected to recognize a portion of the p53 recognition sequence12,15. All 20 of the sequenced prey from the selection were unique and each contained a ZnFp53 recognition element (Fig. 2c).
The one-step selection procedure was also successfully applied to three transcription factors, Dorsal, LAG-1, and Paired, which contain other types of DNA-binding domains. Dorsal from D. melanogaster contains a Rel homology region (RHR) DNA-binding domain and it recognizes DNA as a homodimer. The binding site motif constructed from sequences isolated using the Dorsal bait (Fig. 2d) is similar to the specificity previously determined by SELEX for this domain16. LAG-1 from C. elegans contains a CSL-type DNA-binding domain17. The binding site motif constructed from sequences isolated using the LAG-1 bait (Fig. 2e) is similar to the previously defined DNA-binding specificity of the mouse homolog RBP-Jκ18. The D. melanogaster protein Paired contains a bipartite DNA-binding domain consisting of an N-terminal paired domain and a C-terminal homeodomain. The binding site motif complied from sequences isolated using the Paired bait (Fig. 2f) includes an element that is similar to the previously described recognition sequence for the isolated paired domain determined by SELEX19. For these three factors 70 to 90% of the isolated sequences contained the recognition motif based on computational anaylsis (Supplementary Table 2).
The Drosophila CBFα/β factors Runt and Big-brother (Bgb) were used to test the feasibility of performing selections on a heterodimeric complex. The B1H system should be able to accommodate a heterodimer when both partners are expressed as independent fusions to the alpha subunit of RNA polymerase, since two copies of alpha are present in each polymerase complex (Fig. 1a). Selections performed at low stringency (1 mM 3-AT) using the Runt and Bgb baits and the purified library yielded a few hundred colonies, but only one of eight sequenced clones contained a CBFα/β recognition element (Supplementary Table 2). The high background, which presumably consists of self-activating clones, was removed by an additional counterselection step (Fig. 1b). All of the clones (18 of 18) following the counterselection contained a motif that matches the CBFα/β recognition sequence determined previously by SELEX20 (Fig. 2g). The Runt/Bgb complex binds efficiently to this recognition sequence in vitro, whereas Runt in the absence of Bgb has a lower affinity for its target sequence21. Activation of a reporter containing a representative CBFα/β binding site is dependent on the presence of both members of the heterodimer (Supplementary Fig. 2).
Next, the DNA-binding specificity of a previously uncharacterized transcription factor encoded by the Drosophila gene odd-skipped (odd) was determined. Initial attempts to express the four Cys2His2 zinc fingers of Odd as a B1H bait failed (data not shown). This problem was resolved by changing codons in odd that are poorly utilized in E. coli to preferred synonymous codons. B1H selections using the recoded Odd bait were performed at three different stringencies (1.5, 2.5 and 5 mM 3-AT). Each stringency produced roughly the same 9 base pair motif (Fig. 3a). However, the results suggest different tolerances to base substitutions within the consensus sequence. In particular, A is absolutely conserved at position 4 in the motif generated at 5 mM 3-AT while both A and T are recovered at this position in the motifs generated at 2.5 or 1.5 mM 3-AT.
Figure 3.
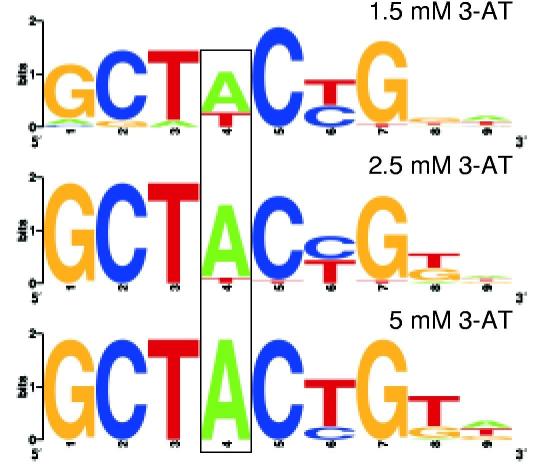

Analysis of the DNA-binding specificity of Odd determined using the B1H system. a) The binding site motifs obtained at 1.5, 2.5 and 5 mM 3-AT were each compiled from more than 20 sequences that contained an overrepresented sequence motif identified by MEME11. The tolerance of Odd for both A and T at position 4 (boxed) in its binding site becomes apparent at lower selection stringencies. Raw sequences of the prey from each selection are listed in Supplementary Table 2. b) Effect of point mutations on the recognition by Odd of its consensus sequence. i) Gel shift assay examining the effect of different DNA competitors on formation of a complex between Odd and its labeled consensus binding site. The presence of Odd and the type of cold competitor in each binding reaction is indicated above each lane of the gel. wt = contains the consensus Odd sequence GCTACTGTA. The other competitors have mutations at each of the first five positions of the consensus sequence where the number represents the position and the letter represents the substitution. For example, 1c = cCTACTGTA. ii) Growth rates for bacteria assayed at 2 mM 3-AT containing the Odd bait and either the wild type or a mutant prey corresponding to DNA competitors used in the gel shift analysis. Growth rates were defined for each bait-prey combination based on serial dilutions of cells harboring these vectors on plates containing 3-AT (Supplementary Fig. 3). The growth rates for cells containing the various bait-prey combinations are qualitatively similar to the degree of competition observed in the gel shift experiments.
The effect of point mutations at each of the first five positions within the Odd consensus sequence on the bait-prey interaction in the B1H system was examined directly. The survival and growth rates of cells containing the Odd bait and each mutant prey were compared to cells containing the consensus Odd prey at various 3-AT concentrations. The effect of individual mutations on the strength of the bait-prey interaction was striking (Fig. 3b and Supplementary Fig. 3). For example, at 2 mM 3-AT only prey containing the conservative A to T mutation at position 4 displayed a similar growth rate to the consensus site. Prey containing a mutation at position 1 or 3 displayed significantly reduced survival while prey containing a mutation at position 2 or 5 were not viable.
in vitro gel shift assays were used to validate the Odd binding site motif. Purified Odd protein binds specifically to a labeled oligonucleotide containing its consensus sequence (Fig. 3b). Excess unlabeled DNA containing this sequence effectively competes for binding to the Odd protein. Competition with an identical concentration of unlabeled DNA containing each of the five point mutations assayed above produced results that are consistent with the B1H data. For example, DNA containing the A to T mutation at position 4 was the only competitor that performs similarly to the consensus sequence. The correlation between the growth rates of bacteria containing the Odd bait and different prey with the in vitro affinity of Odd for these sequences suggests that, under appropriate conditions, the data produced by the B1H system reflects the specificity of the DNA-binding domain being assayed.
To demonstrate that the binding site motif for Odd reflects its in vivo specificity, this data was used to predict potential regulatory targets in the fly genome based on the presence of neighboring Odd binding sites. Target Explorer22 was used to search the D. melanogaster and D. pseudoobscura genomes for syntenic 300 base pair segments that contain at least two Odd binding sites. A list of 130 segments satisfying these criteria contains a number of neighboring genes that share biological functions with Odd (Supplementary Table 3). hairy provides a particularly striking example because the CRMs for individual pair rule stripes have been defined23. The predicted pair of Odd binding sites neighboring hairy fall within the stripe 1 CRM. Moreover, Odd sites of similar quality are found in the corresponding genomic region of six other Drosophila species (Supplementary Fig. 4). In addition, ectopic expression of Odd (a transcriptional repressor) has previously been shown to selectively eliminate the expression of the first stripe of hairy24 (Fig. 4). The effects of ectopically expressed Odd on several other predicted targets were investigated by in situ hybridization (Fig. 4). Two genes, gsb and Gsc, that have not been previously defined as direct targets of Odd, displayed dramatically reduced expression shortly following induction of ectopically expressed Odd. However, other predicted targets (e.g. oc and Ubx) did not display strongly altered expression levels or patterns at the developmental stages that were examined (data not shown).
Figure 4.

Altered gene expression following ectopic expression of Drosophila Odd. Probes used for RNA in situ hybridization are indicated to the left of each pair of panels. (upper panels) Wild type embryos showing the head and trunk stripes of odd expression at the pair rule and segment polarity stages. (lower panels) Expression of putative targets of the Odd repressor was examined in wild type (left) or heat-shock odd embryos24 (right) that were fixed 19 minutes following a six minute heat shock. In heat-shock odd embryos: stripe 1 of h disappears (arrow); the even stripes of prd are missing (bracket); the segment polarity stripes of gsb are missing or reduced (bracket); and the head stripe of Gsc is fainter (arrow). For analysis of Gsc, the embryos were co-stained with a probe to Ubx (central stripe), which did not significantly change following ectopic expression of odd. The changes observed in h and prd expression following ectopic expression of odd have been previously described24.
The B1H system may not be suited for determining the specificity of every DNA-binding domain. In preliminary experiments, we observed that a bait containing the Max bHLH domain was toxic in bacteria, and that selections using the bZip domain of Giant did not yield a significant number of colonies. The current library is sufficiently diverse for the analysis of most transcription factors since it provides nearly complete coverage of all possible 12 bp sites. The use of a more complex library may improve the quality of the binding site motif that is produced for DNA-binding domains with larger recognition elements. It should also be possible to increase the number of binding sites characterized per sequencing reaction by concatemerizing sites using a procedure similar to the SELEX-SAGE protocol2. This modification would facilitate the analysis of larger numbers of selected prey to increase the accuracy of the binding site motif that is produced.
In conclusion, we have constructed a B1H system that is capable of identifying the binding site motif of transcription factors. This technology has the advantage that it employs standard molecular biology reagents and techniques, it requires no protein purification, and it can identify target sites in a single round of selection using one selection plate. Using this technology, we successfully characterized four Cys2His2 zinc finger proteins and four other types of DNA-binding domains. These include proteins that homodimerize and heterodimerize for DNA recognition. In the case of Odd, this information led to the successful prediction of some known and potential new regulatory targets. This system should be applicable to transcription factors from a wide range of organisms, and it may be amenable to the high-throughput analysis of factors.
METHODS
Bacteria selection strain. The E. coli selection strain has a deletion in both the hisB and pyrF genes (the bacterial homologs of HIS3 and URA3) and it contains a F' episome bearing the laclq repressor. The construction of this strain and characterization of the URA3 reporter will be described elsewhere (Meng, X; Smith, RM; Joung, JK and Wolfe, SA unpublished results).
Medium. His-selective (positive) NM medium is composed of M9 minimal medium supplemented with 10 μM ZnSO4, 100 μM CaCl2, 1 mM MgSO4, 200 μM adenine-HCI, 10 μg/ml thiamine, 25 μg/ml kanamycin, 10 μM isopropyl β-Dthiogalactoside, 1 to 5 mM 3-AT, 200 μM uracil, and a mixture of 17 amino acids (excluding histidine, methionine, and cysteine)25. 100 μg/ml carbenicillin and/or 30 μg/ml chloramphenicol were used to select for the presence of the appropriate bait plasmid(s). 5-FOA-selective (negative) YM medium is composed of M9 minimal medium supplemented with 10 μM ZnSO4, 100 μM CaCl2, 1 mM MgSO4, 10 μg/ml thiamine, 25 μg/ml kanamycin, 2 mM 5-FOA, 200 μM uracil, 5 mg/ml histidine, 100 μg/ml yeast extract. For selective plates, 1.8% agar was added to the medium mixture.
Plasmids. The reporter plasmid pH3U3 (kanamycin resistance) contains the HIS3 and URA3 genes, each with independent Shine-Dalgarno sequences, under the control of a weak lac promoter (Fig. 1a). The HIS3/URA3 transcription element was derived from Pzif-HIS3-aadA9 by substituting URA3 in place of aadA. A multiple cloning site for inserting the randomized DNA sequences (prey) was introduced upstream of the HIS3/URA3 promoter. The pH3U3 plasmid also contains a phage f1 origin and a pSC101 origin of replication that limits the plasmid copy number to approximately ten per cell26. The bait plasmid, pACL-αgal4 vector9 (chloramphenicol resistance), was used for all of the selections with the DNA-binding domain introduced between the Not I and Avr II restriction sites. This plasmid allows the IPTG-inducible expression of each transcription factor as a direct fusion to the alpha subunit of RNA polymerase. For the Runt/Bgb selections, Bgb was expressed from the pACL-αgal4 vector and Runt was expressed as a direct fusion to the alpha subunit of RNA polymerase using a derivative of pBR-GP-Z123 (ref. 9; ampicillin resistance) where the N-terminal domain of alpha and its lpp/lacUV5 promoter from pACL-αgal4 replace the Gal11P expression cassette. All fusions to the alpha subunit were via a short linker (15 to 23 amino acids depending on the presence of the FLAG tag). In the case of Odd, Dorsal, Paired, Runt and Bgb a FLAG-epitope tag was included within this linker to monitor protein expression levels. The linker sequence used for Odd is AAADYKDDDDKFRTGSKTPPHRS where AAA encodes the Not I cloning site. pGEX-6p-1 (Amersham) was used to express GST fusions to Odd.
DNA-binding domains used in the B1H selections. Zif268 was subcloned directly from pBR-GP-Z123 (ref. 9). ZnFp53 is clone number 3 from the finger 1 reselected p53ZF zinc finger protein12. The seven fingers used in the PLAG1 bait comprised residues 1-244. The four fingers in the Odd bait comprised residues 214-330, which was amplified from genomic DNA. Western blot analysis of the Odd bait generated using this sequence revealed that the expressed fusion protein was almost exclusively truncated (data not shown). Ten underrepresented codons (R215, R227, K255, R258, R259, R264, R267, P275, R311, R316) in the Odd bait were altered to synonymous codons, which dramatically improved the expression of full-length bait protein (data not shown). The LAG-1 bait comprised residues 198 to 674 that span its CSL domain17. The Dorsal bait comprised residues 46 to 340 that span its RHR domain. The Paired bait comprised residues 2 to 250 derived from clone GH22686 that span both the paired and homeodomain DNA-binding domains19. The Runt bait comprised residues 101 to 255 derived from clone GH02614 that span the runt domain. The Bgb bait comprised residues 21 to 174 derived from clone SD08175 that span the CBFβ domain.
Construction of the original and purified prey library. An 18 bp randomized oligonucleotide library was introduced as a cassette between the Not I and Asc I sites in pH3U3 56 bp upstream of the transcription start site. Following ligation, the resulting mixture of plasmids was transformed into XL-1 Blue electrocompetent cells (Stratagene) to generate the prey library. The library size was estimated to be approximately 2 × 107 unique clones based on the serial dilution of transformed cells. These transformed cells were expanded and the prey plasmids were isolated (Qiagen Maxiprep). All five clones sequenced from the library contained a different 18 bp insert.
The purified prey library was generated by transforming the selection strain with the original prey library and challenging these cells to grow on YM plates containing 2 mM 5-FOA. Transformants (7.8 × 10 7) were divided evenly among 10 square plates (245 mm × 245 mm) and then incubated at 37 °C for one day. Surviving cells were washed off the plates by applying 10 ml 2xYT and a small number of sterile glass beads to the plate. The plates were shaken to resuspend the colonies and the cells were collected as a pool. Prey plasmids were isolated from half of the cell volume using a QIAGEN MIDIprep kit to generate the purified prey library.
Binding site selection protocol. A one-step or two-step selection procedure was used to isolate prey containing recognition sequences for each bait. For the positive selection step, electrocompetent cells containing bait vector were transformed with either the original or purified prey library and grown in SOC medium for one hour at 37°C. These cells were pelleted, resuspended in NM medium and grown at 37°C for an additional hour. Finally the cells were washed four times with sterile water, once with NM medium and then resuspended in NM medium and plated on NM positive selection plates containing the desired concentration of 3-AT. Typically between 1 × 107 and 8 × 107 cells containing the bait and prey library were plated on each square plate (245 mm × 245 mm). (For each bait tested, the number of clones screened and the precise selection conditions are summarized in Supplementary Table 1.) Cells were grown for ∼48 hours at 37 °C until well-defined colonies were visible on the plates. For the one-step selection procedure, which used the purified library, prey from individual colonies were isolated and sequenced. For the two-step selection procedure, the cells from the positive selection were harvested as a pool by washing the plate and the plasmid DNA from these cells was isolated (see purified library protocol). The resulting mixture of bait and prey plasmids were digested with Xmn I, which specifically cleaves the bait plasmid. This pool of prey DNA was purified by QIAquick PCR purification column (QIAGEN) and then transformed into the selection strain for the 5-FOA counterselection. Typically 105 to 106 transformants were screened on YM plates (245 mm × 245 mm) containing 2 mM 5-FOA (Supplementary Table 1). After incubation at 37 °C for one day, prey from individual colonies were isolated and sequenced. Additional details for the selection procedure can be found in Supplementary Methods.
MEME and BioProspector analysis of prey sequences. Overrepresented sequence motifs within the randomized region of the isolated prey were identified using the MEME11 algorithm (http://meme.sdsc.edu/meme/website/meme.html). MEME analysis allowed zero or one occurrence of a motif per sequence searching for a motif width of 6 to 18 bp. Otherwise the default parameters were used for the sequence analysis. In all cases the listed motif represents the best motif identified by MEME. The gapped motif present in the PLAG-1 consensus sequence was characterized in more detail using BioProspector27 (http://robotics.stanford.edu/~xsliu/BioProspector/). Two 7 bp blocks were used in the motif analysis with an intervening gap of 0 to 4 bp allowed between these blocks. The input sequences were used as background. The motif was not required to appear in all sequences in the data set. Sequence logos28 were generated from the aligned sequences representing each overrepresented motif using the WebLogo29 server (http://weblogo.berkeley.edu/).
Mutational analysis of Odd binding sites in the B1H system. Point mutations were introduced independently at each of the first five positions of the Odd consensus binding sequence (GCTACTGTA) and cloned into the pH3U3 reporter between the Not I and Asc I sites with a 56 bp gap between the 3' edge of the Odd site and the transcription start site (sequence of consensus site oligonucleotides: top 5'-CGCGCCTATCAGTGCTACTGTATGC-3'; bottom 5'-GGCCGCATACAGTAGCACTGATAGG-3). Each prey was independently transformed into the selection strain containing the Odd bait. These cells were recovered and washed as described for the positive selections above. Next, they were serially diluted in 10-fold steps and then 5 μl drops of each dilution were plated on selective and non-selective plates.
in vitro gel mobility shift assay. The Odd zinc finger domain was expressed as a GST fusion in Rosetta2 (DE3) pLYS cells (Novagen). During the purification, Odd was proteolytically cleaved from the GST tag. The annealed oligonucleotides used to introduce the consensus site (GCTACTGTA) into pH3U3 were end-labeled with [α-32P]-dCTP using Klenow exo− (New England Biolabs) and purified by G-25 column (Amersham). Each binding reaction, which contained approximately 1 μM protein and 66 pM labeled DNA, was carried out in binding buffer [15 mM Hepes-NaOH pH 7.9, 20 mM KCl, 20 mM potassium glutamate, 20 mM potassium acetate, 5 mM MgCl2, 20 μ M ZnSO4, 100 μg/ml BSA, 5% glycerol, 0.1% NP-40, 1 mM DTT] at room temperature for one hour. For the competition reactions the appropriate cold double-stranded oligonucleotide (0.625 μM) was pre-mixed with the labeled consensus site and then incubated with Odd for one hour at room temperature. The competitor oligonucleotides contain the same flanking sequence as the labeled probe. These binding reactions were analyzed on a 10% non-denaturing polyacrylamide gel (0.5 × TBE).
Computational prediction of Odd binding sites in the Drosophila genome. Target Explorer22 was used to generate a position weight matrix (PWM) that describes the DNA-binding specificity of Odd based on the 69 binding sites identified using the B1H system. This PWM was used to search the D. melanogaster genome for 300 base pair segments that contain at least two Odd binding sites that are also found in the D. pseudoobscura genome and are located within 20 kilobases upstream or downstream or within an intron of an annotated transcript. Binding sites within exons were discarded. The PWM score that defined a sequence as an Odd site was set at a threshold (≥7.5) that would generate a short list of segments in the genome containing high affinity Odd sites. Single Odd sites above this threshold occur on average approximately 1 in every 5000 bp of genomic sequence in D. melanogaster. This stringent criteria is likely to exclude some authentic Odd binding sites within the genome. Using Target Explorer22, the cluster of binding sites was judged to be conserved if the 300 bp sequence from the D. melanogaster genome containing the sites is similar based on BLAST analysis (identity > 75%, P-value < 10) to a region of the D. pseudoobscura genome, if these segments belong to a region of synteny between the two species, and if this region from the D. pseudoobscura genome also contains two Odd binding sites that score ≥ 7.5 within a 300 bp window.
in situ hybridization of Drosophila embryos. Both wild type and HSodd2 (transgenic flies expressing Odd under the control of a heat-shock promoter24) embryos were collected from 0 to 4 hours after egg laying and aged for 1 hour at room temperature. These embryos were dechorionated with bleach for 1 min and heat-shocked in a 37 °C water bath for 6 min. After heat-shock, the embryos were rinsed with room temperature water, allowed to recover for 19 min and fixed in a 1:1 mixture of 4% formaldehyde:heptane. Transcripts were detected by whole-mount in situ hybridization using digoxigenin-labeled antisense mRNA and visualized by the alkaline phosphatase-NBT/CIPB reaction (Roche)30.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Keith Joung, Jessica Hurt, Carl Pabo and Hermann Bujard for precursor plasmids and strains. Henry Krause for providing the HSodd2 flies. Lucio Castilla, Sean Landrette, Marian Walhout, Marc Freeman, Tony Ip and the Drosophila Genomic Resource Center for various cDNAs. Nadine McGinnis and Robin Smith for technical support. Marian Walhout and Keith Joung for very helpful discussions. We thank the UCSC genome bioinformatics site and the Institute for Genomic Research, the Genome Sequencing Center at Washington University, Agencourt Bioscience Corporation and HGSC at Baylor College of Medicine for access to and analysis of unpublished Drosophila genome data. S.A.W. and X.M. were supported in part by the Concern Foundation and NIH grant 1R01GM068110, M.H.B. was supported in part by a Basil O'Connor Starter Research Award from the March of Dimes Birth Defects Foundation. This work was supported by NIH grant 1R01GM068110 (S.A.W.) and ACS grant RSG-05-026-01-CCG (M.H.B.).
Footnotes
COMPETING FINANCIAL INTEREST STATEMENT
The authors declare that they have no competing financial interests.
References:
- 1.Berman BP, et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roulet E, et al. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat Biotechnol. 2002;20:831–835. doi: 10.1038/nbt718. [DOI] [PubMed] [Google Scholar]
- 3.Bulyk ML, Huang X, Choo Y, Church GM. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc Natl Acad Sci U S A. 2001;98:7158–7163. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukherjee S, et al. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat Genet. 2004;36:1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Noll DM, Lieb JD, Clarke ND. DIP-chip: rapid and accurate determination of DNA-binding specificity. Genome Res. 2005;15:421–427. doi: 10.1101/gr.3256505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson TE, Fahrner TJ, Johnston M, Milbrandt J. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science. 1991;252:1296–1300. doi: 10.1126/science.1925541. [DOI] [PubMed] [Google Scholar]
- 8.Deplancke B, Dupuy D, Vidal M, Walhout AJ. A gateway-compatible yeast one-hybrid system. Genome Res. 2004;14:2093–2101. doi: 10.1101/gr.2445504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joung JK, Ramm EI, Pabo CO. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci U S A. 2000;97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dove SL, Joung JK, Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386:627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- 11.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 12.Wolfe SA, Greisman HA, Ramm EI, Pabo CO. Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. J. Mol. Biol. 1999;285:1917–1934. doi: 10.1006/jmbi.1998.2421. [DOI] [PubMed] [Google Scholar]
- 13.Voz ML, Agten NS, Van de Ven WJ, Kas K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000;60:106–113. [PubMed] [Google Scholar]
- 14.Walhout AJ, Vidal M. A genetic strategy to eliminate self-activator baits prior to high-throughput yeast two-hybrid screens. Genome Res. 1999;9:1128–1134. doi: 10.1101/gr.9.11.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greisman HA, Pabo CO. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science. 1997;275:657–661. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- 16.Senger K, et al. Immunity regulatory DNAs share common organizational features in Drosophila. Mol Cell. 2004;13:19–32. doi: 10.1016/s1097-2765(03)00500-8. [DOI] [PubMed] [Google Scholar]
- 17.Kovall RA, Hendrickson WA. Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. EMBO J. 2004;23:3441–3451. doi: 10.1038/sj.emboj.7600349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tun T, et al. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994;22:965–971. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jun S, Desplan C. Cooperative interactions between paired domain and homeodomain. Development. 1996;122:2639–2650. doi: 10.1242/dev.122.9.2639. [DOI] [PubMed] [Google Scholar]
- 20.Melnikova IN, Crute BE, Wang S, Speck NA. Sequence specificity of the core-binding factor. J Virol. 1993;67:2408–2411. doi: 10.1128/jvi.67.4.2408-2411.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golling G, Li L, Pepling M, Stebbins M, Gergen JP. Drosophila homologs of the proto-oncogene product PEBP2/CBF beta regulate the DNA-binding properties of Runt. Mol Cell Biol. 1996;16:932–942. doi: 10.1128/mcb.16.3.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sosinsky A, Bonin CP, Mann RS, Honig B. Target Explorer: An automated tool for the identification of new target genes for a specified set of transcription factors. Nucleic Acids Res. 2003;31:3589–3592. doi: 10.1093/nar/gkg544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riddihough G, Ish-Horowicz D. Individual stripe regulatory elements in the Drosophila hairy promoter respond to maternal, gap, and pair-rule genes. Genes Dev. 1991;5:840–854. doi: 10.1101/gad.5.5.840. [DOI] [PubMed] [Google Scholar]
- 24.Saulier-Le Drean B, Nasiadka A, Dong J, Krause HM. Dynamic changes in the functions of Odd-skipped during early Drosophila embryogenesis. Development. 1998;125:4851–4861. doi: 10.1242/dev.125.23.4851. [DOI] [PubMed] [Google Scholar]
- 25.Serebriiskii I, Joung J. In: Protein-Protein Interactions: A Molecular Cloning Manual. Golemis E, editor. Cold Spring Harbor Laboratory Press; 2001. pp. 93–142. [Google Scholar]
- 26.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput. 2001:127–138. [PubMed] [Google Scholar]
- 28.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.