

Abstract
A competitive PCR (cPCR) assay targeting 16S ribosomal DNA was developed to enumerate growth of a Dehalococcoides-like microorganism, bacterium VS, from a mixed culture catalyzing the reductive dehalogenation of cis-1,2-dichloroethene (cDCE) and vinyl chloride (VC), with hydrogen being used as an electron donor. The growth of bacterium VS was found to be coupled to the dehalogenation of VC and cDCE, suggesting unique metabolic capabilities. The average growth yield was (5.2 ± 1.5) × 108 copies of the 16S rRNA gene/μmol of Cl− (number of samples, 10), with VC being used as the electron acceptor and hydrogen as the electron donor. The maximum VC utilization rate (q̂) was determined to be 7.8 × 10−10 μmol of Cl− (copy−1 day−1), indicating a maximum growth rate of 0.4 day−1. These average growth yield and q̂ values agree well with values found previously for dechlorinating cultures. Decay coefficients were determined with growth (0.05 day−1) and no-growth (0.09 day−1) conditions. An important limitation of this cPCR assay was its inability to discriminate between active and inactive cells. This is an essential consideration for kinetic studies.
The widespread use and disposal of tetrachloroethene (PCE) and trichloroethene (TCE) have resulted in significant groundwater contamination (26). Remediation of such contaminated sites is often required because of the toxic nature of these chlorinated ethenes. A potential remediation strategy is the biological anaerobic reductive dechlorination of PCE and TCE to ethene, whereby the chlorinated ethene serves as the electron acceptor and hydrogen serves as the electron donor for the dehalogenating microorganism's energy metabolism. However, incomplete reduction to ethene is a problem due to the accumulation of the intermediates cis-1,2-dichloroethene (cDCE) and vinyl chloride (VC). Therefore, the removal of cDCE to yield ethene is often the limiting factor in utilizing reductive dechlorination as an approach for remediation. Understanding factors affecting the transformation of these lesser-chlorinated ethenes is hence critical for evaluating natural attenuation and for establishing engineering procedures for chlorinated-solvent remediation. The only reported pure culture isolated to date that can dechlorinate PCE completely to ethene is Dehalococcoides ethenogenes strain 195 (18). This microorganism is able to grow via reductive dehalogenation by using PCE, TCE, or cDCE as the catabolic electron acceptor, but the dechlorination of VC is a slow and possibly cometabolic process (18). Due to this limitation, investigating the final reduction of VC to ethene has become especially important, particularly as VC is a known human carcinogen.
Previous research on an enriched anaerobic culture seeded with aquifer material from a PCE-contaminated site in Victoria, Tex., indicated a high transformation rate for VC and cDCE (18 nmol min−1 mg of protein−1), a low transformation rate for TCE, but no PCE transformation (22). This research suggested that the reductive dehalogenation of VC and cDCE by this mixed culture was different from the previously reported reductive dehalogenation of PCE and TCE, with the high rates being indicative of growth coupled to VC reduction. Analysis of 16S ribosomal DNA (rDNA) from this contaminated site (GenBank accession number AF388550 [11]) and our culture revealed the presence of a Dehalococcoides-like microorganism. It is likely that the high dechlorination rate is linked to this microorganism, which we suggest naming bacterium VS (stands for Victoria Stanford). As isolation of bacterium VS in pure culture proved difficult, we developed a competitive PCR (cPCR) assay to quantify bacterium VS cell numbers during VC and cDCE reduction in mixed culture.
cPCR was chosen for these investigations due to its many merits as reported by others. The results of cPCR are proportional to viable cell counts (16) and selective agar plate counts (13). Also, good reproducibility was found (19) with low coefficients of variation between replicate samples (averaging 2.5%) (21). In addition, the method has been applied to a range of environmental samples, including soil (3, 13, 14, 20), estuarine sediments (20), rumen (21), skim milk (16), and microcosms (15).
cPCR quantifies copy numbers of specific DNA templates, for example, 16S rDNA or functional genes (3, 13, 19, 20, 23). PCR bias is avoided by coamplifying the target fragment with an internal standard (competitor DNA) (15, 21). The competitor DNA fragment acts or competes for primers and DNA polymerase in a manner similar to that of the target DNA in the PCR (7). The cPCR technique involves PCRs containing a mixture of a known competitor DNA concentration and of the target DNA. In order to quantify the target DNA concentration, a series of parallel PCRs where only the competitor DNA concentration is varied is performed. The amplified DNAs are then separated and quantified on agarose gels. As the PCR amplifies both the sample template and the competitor DNA with the same efficiency, the ratios of the final amplification products (target to competitor DNA) reflect the ratios between the initial amounts of the two templates, allowing the amount of initial target DNA to be determined (7). We have developed a cPCR assay to determine cell numbers of the Dehalococcoides-like microorganism bacterium VS and applied this assay for the first quantification of growth of a microorganism by means of the reductive dehalogenation of VC.
MATERIALS AND METHODS
Chemicals.
Liquid cDCE (Aldrich Chemical Co., Milwaukee, Wis.) and gaseous VC (≥99%; Scott Speciality Gases, Alltech Associates, Inc., Deerfield, Ill.) were used to prepare stock solutions and analytical standards. In addition, ethene (≥99%; Scott Speciality Gases, Alltech Associates) was used as an analytical standard, benzoate (sodium salt, 99%; Aldrich Chemical Co.) was used as a substrate, and sodium sulfide (Aldrich Chemical Co.) was used as a reducing agent.
Culture and growth conditions.
A dehalogenating source culture was maintained in a closed continuously stirred tank reactor (CSTR) initially seeded with aquifer material from a PCE-contaminated site in Victoria, Tex. The reactor was anaerobically maintained, as indicated by a redox indicator (resazurin), at 20 (±2)°C. A continuous anaerobic feed consisting of 5.9 mg of Na2S liter−1 as a reductant, 1.7 mM sodium benzoate, 20 mg of yeast extract liter−1, 0.5 mM TCE, and trace nutrients in a basal medium was delivered by a syringe pump at 67 ml/day, resulting in a 57-day retention time. The basal medium was identical to that previously described (27), except that the chloride salts in the mineral solution were replaced with bromide salts (10 g of NaBr, 20 g of NH4Br, 8 g of KBr, 7 g of MgBr2 · 6H2O, and 2.5 g of CaBr2 liter−1). Every 3 days, 200 ml was removed to bring the liquid volume within the reactor to 3.8 liters. Complete dehalogenation of TCE to ethene occurred with all chloroethenes in the reactor remaining below 1 μM. DNA was periodically extracted (see DNA isolation) from the CSTR culture and subjected to cPCR to determine variations in culture concentration over time.
VC growth curve and yield determination.
In an anaerobic chamber (10% H2, 10% CO2, 80% N2), two batch bottles (120 ml) were filled with 59 ml of anaerobic medium (without sodium benzoate or TCE, as previously described above). Each bottle was inoculated with 1 ml of inoculum of a rapidly dechlorinating culture grown from a small inoculum (1 ml of CSTR culture) under optimal conditions (>3% H2 and >40 μM VC). Two septum liners (TFE-silicone, part number 95302; Alltech Associates) were crimped on each bottle, allowing the diffusion of hydrogen from the anaerobic chamber (20 ± 2°C) into the bottles (with minimal diffusion of chloroethenes or ethene occurring), resulting in a continuous hydrogen supply over the incubation period. VC (99.5%) was added with a gas-tight syringe on day 0 (130 μl) and day 15 (250 μl). An abiotic control with medium and VC only was included to demonstrate that the culture was required for dechlorination. A control that contained medium and culture only, without VC, was also included to illustrate that the presence of VC was required for growth. The bottles were kept in an anaerobic chamber on a shaker. cPCR was carried out on the inoculum (in triplicate with 1-ml culture samples) and on both the sample cultures and the VC control on days 12, 15, 17, 18, and 19 (single 1-ml culture samples).
Batch study with a range of inocula.
Samples of CSTR culture were taken anaerobically from the reactor and purged with a mixture of 80% N2 and 20% CO2 to displace ethene. Batch studies were conducted with dilutions of the CSTR culture (0, 0.5, 1, 3, and 9 ml for samples with VC added and 0, 1, and 9 ml for samples with cDCE added). In an anaerobic chamber, sufficient anaerobic medium (as described above) was added to bring the final volume to 60 ml. VC (125 μl of 99.5% VC) was added with a gas-tight syringe, and cDCE (60 μl of cDCE solution) was added with a Hamilton syringe. Abiotic controls consisted of 60 ml of medium (as described above) plus VC or cDCE only, and controls containing culture (1 ml) and medium (59 ml) but no VC or cDCE were also included. The bottles were maintained as described above. cPCR was performed on the original seed culture and within 1 to 2 days after dechlorination was complete.
Following complete dechlorination, three of the batch bottles were filled with a gas mixture (10% H2, 10% CO2, 80% N2) and a 2-ml vial (containing the same gas mixture) with a septum allowing the diffusion of hydrogen from the vial was also added to each bottle. The 2-ml vial was added to supply an extra source of hydrogen to the culture, thereby promoting maximum dechlorination rates. In addition, more VC was added (day 40) and cPCR was again performed within 1 to 2 days after dechlorination was complete.
Rate determinations.
Monod kinetics (equations 1 and 2) were fitted by nonlinear least-squares analysis (25) to the VC dechlorination data to determine values for the maximum utilization rate (q̂) and decay rate (b) as follows:
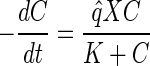 |
(1) |
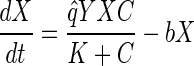 |
(2) |
where C is the solution concentration of VC (in micromolar units), X is the bacterium VS concentration (in number of chromosome copies per liter), q̂ is in micromoles per copy per day, and K is the VC half-velocity coefficient (2.6 μM) (10). In addition, Y is the yield (number of copies per micromole) and decay rate is per day. Properties of the nonlinear least-squares estimates of q̂ VC and decay rate were analyzed according to the method of Smith et al. (25).
Decay rate measurements.
For a more direct measurement of the decay coefficient, a batch study was conducted to measure maximum dechlorination rates in cultures lacking an electron acceptor (i.e., no-growth conditions) as a function of the time of culture incubation. Eight bottles were filled with 60 ml of mixed culture, and four abiotic controls consisted of 60 ml of medium only. At days 0, 8, 14, and 21, the gas spaces within duplicate culture bottles and one control were filled with a gas mixture (10% H2, 10% CO2, 80% N2) and VC (70 μl of 99.5%) was added to each duplicate bottle with a gas-tight syringe. VC removal and ethene formation were then monitored over time. Hydrogen and VC levels remained high (>3% H2 and [VC] ≫ K) throughout the 1.25-day time period. cPCR was carried out on the day 0 and 21 culture samples.
Analytical methods.
Analyses of ethene, VC, and cDCE (250 μl of headspace) were performed with a temperature gradient of 40 to 220°C and with a model 5890 series II gas chromatograph equipped with a flame ionization detector (Hewlett-Packard) and a GS-Q fused-silica capillary column (length, 30 m; inside diameter, 0.53 mm; J&W Scientific). Total mass values were determined with Henry's law constants (8).
DNA isolation.
Genomic DNA was extracted from 1 ml of culture samples with a DNeasy tissue system (Qiagen, Inc.) by following the manufacturer's instructions (final volume, 200 μl). Replicate samples were taken from each culture to ensure consistency of extraction.
Amplification and cloning of the D. ethenogenes 16S rRNA sequence.
PCR was carried out on DNA extracted from the culture with forward primer DeF and reverse primer DeR (Table 1). Amplification reactions were performed in a total volume of 20 μl with a PTC-200 Peltier thermal cycler (MJ Research). The reaction mixtures contained a 0.5 μM concentration of each primer (Operon), a 200 μM concentration of each deoxynucleoside triphosphate (Qiagen, Inc.), 1.5 U of Taq polymerase (Promega), PCR buffer [Tris-Cl, KCl, (NH4)2SO4, MgCl2] (Qiagen, Inc.), Q solution (Qiagen, Inc.), and 1 μl of extracted DNA from the culture. The following PCR program was used: 95°C for 5 min; 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and 72°C for 5 min. The PCR products were resolved by electrophoresis in 2% (wt/vol) agarose gels (Sigma) in Tris-borate-EDTA buffer, stained in ethidium bromide solution (5 μg/ml) for 20 min, and destained in water for 40 min. The band, visualized upon UV excitation, was found to be of the appropriate size with a 1-kb DNA ladder (Gibco). The remaining PCR product was purified from contaminants (Qiagen, Inc.) and cloned into Escherichia coli with a TOPO TA cloning kit (Invitrogen Corporation, Carlsbad, Calif.). Plasmids were extracted from the cloned cells with a QIAprep miniprep system (Qiagen, Inc.), and the insertion was sequenced at Stanford University’s Protein and Nucleic Acid Facility (PAN).
TABLE 1.
Primers used in this study
| Primer | Sequence (5′ to 3′) | Target(s)a |
|---|---|---|
| DeR | ACTTCGTCCCAATTACC | 1406-1422, D. ethenogenes 16S rDNA |
| DeF | GCAATTAAGATAGTGGC | 49-65, D. ethenogenes 16S rDNA |
| VicF | TCACAGGGAAGAATAATGAC | 402-422, D. ethenogenes 16S rDNA |
| CDeR | ACTTCGTCCCAATTACCGTCTCGCTAGAAAATTTAAC | 1406-1422 and 1067-1086, D. ethenogenes 16S rDNA |
Numbers refer to nucleotide positions of the D. ethenogenes 16S rDNA (GenBank accession number AF004928).
The resulting 16S rRNA gene sequences (1,373 bp) obtained from the mixed culture match (100%) a sequence previously submitted to GenBank (GenBank accession number AF388536 [11]) and are similar to the sequence found at Victoria, Tex. (GenBank accession number AF388550 [11]). The two sequences, differing by only 2 bp, have been placed into the Victoria sequence subgroup (11) and contain 14- to 16-bp differences from D. ethenogenes (GenBank accession number AF004928).
Construction of competitor and target standards.
The competitor DNA fragment was constructed according to the method of Celi et al. (4) with primers VicF and CDeR (Table 1). The resulting amplicon contained the same priming sites as the target with a 318-bp deletion near the 3′ end. The reverse primer CDeR contained a sequence of the 3′ end (positions 1406 to 1422) followed by a sequence from within the target (positions 1067 to 1086), allowing the amplification product to contain a deletion of 318 bp. PCR with the VicF and DeR primers resulted in 703 (competitor DNA)- and 1,021 (target DNA)-bp amplicons.
The competitor DNA fragment was cloned into E. coli with a TOPO TA cloning kit (Invitrogen Corporation). Plasmids were extracted from the cloned cells using a QIAprep miniprep system (Qiagen, Inc.) and were PCR amplified with the primers VicF and DeR. The PCR product was then extracted from agarose gel with a QIAquick gel extraction kit (Qiagen, Inc.), stored at −20°C, and kept as a stock solution for cPCRs. Target DNA was amplified from the dehalogenating culture DNA and, as with the competitor standard, the PCR products were cloned, extracted from plasmids, PCR amplified, extracted from gel, and stored at −20°C. Both stock solutions of competitor and target DNAs were quantified with a DyNA Quant 200 fluorometer (Hoefer Pharmacia Biotech, San Francisco, Calif.) calibrated with calf thymus and coumarin 311.
Primer design.
The specificity of the selected primer set was examined with the PROBE-MATCH program of the Ribosomal Database Project (17) and BLAST in GenBank (2). The VicF and DeR primers were used for selective amplification of the 16S rDNA of bacterium VS from samples. The maximum annealing temperature to optimally amplify the target was empirically found to be 55°C (data not shown).
cPCR.
Amplification reactions were performed as described above with primers VicF and DeR, 1 μl of extracted DNA from the sample, and 1 μl of competitor DNA. The PCR products were resolved in 2% (wt/vol) agarose gel (Sigma) in Tris-borate-EDTA buffer, stained in ethidium bromide solution (5 μg/ml) for 20 min, and destained in water for 40 min. The bands were visualized by UV excitation and then digitally photographed (Kodak DC290 zoom digital camera). The images were analyzed with Kodak ID image analysis software.
Sequencing of cPCR primer amplicons.
Amplicons from the mixed culture with VicF and DR primers were separately cloned into E. coli with the TOPO TA cloning kit (Invitrogen Corporation). Plasmids were extracted from the cloned cells with the QIAprep miniprep system (Qiagen, Inc.) and sequenced (PAN facility). The sequences were analyzed with DNASTAR MegAlign software.
RESULTS AND DISCUSSION
Quantification of Dehalococcoides-like bacterium VS.
To study the microorganism responsible for high VC and cDCE dechlorination rates (22) in mixed culture, we developed an assay to quantify 16S rRNA gene copies of this Dehalococcoides-like microorganism. The ability to enumerate this microorganism in mixed culture with cPCR provided an opportunity to study its growth and kinetics with greater ease and in the natural environment of a mixed culture.
Chromosomal DNA was typically extracted from three 1-ml samples of culture, and cPCR was carried out on all three samples. To determine assay reproducibility, the coefficients of variation (number of assays, 15) for a single measurement (13%) and for triplicate samples (7%) were calculated, illustrating consistency in both DNA extraction and the cPCR assay itself. Figure 1 provides an example of the assay; the final copy number here was estimated to be (2.6 ± 0.1) × 107 per ml of mixed culture (for triplicate samples) (data not shown).
FIG. 1.

Example of the cPCR assay. (A) A constant amount of extracted DNA was coamplified with the serially diluted competitor sequences (2.9 × 105, 1.4 × 105, 0.7 × 105, and 0.4 × 105 copies/μl) and subjected to gel electrophoresis. (B) The ratios of the intensities of the target to the competitor (diamonds) are plotted against the concentrations of the competitor on a log scale. The target copy number equals the competitor number when the log ratio equals zero.
cPCR is dependent on the equal amplification efficiencies of the target and competitor sequences. Amplification efficiencies were investigated for a range of PCR cycles (25 to 30) by starting with equal copy numbers of target and competitor sequences (both from stock solutions) and then determining the final ratio at cycle numbers 25, 26, 27, 28, 29, and 30. At all cycles, with equal initial concentrations of competitor and target sequences, the competitor was amplified more efficiently than the target (data not shown). Preferential amplification of a competitor or target can occur due to different lengths or because of differential denaturation (5). cPCR was carried out on a range of target copy numbers to quantify and correct for the bias in amplification efficiency between the target and competitor sequences. When the expected copy number based on the concentration of the cloned 16S rDNA was plotted versus the copy number calculated by the cPCR assay, the copy number was underestimated, but the relationship was linear, with a slope of 0.76 (standard error ± 0.037) (Fig. 2). This value was utilized as a correction factor for all cPCR assays. Such correction factors have been successfully used in other cPCR systems (6, 14).
FIG. 2.

Quantification of 16S rDNA copies. Numbers of determined copies were counted via cPCR and compared to actual copy numbers (previously quantified with a fluorometer—see Materials and Methods). The lighter line represents a 1:1 ratio of actual numbers of copies to determined numbers of copies.
To evaluate primer pair specificity, PCR (with primers VicF and DR) was carried out with the mixed-culture DNA, the PCR products were cloned, and 10 were randomly selected for sequencing. Nine sequences exhibited 100% identity with the 16S rDNA previously obtained from the culture, indicating that the primers are specific to the target 16S rDNA. The remaining sequence contained one base pair change, which was probably a result of PCR error. These results indicate that the cPCR primers VicF and DR are selective for the 16S rDNA of bacterium VS from the CSTR culture. It is important to note that these primers may not be selective for DNA obtained from other environmental samples containing microorganisms not present in this CSTR culture. Although the primers were checked for specificity against sequences in the current NCBI database, perhaps there are microorganisms with 16S rDNA that may hybridize to these primers but are not yet documented in the NCBI database. Notably, the primers may amplify other Dehalococcoides strains. Therefore, before this assay is applied to samples from different environments, amplicons should be initially sequenced to ensure specificity, as was done in this study. The detection limit of the assay performed with 30 cycles was determined to be 2,000 copies, equivalent to 4 × 105 copies/ml of mixed culture.
cPCR was carried out with six duplicate or triplicate 1-ml samples of CSTR culture over 10 months. The resulting concentrations of bacterium VS remained relatively constant over this time at (1.2 ± 0.4) × 107 copies/ml. The constancy in CSTR cell concentration provided confidence in both the consistency of the DNA extraction method and the assay itself.
Growth of bacterium VS through reductive dehalogenation.
A growth curve with the corresponding change in mass of VC and ethene was obtained (Fig. 3). The lines in Fig. 3A and B represent numerical simulations of data discussed later. Mass balances ranged from 73 to 99% (depending on incubation time), with loss being attributed to VC leakage as the abiotic control also exhibited VC loss. For all studies, no dechlorination occurred in the abiotic controls and no increase in copy numbers above detection limits (4 × 105 copies/ml) was found in control cultures without chlorinated ethene addition, but copy numbers were greater than 8 × 106 per ml in all bottles incubated with VC or cDCE. As would be expected from growth via VC dehalorespiration, a clear correlation (r2 = 0.93) resulted between cell numbers and VC dechlorination (Fig. 3C). These data indicate a growth yield of 5.1 × 108 copies/μmol of Cl− released.
FIG. 3.

VC (filled squares) removal and ethene (open squares) formation (A) with the resulting increase in copy number (filled circles) (B). Additional VC (10.5 μmol) was added on day 15. Points are measured values, and lines are those predicted by a nonlinear least-squares fit to the model (equations 1 and 2) to determine decay rate and q̂. An increase in the number of copies is correlated with the formation of ethene, resulting in a yield coefficient of 5.1 × 108 copies/μmol of Cl− (r2 = 0.93) (C). The duplicate culture behaved similarly, with a yield coefficient of 5.9 × 108 copies/μmol of Cl− (r2 = 0.83) (data not shown).
Dechlorination kinetics.
Using the determined growth yield, experimental data (concentrations of VC and ethene and copy numbers) (Fig. 3) were fit to equations 1 and 2 by nonlinear least-squares analysis, yielding a q̂ coefficient of 7.8 × 10−10 μmol of Cl− (copy−1 day−1) and a decay coefficient of 0.05 day−1, resulting in a growth rate of 0.4 day−1. This model, when tested for a range of different cell inoculum concentrations (Fig. 4), predicted the data well. Here, the initial inoculum concentration was adjusted for best fit by the least-squares method. This best-fit inoculum concentration was only 8% of that measured by cPCR. As indicated later, the cPCR analysis was found to measure both viable and nonviable cells, and the CSTR cells were found to consist of many nonviable cells. Time for complete dechlorination was dependent on the initial inoculum volume, and hence the number of cells, in both VC (Fig. 4)- and cDCE (data not shown)-amended bottles, and the resulting VC utilization curves were consistent with growth-linked reductive dehalogenation. When the cultures were refed with VC (day 40), we observed a dechlorination rate that was higher than initial rates, indicating the presence of more dechlorinating cells. Copy numbers were obtained both after the initial VC dechlorination and after the additional VC dechlorination; on both occasions, an increase in cells was found (data not shown), again providing evidence of growth. Copy numbers after the additional VC dechlorination occurred were compared to those predicted by the model, resulting in an average ratio (values obtained by cPCR to predicted values) and standard deviation of 0.83 ± 0.29 (number of assays, 3).
FIG. 4.

Application of model (lines) to cultures containing a range of initial inocula. The points are the measured values with 0.5 ml (triangles) (A), 1 ml (diamonds) (B), 3 ml (squares) (C), and 9 ml (circles) (D) of initial inoculum (8 × 106 copies/ml). Additional VC (5.6 μmol) was added on day 40 to the 0.5-ml (A), 1-ml (B), and 9-ml (D) samples.
To determine the relationship between dechlorination activity and copy numbers and to more directly investigate decay rate, maximum dechlorination activity was monitored in cultures lacking an electron acceptor for different periods of time. While there was a significant decrease in dechlorination activity over 3 weeks (Fig. 5), there was no significant change in cell copy numbers [(3.4 ± 0.4) × 1010 versus (3.7 ± 0.4) × 1010 copies/liter at days 0 and 21, respectively], confirming that the assay measured both active and inactive cells. Li and Drake (16) hypothesized that DNA from dead cells was responsible for higher-than-predicted bacterial numbers. Our data demonstrate this phenomenon. These results suggest that cPCR is not a good measure of active cells under conditions in which the growth rate is low and decay has been an important factor. The CSTR was maintained with low hydrogen levels (2 nM), resulting in an environment in which the growth rate was low and long-term decay was important. Inactive cells likely artificially increased the original inoculum concentration estimates, as already discussed. In contrast, the inoculum for the experiment whose results are shown in Fig. 3 was an actively dechlorinating culture growing under optimal conditions (with high concentrations of the electron donor and acceptor); therefore, the cell concentration determined via cPCR, and the resulting maximum utilization and decay coefficients for this case were considered accurate.
FIG. 5.

Ethene formation following VC addition to duplicate cultures lacking an electron acceptor for 0, 8, 14, and 21 days. Points (diamonds) are measured values, and lines are those predicted by a nonlinear least-squares fit to the model (equations 1 and 2) to determine decay rate.
Equations 1 and 2 were fit to the decay data in Fig. 5 by nonlinear least-squares analysis, giving a decay coefficient of 0.09 day−1. The cell concentration from cPCR [(3.4 ± 0.4) × 1010 copies/liter] was not used in the model but was estimated [(2.4 × 1010) copies/liter] from the initial dechlorination rate (19.9 μmol of VC liter−1 day−1) and the maximum specific dechlorination rate found previously (7.8 × 10−10 μmol of VC copy−1 day−1), in order to circumvent the problem of measuring inactive cells. Although it is not clear why the decay coefficient determined with resting cells is higher than that obtained with actively growing cells (0.05 day−1), this finding demonstrates that decay is an important factor for bacterium VS.
In a previous study with an enriched bacterium VS culture (22), a strong correlation between an increase in cell mass and the VC dechlorination rate was found, suggesting that growth is coupled to VC reduction. We have now quantified the growth of the Dehalococcoides-like microorganism in culture and correlated it to VC dehalogenation. We can convert yield and q̂ to the more usual cell mass units by assuming a conversion factor of 1.6 × 10−14 g of cells/copy, based on estimates of 0.2 g of cells (dry weight)/g of cells (wet weight), a cell diameter of 0.5 μm (from an electron micrograph of D. ethenogenes [18]) and one 16S rRNA gene copy/cell (from the D. ethenogenes sequence in GenBank). We further assumed that the bacterium VS cell shape is spherical, based on microscopic observation of the mixed and enriched culture containing bacterium VS and on literature describing Dehalococcoides-like microorganisms as irregular coccoid bacteria (1, 18). The resulting growth yield of 4.9 g of protein/mol of Cl− released and q̂ of 56 nmol min−1 mg of protein−1 are consistent with similar coefficients for other organohalide-respiring cultures (Table 2) and strongly suggest that VC reduction in bacterium VS, with hydrogen as an electron donor, is a catabolic reaction. Notably, reductive dechlorination of VC is a thermodynamically favorable process with hydrogen as the electron donor (ΔGo′ = −149 kJ/mol of VC) (22). The maximum growth rate with VC (0.40 day−1) was similar to that previously estimated by Haston (9) for this culture (0.21 to 0.45 day−1) from dechlorination curves from a range of initial culture inocula.
TABLE 2.
Comparison of growth yields and chloroethene utilization rates of dechlorinating cultures
| Dechlorinating culture | Yield (g of protein/mol of CI−) | Utilization rate (nmol/min · mg of protein) | Reference | Electron donor | Electron acceptors |
|---|---|---|---|---|---|
| Dehalobacter restrictus | 2.1 | 12 | Hydrogen | PCE, TCE | |
| Dehalospirillum multivorans | 1.4 | 50 | 24 | Hydrogen | PCE, TCE |
| Dehalococcoides ethenogenes | 4.8 | 69 | 18 | Hydrogen | PCE, TCE, cDCE |
| Bacterium VSa | 4.9 | 56 | This study | Hydrogen | cDCE, VC |
Values assume 0.6 g of protein/g of cells and 1.6 × 10−14 g of cells/copy.
An important finding through the use of this assay is the ability of bacterium VS to grow on VC, in contrast to the cometabolic VC dehalogenation exhibited by D. ethenogenes strain 195 (18). It appears that the different cDCE- and VC-dehalogenating strains with 16S rRNA similar to D. ethenogenes strain 195 have varying abilities for dechlorination and growth: bacterium VS exhibits high rates of cDCE and VC dechlorination with little TCE dechlorination and no PCE dechlorination (22), whereas D. ethenogenes strain 195 dechlorinates cDCE, TCE, and PCE well but with slow, only cometabolic, VC dechlorination (18). Such a varying dechlorinating ability has been previously noted for another strain called CBDB1, sharing 98.3% similarity with D. ethenogenes strain 195 (1). However, in this case, chlorobenzene was used as the electron acceptor and hydrogen was used as the electron donor in order to obtain growth (1).
Microorganisms with 16S rRNA similar to that of D. ethenogenes are frequently found at contaminated sites and in mixed cultures undergoing complete dechlorination (11). Thus far, different strains can grow on the more chlorinated ethenes (D. ethenogenes strain 195), the less chlorinated ethenes (bacterium VS), or chlorobenzenes (1). We can therefore speculate that there are many different strains of Dehalococcoides with the ability to grow on a variety of different chlorinated compounds. The difficulty in isolating these microorganisms has hindered researchers in understanding their distinctive characteristics. The ability to enumerate these similar strains from mixed culture by cPCR should enable researchers to better identify and quantify these unique growth abilities.
Acknowledgments
We thank Yanru Yang, Jochen Müller, Dale Pelletier, Mandy Ward, and Gypsy Achong for their useful discussions and technical support. We also extend our thanks to Edwin Hendrickson for his advice on primers.
This study was supported through grant CU-1169 by the Strategic Environmental Research and Development Program, sponsored by the U.S. Department of Defense, the U.S. Department of Energy, and the U.S. Environmental Protection Agency, and by E. I. DuPont de Nemours, Inc., through the U.S. Environmental Protection Agency-sponsored Western Region Hazardous Substance Research Center.
REFERENCES
- 1.Adrian, L., U. Szewzyk, J. Wecke, and H. Görisch. 2000. Bacterial dehalorespiration with chlorinated benzenes. Science 408:580-583. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Baek, J.-M., and C. M. Kenerley. 1998. Detection and enumeration of a genetically modified fungus in soil environments by quantitative competitive polymerase chain reaction. FEMS Microbiol. Ecol. 25:419-428. [Google Scholar]
- 4.Celi, F. S., M. E. Zenilman, and A. R. Shuldiner. 1993. A rapid and versatile method to synthesize internal standards for competitive PCR. Nucleic Acids Res. 21:1047.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandler, D. P. 1998. Redefining relativity: quantitative PCR at low template concentrations for industrial and environmental microbiology. J. Ind. Microbiol. Biotechnol. 21:128-140. [Google Scholar]
- 6.Dionisi, H. M., A. C. Layton, G. Harms, I. R. Gregory, K. G. Robinson, and G. S. Sayler. 2002. Quantification of Nitrosomonas oligotropha-like ammonia-oxidizing bacteria and Nitrospira spp. from full-scale wastewater treatment plants by competitive PCR. Appl. Environ. Microbiol. 68:245-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diviacco, S., P. Norio, L. Zentilin, S. Menzo, M. Clementi, G. Biamonti, S. Riva, A. Falaschi, and M. Giacca. 1992. A novel procedure for quantitative polymerase chain reaction by coamplification of competitive templates. Gene 122:313-320. [DOI] [PubMed] [Google Scholar]
- 8.Gossett, J. M. 1987. Measurement of Henry's law constants for C1 and C2 chlorinated hydrocarbons. Environ. Sci. Technol. 21:202-208. [Google Scholar]
- 9.Haston, Z. C. 1999. Ph.D. thesis. Stanford University, Stanford, Calif.
- 10.Haston, Z. C., and P. L. McCarty. 1999. Chlorinated ethene half-velocity coefficients (Ks) for reductive dehalogenation. Environ. Sci. Technol. 33:223-226. [Google Scholar]
- 11.Hendrickson, E. R., J. A. Payne, R. M. Young, M. G. Starr, M. P. Perry, S. Fahnestock, D. E. Ellis, and R. C. Ebersole. 2002. Molecular analysis of Dehalococcoides 16S ribosomal DNA from chloroethene-contaminated sites throughout North America and Europe. Appl. Environ. Microbiol. 68:485-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holliger, C., D. Hahn, H. Harmsen, W. Ludwig, W. Schumacher, B. Tindall, F. Vazquez, N. Weiss, and A. J. B. Zehnder. 1998. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch. Microbiol. 169:313-321. [DOI] [PubMed] [Google Scholar]
- 13.Johnsen, K., Ø. Enger, C. S. Jacobsen, L. Thirup, and V. Torsvik. 1999. Quantitative selective PCR of 16S ribosomal DNA correlates well with selective agar plating in describing population dynamics of indigenous Pseudomonas spp. in soil hot spots. Appl. Environ. Microbiol. 65:1786-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee, S.-Y., J. Bollinger, D. Bezdicek, and A. Ogram. 1996. Estimation of the abundance of an uncultured soil bacterial strain by a competitive quantitative PCR method. Appl. Environ. Microbiol. 62:3787-3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leser, T. D., M. Boye, and N. B. Hendriksen. 1995. Survival and activity of Pseudomonas sp. strain B13(FR1) in a marine microcosm determined by quantitative PCR and an rRNA-targeting probe and its effect on the indigenous bacterioplankton. Appl. Environ. Micobiol. 61:1201-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, W., and M. A. Drake. 2001. Development of a quantitative competitive PCR assay for detection and quantification of Escherichia coli O157:H7 cells. Appl. Environ. Microbiol. 67:3291-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maidak, B. L., J. R. Cole, C. T. J. Parker, G. M. Garrity, N. Larson, B. Li, T. G. Liburn, M. J. McCaughey, G. J. Olsen, R. Overbeek, S. Pramanik, T. Schmidt, J. Tiedje, and C. R. Woese. 1999. A new version of the RDP (Ribsomal Database Project). Nucleic Acids Res. 27:171-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maymo-Gatell, X., Y. Chien, J. M. Gossett, and S. H. Zinder. 1997. Isolation of bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276:1568-1571. [DOI] [PubMed] [Google Scholar]
- 19.Mesarch, M. B., C. H. Nakatsu, and L. Nies. 2000. Development of catechol 2,3-dioxygenase-specific primers for monitoring bioremediation by competitive quantitative PCR. Appl. Environ. Microbiol. 66:678-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips, C. J., E. A. Paul, and J. I. Prosser. 2000. Quantitative analysis of ammonia oxidizing bacteria using competitive PCR. FEMS Microbiol. Ecol. 32:167-175. [DOI] [PubMed] [Google Scholar]
- 21.Reilly, K., and G. T. Attwood. 1998. Detection of Clostridium proteoclasticum and closely related strains in the rumen by competitive PCR. Appl. Environ. Microbiol. 64:907-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosner, B. M., P. L. McCarty, and A. M. Spormann. 1997. In vitro studies on reductive vinyl chloride dehalogenation by an anaerobic mixed culture. Appl. Environ. Microbiol. 63:4139-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudi, K., O. M. Skulberg, F. Larsen, and K. S. Jakobsen. 1998. Quantification of toxic cyanobacteria in water by use of competitive PCR followed by sequence-specific labeling of oligonucleotide probes. Appl. Environ. Microbiol. 64:2639-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholz-Muramatsu, H., A. Neumann, M. Messmer, E. Moore, and G. Diekert. 1995. Isolation and characterization of Dehalospirillum multivornas gen. nov., sp. nov., a tetrachloroethene-utilizing, strictly anaerobic bacterium. Arch. Microbiol. 163:48-56. [Google Scholar]
- 25.Smith, L. H., P. L. McCarty, and P. K. Kitanidis. 1998. Spreadsheet method for evaluation of biochemical reaction rate coefficients and their uncertainties by weighted nonlinear least-squares analysis of the integrated Monod equation. Appl. Environ. Microbiol. 64:2044-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westrick, J. J., J. W. Mello, and R. F. Thomas. 1984. The groundwater supply survey. J. Am. Water Works Assoc. 76:52-59. [Google Scholar]
- 27.Yang, Y., and P. L. McCarty. 1998. Competition for hydrogen within a chlorinated solvent dehalogenating anaerobic mixed culture. Environ. Sci. Technol. 32:3591-3597. [Google Scholar]