

Abstract
Using periplasmic penicillin amidase (PA) from Escherichia coli ATCC 11105 as a model recombinant protein, we reviewed the posttranslational bottlenecks in its overexpression and undertook attempts to enhance its production in different recombinant E. coli expression hosts. Intracellular proteolytic degradation of the newly synthesized PA precursor and translocation through the plasma membrane were determined to be the main posttranslational processes limiting enzyme production. Rate constants for both intracellular proteolytic breakdown (kd) and transport (kt) were used as quantitative tools for selection of the appropriate host system and cultivation medium. The production of mature active PA was increased up to 10-fold when the protease-deficient strain E. coli BL21(DE3) was cultivated in medium without a proteinaceous substrate, as confirmed by a decrease in the sum of the constants kd and kt. The original signal sequence of pre-pro-PA was exchanged with the OmpT signal peptide sequence in order to increase translocation efficiency; the effects of this change varied in the different E. coli host strains. Furthermore, we established that simultaneous coexpression of the OmpT pac gene with some proteins of the Sec export machinery of the cell resulted in up to threefold-enhanced PA production. In parallel, we made efforts to increase PA flux via coexpression with the kil gene (killing protein). The primary effects of the kil gene were the release of PA into the extracellular medium and an approximately threefold increase in the total amount of PA produced per liter of bacterial culture.
Escherichia coli is the most frequently used prokaryotic expression system for high-level expression of homologous and heterologous proteins. However, despite its advantages, it has been commonly observed that the overexpression of foreign proteins in E. coli triggers a large metabolic burden and leads to undesired metabolic responses, including changes in the central metabolism and regulatory functions (36), cell growth retardation (1), and ribosome destruction and cell death (8). The expression of heterologous genes in recombinant E. coli is associated with increased activity of the energy-generating dissimilatory pathway (41), and the fate of foreign proteins expressed in E. coli is determined in part by the degradative activities of the host cells (15). Moreover, the efficient expression of different genes in E. coli is not a routine matter, as the regulatory network of protein expression at the posttranslational level is very complex and must be considered in order to attain a high level of protein expression. Recent developments in DNA recombinant technology have contributed to significant progress in protein overexpression. Genetic manipulations improving biosynthesis at the transcriptional level or optimization of fermentation strategies developed to enhance the yield of recombinant proteins of interest lead to considerable increases in their production. Nevertheless, higher levels of production are still desired to satisfy the requirements for economically attractive process.
Using an industrially important enzyme, penicillin amidase (PA), from E. coli ATCC 11105 as a model system, we investigated the posttranslational yield-limiting steps influencing its production and developed a biochemical engineering approach for enhancing its overexpression in recombinant E. coli strains. The nascent polypeptide precursor is synthesized as 96-kDa pre-pro-PA (ppPA), containing, at its N terminus, a signal peptide which mediates translocation into the periplasm via the Tat pathway (18). Thereafter, translocated pro-PA (pPA) is further processed into two chains (A, 23 kDa, and B, 63 kDa) by various intra- and intermolecular autoproteolytic reactions (16, 21). Due to this complex regulation mechanism for biosynthesis, PA serves as a good model system for studying the relative influences of all of these processes in order to develop efficient strategies for improving recombinant prokaryotic protein production.
The emerging details of the structure and regulation of the pac gene (6, 32, 40) have allowed the manipulation of transcriptional and translational efficiencies via the development of appropriate host-vector systems (5). While PA activity was significantly improved, the processes leading to decreased yields posttranslationally were not completely understood. Yields of active PA are restricted by the following posttranslational processes: (i) nonspecific intracellular proteolytic degradation by host-specific cytoplasmic endopeptidases, (ii) inclusion body formation, (iii) transport through the cytoplasmic membrane, (iv) maturation, and (v) nonspecific proteolysis in the periplasm (17). The extent to which yields are affected by the posttranslational bottleneck steps mentioned above suggests strategies for improvement of the overexpression of a target protein.
In the present report, both translocation and intracellular proteolysis were manipulated in order to increase PA expression at the molecular level in recombinant E. coli strains. Intracellular proteolysis was modulated either by the composition of the medium or by the host strain used for overexpression. The exchange of the original signal sequence of ppPA with the Sec-dependent OmpT signal peptide sequence varied in the different host strains. The overproduction of SecA, SecB, and SecF improved transport efficiency and enhanced up to threefold the level of expression of an OmpT-PA fusion in the periplasm. Parallel efforts were made to increase PA flux via coexpression with the kil gene (killing protein). The controlled release of PA into the extracellular medium, leading to increased PA production, was only temporary due to the decreased viability of the host cells.
MATERIALS AND METHODS
DNA techniques.
The pac gene was amplified with its ribosome binding site (RBS) and Shine-Dalgarno sequence upstream of the start codon by using chromosomal DNA from E. coli ATCC 11105 (Deutsche Sammlung von Microorganismen und Zellkulturen [DSMZ], Braunschweig, Germany) as a template, double digested with EcoRI and KpnI, and cloned into the polylinker of the low-copy-number vector pMMB207 (Cmr) (kindly provided by V. de Lorenzo); this procedure yielded plasmid pPAEC. Plasmid pPAOT, used for the in vivo expression of OmpT-PA, was constructed as described by Ignatova et al. (18). The pac gene in pPAEC or the OmpT pac gene in pPAOT was cloned under the control of the tac promoter; therefore, the expression of these genes was induced with isopropyl-β-d-thiogalactopyranoside (IPTG) (0.5 mM). For all genetic manipulations, plasmids were transformed in E. coli DH5α cells. Transformations were carried out according to the standard transformation procedure described elsewhere (30) or by using an electroporator (PeqLab, Erlangen, Germany). pac-containing clones were selected on chloramphenicol (25 μg/ml)-containing Luria-Bertani (LB) agar plates and screened phenotypically for PA activity (48).
The secB, secA, and secF genes were amplified from E. coli K5 chromosomal DNA isolated by standard procedures as described previously (30). The KpnI-BamHI-digested secB and secF genes were inserted into the polylinker of pPAOT immediately after the stop codon of the OmpT pac gene, resulting in plasmids pPAOTsecB and pPAOTsecF, respectively. The XbaI-BamHI-digested PCR fragment of secA was also subcloned into pPAOT, yielding plasmid pPAOTsecA.
The kil gene from the ColE1 plasmid (DSMZ) was amplified by using 3-s dwell times with AGSGold polymerase on a Hybaid PCR-Sprint apparatus with active tube control according to the manufacturer's instructions (AGS-Hybaid GmbH, Heidelberg, Germany). The KpnI-BamHI-digested kil amplification product was cloned into pPAEC after the stop codon of the pac gene, resulting in plasmid pPAK2.
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. Transformed E. coli host strains were grown in LB medium, in M9 minimal medium (35) containing 2.5 g of glucose/liter (initial concentration), or in modified minimal medium (M9Y medium) supplemented with 2.5 g of yeast extract/liter and 1 g of glucose/liter. The selection marker for the plasmids tested was chloramphenicol at 25 μg/ml. All experiments were performed at 28°C because of the temperature dependence of intracellular proteolysis and its limitation on PA expression.
TABLE 1.
E. coli strains used in this study
| Strain | Description | Source |
|---|---|---|
| DH5α | Φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17thi-1 gyrA relA1 | DSMZ |
| K5 | thr thi | DSMZ |
| JM109 | endA1 recA1 gyrA96 thi hsdR17relA1 supE44 Δ(lac-proAB) (F′ traD36 proAB lacZg ΔM15) | Promega, Mannheim, Germany |
| BL21(DE3) | ompThsdSdcm+ Tetrgal(DE3) endA Hte | Stratagene, Amsterdam, The Netherlands |
| MC4100 | araD139 Δ(argF-lac)U169 rpsL1 relA1 deoC1 ptsF25 rboR | DSMZ |
Pulse-chase experiments.
Overnight E. coli K5 cultures were diluted 1:25 into M9 (as described above) chase medium with a methionine-free amino acid mixture (50 μg/ml each) (pH 7.4) and cultivated at 28°C. As a selection marker, chloramphenicol was added at 25 μg/ml. At an A600 of 0.4, the expression of ppPA and OmpT-PA was induced with 0.5 mM IPTG. At 10 min after induction, cells were pulse-labeled with [35S]methionine (100 μCi/ml; Amersham Buchler) for 1 min at 28°C, followed by the addition of excess unlabeled methionine (500 μg/ml) and chloramphenicol (100 μg/ml) (46). Mature PA was quantitatively isolated by immunoaffinity chromatography as described previously (18).
Cell fractionation.
After the cells were harvested by centrifugation at 1.93 × g for 20 min, they were disrupted by sonication in phosphate buffer (pH 7.5; I = 0.01) (Branson sonifier W 450 for 10 min with a 50% duty cycle at 4°C) or by a cold mild osmotic shock procedure (17). The periplasmic fraction was separated from the spheroplast suspension by centrifugation at 8,000 rpm for 40 min at 4°C. The spheroplasts were sonicated in distilled water, and cell debris was removed by centrifugation at 13,000 rpm for 10 min. The clarified supernatant was designated the cytoplasmic fraction.
Measurement of in vivo proteolysis.
To study the rate of intracellular proteolysis in vivo, E. coli host strains transformed with plasmid pPAEC were grown under the same conditions as those described above. At 3 h after induction with 0.5 mM IPTG, rifampin was added to the culture to a final concentration of 500 μg/ml to inhibit transcription initiation and to stop protein synthesis (37). Five-milliliter samples of this culture were removed at different intervals and sonicated. Each sample was analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Western blotting after centrifugation and cell pellet sonication.
Electrophoresis and immunoblotting.
SDS-PAGE of proteins with 10 or 12% polyacrylamide gels was done according to the method of Laemmli (24). Protein bands were detected by Coomassie blue staining and immunoblotting (Immun-blot assay kit; Bio-Rad). Proteins were transferred to polyvinylidene difluoride membranes and detected with a monoclonal antibody for an epitope of the B chain of PA. The secondary antibody used was goat anti-mouse immunoglobulin G (heavy and light chains) conjugated to alkaline phosphatase (Bio-Rad). Detection was accomplished with 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium. PA with an isoelectric point of 7.0 (0.1 mg/ml) and a pure mutant (Thr263Gly) pPA precursor (0.06 mg/ml) were purified as described previously (20, 21) and used as standards. Calculation of the relative optical densities of the bands of interest from the immunoblots, in relation to the intensity of the standards, was performed by using the program ONE-Dscan (Scananalytics, a division of CSPI, Billerica, Mass.).
Determination of enzyme activity.
PA activity in the homogenate or purified periplasmic fraction was determined by a spectrophotometric assay with the chromogenic substrate 6-nitro-3-phenylacetamido benzoic acid. The change in the A380 per minute was converted into benzylpenicillin units (BPU). Under these conditions for pure PA (1 mg/ml), the change in the A380 was 3.0 per min, a value which corresponded to 42 BPU/ml (20).
RESULTS
Influence of the medium on pac expression.
Cultivation of parental E. coli ATCC 11105 expressing the chromosomal pac gene in M9 minimal medium led to a 1.7-fold increase in PA yields compared to cultivation in complex LB medium (Table 2). The cells grew slowly in M9 minimal medium, and the cell density achieved was about twofold lower than that in LB medium (data not shown). Immunoblot analysis of E. coli ATCC 11105 cells expressing PA showed an increased intensity of the B-chain band corresponding to active PA in the periplasmic fraction in M9 minimal medium compared to LB medium (Fig. 1). In the cytoplasmic fraction, some fragments (in the range of 60 to 30 kDa) still possessing the epitope for the monoclonal antibody used as the first antibody in the Western blots were detected. N-terminal amino acid analysis confirmed their origin as ppPA (data not shown). The 50-kDa proteolytic band dominated and was used in further experiments as a representative example to monitor the changes in the degradation activity of host cells under the studied conditions. N-terminal amino acid analysis of the 50-kDa band revealed the sequence AEKPGYYL, which overlaps amino acids 347 to 354 in the B chain (numbering is according to the published primary amino acid sequence of pPA) (42) and corresponds to the calculated molecular mass of 53.6 kDa for the fragment Ala347-Arg820. A significantly decreased intensity of the 50-kDa band was observed in M9 minimal medium (Fig. 1B) compared to the intensity in the cytoplasmic fraction of cells cultivated in complex LB medium (Fig. 1A). The intensity ratio of mature PA to proteolytic products increased for M9 minimal medium compared to LB medium when larger parts of the newly synthesized precursor underwent intracellular proteolytic degradation, leading to increased intensity of the main proteolytic band (50 kDa).
TABLE 2.
PA expression levels in various E. coli host cellsa
| Plasmid | E. coli host | Medium | PA expression level (BPU/g of cell dry wt) |
|---|---|---|---|
| None | ATCC 11105 | LB | 80 |
| M9 | 140 | ||
| ρPAEC | DH5α | LB | 50 |
| M9Y | 130 | ||
| K5 | LB | 130 | |
| M9Y | 295 | ||
| M9 | 364 | ||
| MC4100 | LB | 147 | |
| M9Y | 250 | ||
| M9 | 390 | ||
| JM109 | LB | 190 | |
| M9Y | 455 | ||
| BL21(DE3) | LB | 110 | |
| M9Y | 610 | ||
| M9 | 1,170b |
PA activity was measured 6 h after induction with 0.5 mM IPTG. Cultivation was carried out with 40 ml of medium at 28°C. Experiments with chromosomal pac in parental strain E. coli ATCC 11105 served as a control. M9, M9 minimal medium.
PA activity was measured 10 h after induction with IPTG due to the slower growth and the longer time necessary to achieve the stationary phase.
FIG. 1.

Influence of the medium on PA biosynthesis in parental strain E. coli ATCC 11105. Cells were cultivated in LB medium (A) or M9 minimal medium (B) at 28°C. Aliquots were removed at the indicated times after the addition of phenylacetic acid (1 g/liter), separated into cytoplasmic (C) and periplasmic (P) fractions, resolved by SDS-PAGE (12.5% gel), and immunoblotted. For each time point, the same number of cells was analyzed. Pure PA at a concentration of 0.1 mg/ml (B chain) and the slowly processed Thr263Gly mutant PA precursor (pPA) at a concentration of 0.06 mg/ml were used as standards.
Screening for an appropriate expression system.
For selection of an appropriate host expression system, different E. coli strains (Table 1) were cultivated in complex and minimal media (Table 2). Previous experiments showed that PA production correlates with cell growth and that PA biosynthesis stops in the early stationary phase (17). pac expression was induced in the middle of the logarithmic phase; at approximately 6 h after induction, the cells reached the stationary phase. The measured specific activity at this point was used for quantitative comparisons (Table 2). The levels of expression of PA in all strains tested (except for DH5α) harboring plasmid pPAEC in LB medium differed up to twofold from that in E. coli ATCC 11105. In the proteinase-deficient host strain E. coli BL21(DE3), the measured PA activity was comparable to that in control strain E. coli ATCC 11105 cultivated in M9 minimal medium (Table 2). A substantial increase in PA activity was measured when E. coli BL21(DE3) cells were cultivated in medium without proteinaceous substrates, i.e., M9 minimal medium (Table 2). This expression level was approximately 10-fold higher than that in control strain E. coli ATCC 11105 cultivated in the same medium. E. coli BL21(DE3) cells grew more slowly in M9 minimal medium than the other host strains and had an extended lag phase in M9 minimal medium. The achievement of the stationary phase in shake flasks occurred approximately 10 h after induction, necessitating longer cultivation of these host cells.
In order to quantify the influence of posttranslational processes on protein flux, rate constants for intracellular proteolysis (kd) and for transport through the membrane (kt) must be determined. The kinetic equation for the in vivo fate of an intracellular protein precursor (P) can be expressed as follows:
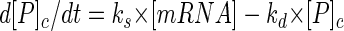 |
(1) |
where ks is the rate constant for the synthesis of mRNA, kd is the rate constant for intracellular proteolytic degradation, and [P]c is the protein concentration in the cytoplasmic at any time. In all degradation processes, proteolytic enzymes from the destructive machinery of the cell are involved. Therefore, the apparent first-order rate constant kd is a summarizing constant that includes the Michaelis-Menten constant Km, the turnover number kcat, and the concentration of peptidases participating in this process, assuming that the intracellular peptidases are not saturated.
For proteins which are not localized in the cytoplasmic space and which are additionally translocated through the membrane, such as PA, the equation is changed as follows:
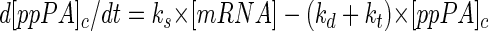 |
(2) |
where kt is the apparent first-order rate constant for transport through the plasma membrane and [ppPA]c is the precursor concentration in the cytoplasm at any time. Assuming that the translocation machinery is not saturated, i.e., a first-order process, kt depends on the concentration of ppPA and on the binding and efficiency of the translocation system.
The sum of the apparent first-order rate constants for proteolytic degradation and transport (kd plus kt) can be determined after the inhibition of de novo protein synthesis. Equation 1 is thus transformed as follows:
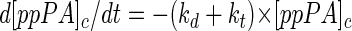 |
(3) |
For this experiment, de novo protein synthesis was blocked with rifampin; at regular intervals, samples were withdrawn and subjected to immunoblot analysis (Fig. 2). Protein synthesis flux was not abolished immediately due to a delay in the diffusion of the antibiotic into the cells, leading to a slight increase in the intensity of the ppPA band in the first 20 min. Thereafter, the intensity of the ppPA band decreased continuously parallel to the observed concomitant increase in the intensity of the main proteolytic band (50 kDa) (Fig. 2). The degradation products obtained from the intracellular breakdown of the ppPA precursor resulted from both primary and secondary cleavages involving intracellular peptidases. Proteolytic fragments still containing the N-terminal signal peptide of ppPA might have competed for the translocation pores in the membrane and influenced the estimation of kt. Thus, the sum of kt and kd is the only quantitative measure for comparing the strains used in this study. The similar PA activities measured for host strains E. coli K5, E. coli MC4100, and E. coli BL21(DE3) cultivated in LB medium (Table 2) correlated with the calculated similar values for the sum of kd and kt for that medium (Table 3). In this case, the intensity of the main protein degradation band (50 kDa) was lower in E. coli BL21(DE3) cells (Fig. 2A) than in E. coli K5 cells (Fig. 2C) or in E. coli MC4100 cells (Fig. 2B), indicating reduced intracellular proteolysis in E. coli BL21(DE3) cells.
FIG. 2.


In vivo proteolysis of ppPA in E. coli host cells. E. coli BL21(DE3) (A), E. coli MC4100 (B), and E. coli K5 (C) were used as host cells. Residual concentrations of ppPA and the 50-kDa proteolytic product were determined by immunoblot analysis after the inhibition of de novo protein synthesis by rifampin. The relative optical densities (dens.) of the bands of interest from the Western blot were quantified by using ONE-Dscan. ppPA density was related to the density of the pPA standard, and the density of the 50-kDa proteolytic band was related to the density of the PA standard (for details, see Materials and Methods). LB, LB medium; M9, M9 minimal medium.
TABLE 3.
Rate constants for intracellular proteolysis and transport in different strainsa
| E. coli strain | Medium | (kd + kt), h−1 |
|---|---|---|
| ATCC 11105 | LB | 1.00 ± 0.11 |
| K5 | LB | 0.91 ± 0.11 |
| M9 | 0.62 ± 0.07 | |
| MC4100 | LB | 0.90 ± 0.08 |
| M9 | 0.47 ± 0.03 | |
| BL21(DE3) | LB | 0.67 ± 0.07 |
| M9Y | 0.37 ± 0.04 | |
| M9 | 0.28 ± 0.04 |
M9, M9 minimal medium.
The omission of the proteinaceous substrates by changing the medium to M9 minimal medium led to a significant reduction in the amount of the main proteolytic band (50 kDa) in E. coli BL21(DE3) harboring pPAEC (Fig. 2A) and caused a decrease in the sum of kd and kt to 0.28 h−1 (Table 3). The effect of the decreased sum of kd and kt can quantitatively explain the observed increase in PA activity in E. coli BL21(DE3) harboring (pPAEC) compared to wild-type E. coli ATCC 11105 (Tables 2 and 3).
Improvement of the translocation capacity of the host cell.
Recently published investigations indicated that the parental signal peptide of E. coli PA directs ppPA to the Tat translocation pathway (18). Compared to the velocity of Sec-dependent export of proteins, the translocation of proteins by Tat-mediated transport is slower (2, 7). In this context, in order to accelerate export and to decrease the exposure of the ppPA precursor in the cytoplasm, we exchanged the original signal peptide of ppPA with a Sec-dependent signal peptide (from the OmpT peptide); this procedure efficiently rerouted the OmpT-PA fusion to the Sec pathway (18). In E. coli K5, the kinetics of translocation of the OmpT-PA precursor were linear over the first 2 min, and translocation was accomplished after 5 min (Fig. 3B and C) The translocation of OmpT-PA was about threefold faster than the transport of ppPA (Fig. 3A). Similar translocation kinetics for both precursors were also observed for host strain E. coli MC4100 (18).
FIG. 3.

Translocation kinetics for ppPA and OmpT-PA. (A and B) E. coli K5 carrying plasmid pPAEC or pPAOT was grown at 28°C in the chase medium. At 10 min after induction, the cells were pulse-labeled with [35S]methionine for 1 min and then chased with nonradioactive methionine. At the times indicated, samples (500 μl) were withdrawn, subjected to immunoaffinity chromatography for the quantitative isolation of mature PA, and assessed by SDS-PAGE followed by autoradiography. The position of the B-chain PA band was verified by using a prestained protein marker (Bench Mark; Gibco BRL). (A) ppPA. (B) OmpT-PA. (C) The intensity of the B chain of PA from panel A (squares) and panel B (circles) was quantified by using ONE-Dscan and is represented graphically as a function of the chase time. The highest intensity reached in each blot was taken as 1. Error bars indicate standard deviations.
As has been already shown for the isolated main degradation product (50-kDa band), the intracellular proteolytic attack on ppPA occurs at residues localized in the B chain. ppPA and OmpT-PA differ only in their signal sequences, allowing us to assume that the same intracellular proteolytic degradation occurs for PA with different signal peptides. To examine the influence of the signal peptide on intracellular proteolysis, we transformed plasmid pPAOT into different E. coli host strains (Table 4). A comparison of the final PA activities achieved by expressing plasmid pPAOT (Table 4) with the pPAEC vector (Table 2) revealed increased enzyme activities only in E. coli DH5α, E. coli K5, and E. coli MC4100. In contrast, the levels of expression of OmpT-PA in E. coli JM109 decreased by 50% in LB medium and by 11% in M9Y medium. The situation was less clear for E. coli BL21(DE3), for which we detected approximately a twofold reduction in PA activity in all media tested (Table 4).
TABLE 4.
OmpT-PA expression levels in various E. coli host cellsa
| E. coli host | Medium | OmpT-PA expression level (BPU/g of cell dry wt) |
|---|---|---|
| DH5α | LB | 115 |
| M9Y | 190 | |
| K5 | LB | 200 |
| M9Y | 430 | |
| JM109 | LB | 95 |
| M9Y | 405 | |
| MC4100 | LB | 182 |
| M9Y | 324 | |
| BL21(DE3) | LB | 80 |
| M9Y | 335 | |
| M9 | 510b |
The host strains were transformed with plasmid pPAOT (Cm) and cultivated in different media at 28°C PA activity was measured 6 h after induction with 0.5 mM IPTG. M9, M9 minimal medium.
PA activity was measured 10 h after induction with IPTG.
Another approach to manipulating translocation is to increase the export efficiency of the cell. To determine whether the overexpression of Sec proteins might enhance the PA expression level, E. coli K5 cells were transformed with a plasmid bearing additional copies of either secA, secB, or secF cloned behind the OmpT pac gene under the control of the same tac promoter. The overproduction of SecA had no significant influence on PA production. Although an increase in PA activity in the periplasmic fraction was measured (Fig. 4A), the intensity of the PA band in the immunoblot was comparable to that in the control experiment (Fig. 4B). The coexpression of OmpT-PA with SecB and SecF led to three- and twofold-enhanced PA expression, respectively (Fig. 4).
FIG. 4.

Coexpression of OmpT-PA with some of the compounds of the Sec machinery. (A) E. coli K5 host cells transformed with the indicated plasmids were grown in LB medium supplemented with chloramphenicol (25 μg/ml) at 28°C to an A600 of 0.6. IPTG (0.5 mM) was added, and incubation was continued for 6 h. The activity of PA in the control E. coli K5 culture transformed with pPAOT was taken as 1. Error bars indicate standard deviations. (B) Western blot of the periplasmic fraction of the same samples as those used for activity measurements in panel A. The protein concentration load per lane corresponds to the same amount of cells verified by optical density measurements. Pure PA (0.1 mg/ml) was used as a standard (PAst).
Excretion of PA into the extracellular medium.
Several homologous and heterologous proteins of different origins are efficiently released into the culture medium by the action of the kil gene of the ColE1 plasmid. The small protein product of that gene (6 kDa) resembles the E. coli membrane lipoprotein causing the release of periplasmic proteins (44). We assumed that by guiding protein flux into the extracellular medium, the cellular machineries (e.g., translocation pore and periplasm with periplasmic inclusion bodies) saturated by the overproduced protein could be desaturated. Therefore, there were reasons to believe that the controlled release of PA into the medium could also enhance its overproduction. To examine this notion, we placed both the target pac gene and the kil gene in the same plasmid, pPAK2, under the control of the tac promoter. Cultures of E. coli K5(pPAK2) and E. coli K5(pPAEC) as a kil-negative control were grown at 28°C in LB medium. Over the first 3 h after induction, the increase in PA activity in the cells and this released into the periplasm was linear and almost threefold higher than that in the control (Fig. 5A). Over the next 3 h, PA activity measured in the extracellular medium continued to increase, whereas cellular PA activity dropped and the total amount of PA produced per liter of bacterial culture was 25% higher than that measured in the control. At that time, kil-bearing cells stopped de novo ppPA synthesis and released the already synthesized mature PA from the periplasm into the culture broth. During the last 3 h, cell growth was inhibited and the number of viable cells decreased slightly, as determined by counting of colonies on selective agar plates (data not shown).
FIG. 5.

Release of PA into the extracellular medium. (A) E. coli K5 cells transformed with pPAEC (control) and pPAK2 were grown at 28°C in LB medium supplemented with 25 μg of chloramphenicol/ml. Plasmid-carried pac and kil genes were induced with 0.5 mM IPTG; thereafter, at the times indicated, 1-ml samples were withdrawn. The PA activity in kil-expressing cells was measured in homogenized cells (periplasm) and in the culture supernatant (extracellular). (B) E. coli K5(pPAK2) cells were cultivated in different media as described for the cultivation in panel A. At 6 h after IPTG induction, PA activity was measured in the supernatant (extracellular) and in homogenized cells (periplasm). LB, LB medium; M9, M9 minimal medium.
To test whether the beneficial effect of the media on PA biosynthesis (Table 2) might have a similar influence on cells bearing the kil gene, E. coli K5(pPAK2) cells were also cultured in M9 minimal medium and M9Y medium. The change in the cultivation medium led to a moderate release of extracellular PA (Fig. 5B) and, unexpectedly, the detected total PA activity was lower than the activity measured for E. coli K5(pPAEC) (Table 2). Despite that result, the viability of the kil-induced cells was not reduced.
DISCUSSION
A primary goal of industrial technology for the overexpression of proteins in recombinant E. coli hosts is the cost-effective increase in the production of the desired protein. This can be achieved by increasing the production of the active protein per cell and/or by manipulation of the fermentation conditions. Optimization of the fermentation media and operational strategies or a combination of both succeeded in enhancing the production of some recombinant proteins (15, 19, 47). Increasing protein production by means of increasing cell density for recombinant E. coli host strains has reached its limits (47).
Alternatively, genetic engineering methods influencing the levels of expression of genes offer more possibilities for improving the biosynthesis of recombinant proteins. Some are specific for each gene, such as mRNA stability, codon usage, and transcriptional regulation in the 5′- and 3′-flanking regulatory regions. These parameters must be optimized individually. Another, more general method for improving productivity is the use of strong and tightly regulated promoters (e.g., tac and rhaBAD). However, this method often leads to an imbalance of protein flux, causing aggregation of the product in insoluble inclusion bodies. The problem with cytoplasmic inclusion bodies can be overcome by the application of low-copy-number plasmids for the overexpression of proteins. For periplasmic proteins, insoluble inclusion bodies composed of protein precursors are localized in the cytoplasm and periplasm (4, 25). The amounts of periplasmic inclusion bodies can be significantly reduced by coexpression with periplasmic proteases (27).
Besides optimizing recombinant protein production at the translational or transcriptional level, many posttranslational factors (e.g., folding and maturation of the protein and physiological activities of the host) influence the level of biosynthesis. The expression of foreign proteins in E. coli causes a metabolic burden often resulting in increased degradative activities of the host. The biotechnological consequences of intracellular host-dependent proteolysis are usually loss of the product and its contamination by proteolytic products. A variety of strategies have been developed to reduce the intracellular proteolytic destruction of recombinant proteins, e.g., reduction of the growth rate of cells, protective fusions, and replacement of specific residues to eliminate cleavage sites (for a review, see reference 13). For the system used in this study, PA, intracellular proteolysis causes more than an 80% loss of a newly synthesized ppPA precursor for parental E. coli ATCC 11105 (17). Thus, the use of a proteinase-deficient strain as a host strain for pac expression seemed to be a successful strategy for improvement of the yield of proteolytically sensitive precursors. For this purpose, we chose commercially available E. coli BL21(DE3), which has natural deficiencies of ATP-dependent proteinase Lon and outer membrane proteinase OmpT (14). Lon and two other proteases (ClpAP and ClpXP) are responsible for 70 to 80% of the energy-dependent protein degradation in E. coli (31). Nevertheless, attempts to increase sufficiently PA production in BL21(DE3) as a host strain cultivated in complex LB medium failed. The protease activity of the host is influenced by the medium composition, and cultivation of E. coli BL21(DE3) harboring pPAEC in medium without proteinaceous substrates increased PA production by up to 10-fold. Moreover, the amount of the main proteolytic band (50 kDa) was reduced, and the calculated rate constants for transport and intracellular proteolysis (kd and kt) decreased to 0.28 h−1. The intracellular proteolytic breakdown could not be circumvented completely but was reduced drastically. Similar results have been observed in expression studies with Bacillus subtilis, showing that even with strains missing extracellular proteases, proteolytic degradation reduces the yield of the protein of interest (3). In some recently published studies, it was observed that PA production is sensitive to complex proteinaceous substrates and that yields can be raised when minimal medium with different carbon sources is used (29, 32, 43).
For proteins destined to be exported into the periplasm, the translocation machinery of the host markedly restricts their yields (45). The efficient transfer of preproteins through membranes requires an intact translocation system. However, when a protein is overexpressed, the export machinery becomes overloaded and its efficiency decreases significantly, and some advantages of the system are lost. An approach for influencing the bottleneck steps in secretion is the manipulation of the signal peptide, the sole determinant directing preproteins to the appropriate translocation system (45). Optimization of original signal peptides has led to improved yields of many periplasmic enzymes (12, 28). In experiments with the OmpT-PA fusion, the effect varied in the different host strains, and no consistent influence could be found. The exchange of the whole signal peptide is not an appropriate approach for enhancing extracellular protein production, opposite the use of single mutations in the original signal sequence of the protein of interest (12).
Alternatively, providing a strain with additional copies of components of the translocation system can relieve secretion and enhance the yields of heterologous proteins (38). The overexpression of SecYEG proportionally enhances in vitro Sec translocase activity (9, 10). Perez-Perez et al. (38) reported that supplementation with a plasmid bearing copies of secE and prlA4 (secY allele) increased the periplasmic production of a preOmpA-interleukin-6 fusion about 10 times. The coexpression of an OmpT-PA fusion with some of the components of the Sec machinery (e.g., SecA, SecB, and SecF) led to an overall increase in PA activity in the periplasmic fraction. Both SecA and SecB were chosen based on the assumption that as components of the Sec machinery delivering preproteins to the translocation pore in the membrane (11), they might prevent the cytoplasmic proteolytic breakdown of ppPA. SecD and SecF are important in the release of the protein into the periplasm (39). The most remarkable influence was observed for coexpression with SecB. The SecB protein, which is involved in the early translocation act, maintains presecretory proteins in a translocation-competent state (23). Obviously, for PA, the chaperone function of SecB stabilizes ppPA against intracellular proteolytic degradation, and more precursor is delivered to the translocation pore.
Recently published results demonstrated that controlled flux of a target protein into the extracellular medium by coexpression with the kil gene can increase its total production (34). However, the use of strong inducible promoters causes some problems with the viability of recipient cells. For the model system used in this study, the effect of kil gene expression not only varied in the different media but also depended, as recently reported, on the host strain used for pac expression (26). As a consequence, even though coexpression of the protein of interest with the killing protein could enhance protein yield, the observed drawbacks demanded optimization of the system by use of different conditions to attain the desired goals. The use of simultaneous induction of the kil gene with the gene for the recombinant protein is not an efficient strategy, as the viability of cells after induction is severely reduced. Other promoters, e.g., weak constitutive or growth-phase-dependent promoters (22, 33), enable viability to be maintained efficiently. However, the expression of the kil gene under the control of a stationary-phase-dependent promoter has not given the expected increase in the yield of the target protein, as adaptation to the stationary phase requires the reduction of global gene expression (33). The use of separate and differently controlled promoters, a strong one for the recombinant protein and a moderate one for the killing protein, might prevent early lysis of host cells without the loss of their expression capabilities.
As outlined above, several posttranslational bottlenecks must be considered for achieving improvement of the yields of recombinant proteins in E. coli. Thus, the efficient production of a target protein requires the identification and quantification of specific yield-limiting processes followed by a direct attempt to decrease their influence. Quantification of the rates of proteolysis and transport through the membrane for the model system used here, PA, allowed us to conclude that the use of proteinase-deficient strains as host strains cultivated in medium without proteinaceous substrates is a successful strategy for achieving higher productivity of a proteolysis-susceptible target protein. Furthermore, influencing translocation offers ample possibilities for improving protein yield. Thus, the development of better strategies designed to maximize the yields of recombinant proteins must be based on simultaneous modulation of more than one of the yield-restricting factors.
Acknowledgments
We thank St. Stoeva (Abteilung Physiologie und Biochemie, Universität Tübingen) for performing the N-terminal sequence analysis and P. Zipfel (Jena, Germany) for making it possible for us to perform the pulse-chase experiments in his laboratory.
This work was funded by DFG (KA 505/9-1).
REFERENCES
- 1.Bentley, W. E., N. Mirjalili, D. C. Andersen, R. H., Davis, and D. S. Kompala. 1990. The principal factor in the ′metabolic burden' associated with recombinant bacteria. Biotechnol. Bioeng. 35:668-681. [DOI] [PubMed]
- 2.Berks, B. C., F. Sargent, and T. Palmer. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260-274. [DOI] [PubMed] [Google Scholar]
- 3.Bolhuis, A., H. Tjalsma, H. E. Smith, A. de Jong, R. Meima, G. Venema, S. Bron, and J. M. van Dijl. 1999. Evaluation of the bottlenecks in the late stages of protein secretion in Bacillus subtilis. Appl. Environ. Microbiol. 65:2934-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou, C. P., W.-J. Lin, B.-Y. Kuo, and C.-C. Yu. 2000. Genetic strategies to enhance periplasmic penicillin acylase production in Escherichia coli. Enzyme Microb. Technol. 27:766-773. [DOI] [PubMed] [Google Scholar]
- 5.Chou, C. P., C.-C. Yu, W.-J. Lin, B.-Y. Kuo, and W.-C. Wang. 1999. Novel strategy for efficient screening and construction of host/vector systems to overproduce penicillin acylase in Escherichia coli. Biotechnol. Bioeng. 65:219-226. [PubMed] [Google Scholar]
- 6.Chou, C. P., C.-C. Yu, J.-H. Tseng, M.-I. Lin, and H.-K. Lin. 1999. Genetic manipulation to identify limiting steps and develop strategies for high-level expression of penicillin acylase in Escherichia coli. Biotechnol. Bioeng. 63:263-272. [PubMed] [Google Scholar]
- 7.Clark, S. A., and S. M. Theg. 1997. A folded protein can be transported across the chloroplast envelope and thylakoid membranes. Mol. Biol. Cell 8:923-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong, H., L. Nilsson, and C. G. Kurland. 1995. Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol. 177:1497-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doung, F., and W. T. Wickner. 1997. Distinct catalytic roles of the SecYE, SecG, and SecDFyajC subunits of preprotein translocase. EMBO J. 16:4871-4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douville, K., A. Price, J. Eichler, A. Economou, and W. Wickner. 1995. SecYEG and SecA are the stoichiometric components of preprotein translocase. J. Biol. Chem. 270:20106-20111. [DOI] [PubMed] [Google Scholar]
- 11.Driessen, A. J. M. 1994. How proteins cross the bacterial cytoplasmic membrane. J. Membr. Biol. 142:145-159. [DOI] [PubMed] [Google Scholar]
- 12.Dupuis, L., S. Canaan, M. Riviere, and C. Wicker-Planquart. 1997. Influence of various signal peptides on secretion of mammalian acidic lipases in baculovirus-insect cell system. Methods Enzymol. 284:261-272. [DOI] [PubMed] [Google Scholar]
- 13.Enfors, S. O. 1992. Control of proteolysis in fermentation of recombinant proteins. Trends Biotechnol. 10:310-315. [DOI] [PubMed] [Google Scholar]
- 14.Gottesmann, S. 1996. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 30:465-506. [DOI] [PubMed] [Google Scholar]
- 15.Hanning, G., and S. C. Makrides. 1998. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 16:54-60. [DOI] [PubMed] [Google Scholar]
- 16.Hewitt, L., V. Kasche, K. Lummer, R. J. Lewist, G. N. Murshudov, G. N., Verma, G. G. Dodson, and K. S. Wilson. 2000. Structure of a slow processing precursor penicillin acylase from Escherichia coli reveals the linker peptide blocking the active cleft. J. Mol. Biol. 302:887-898. [DOI] [PubMed] [Google Scholar]
- 17.Ignatova, Z., S.-O. Enfors, M. Hobbie, S. Taruttis, C. Vogt, and V. Kasche. 2000. The relative importance of intracellular proteolysis and transport on the yield of the periplasmic enzyme penicillin amidase in Escherichia coli. Enzyme Microb. Technol. 26:165-170. [DOI] [PubMed] [Google Scholar]
- 18.Ignatova, Z., C. Hörnle, A. Nurk, and V. Kasche. 2002. Unusual signal peptide directs penicillin amidase from E. coli to the Tat translocation machinery. Biochem. Biophys. Res. Commun. 291:146-149. [DOI] [PubMed] [Google Scholar]
- 19.Jeong, K. J., and S. Y. Lee. 1999. High-level production of human leptin by fed-batch cultivation of recombinant Escherichia coli and its purification. Appl. Environ. Microbiol. 65:3027-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasche, V., U. Haufler, D. Markowsky, S. Melnyk, A. G. Zeich, and B. Galunsky. 1987. Penicillin amidase from E. coli: enzyme heterogeneity and stability. Ann. N. Y. Acad. Sci. 501:97-102. [DOI] [PubMed] [Google Scholar]
- 21.Kasche, V., K. Lummer, A. Nurk, E. Piotraschke, A. Rieks, S. Stoeva, and W. Voelter. 1999. Intramolecular proteolysis initiates the maturation of penicillin amidase from Escherichia coli. Biochim. Biophys. Acta 1433:76-86. [DOI] [PubMed] [Google Scholar]
- 22.Kato, H., T. Kobayashi, T. Kudo, T. Furusoto, Y. Murakami, T. Tanaka, H. Baba, T. Oishi, E. Ohtsuka, M. Ikehara, T. Yanagida, H. Kato, S. Moriyama, and K. Horikoshi. 1987. Construction of a secretion vector production of human growth hormone from Escherichia coli. Gene 54:197-202. [DOI] [PubMed] [Google Scholar]
- 23.Kumamoto, C. 1990. SecB protein: a cytosolic export factor that associates with nascent exported proteins. J. Bioeng. Biomembr. 22:337-351. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 25.Lin, W.-J., B.-Y. Kuo, and C. P. Chou. 2001. A biochemical and engineering approach for enhancing production of recombinant penicillin amidase. Bioproc. Biosyst. Bioeng. 24:239-247. [Google Scholar]
- 26.Lin, W.-J., S.-W. Huang, and C. P. Chou. 2001. High-level extracellular production of penicillin acylase by genetic engineering of Escherichia coli. J. Chem. Technol. Biotechnol. 76:1030-1037. [Google Scholar]
- 27.Lin, Y.-H., W.-L. Fang, W.-J. Lin, S.-W. Huang, and C. P. Chou. 2001. Improving production of penicillin acylase in Escherichia coli via efficient DegP-mediated processing of precursors in periplasm. Process Biochem. 37:23-30. [Google Scholar]
- 28.Liu, X.-Q., S. Zhang, X.-M. Pan, and C.-C. Wang. 1999. A novel method for increasing production of mature proteins in the periplasm of Escherichia coli. Protein Sci. 8:2085-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, Y.-C., L.-C. Liao, and W.-T. Wu. 2000. Cultivation of recombinant Escherichia coli to achieve high cell density with a high level of penicillin G acylase activity. Proc. Natl. Sci. Counc. Repub. China Part B 24:156-160. [PubMed] [Google Scholar]
- 30.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 31.Maurizi, M. R. 1992. Proteases and protein degradation in Escherichia coli. Experientia 48:178-201. [DOI] [PubMed] [Google Scholar]
- 32.Merino, E., P. Balbas, F. Recillas, B. Bacerril, F. Valle, and F. Bolivar. 1992. Carbon regulation and role in nature of the Escherichia coli penicillin acylase (pac) gene. Mol. Microbiol. 6:2127-2182. [DOI] [PubMed] [Google Scholar]
- 33.Miksch, G., E. Fiedler, P. Dobrowolski, and E. Flaschel. 1997. The kil gene of the ColE1 plasmid in Escherichia coli controlled by a growth-phase-dependent promoter mediates the secretion of a heterologous periplasmic protein during the stationary phase. Arch. Microbiol. 167:143-150. [PubMed] [Google Scholar]
- 34.Miksch, G., R. Neitzel, E. Fiedler, K. Friehs, and E. Flaschel. 1997. Extracellular production of a hybrid β-glucanase from Bacillus by Escherichia coli under different cultivation conditions in shaking cultures and bioreactors. Appl. Microbiol. Biotechnol. 47:120-126. [DOI] [PubMed] [Google Scholar]
- 35.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 36.Oh, M.-K., and J. C. Liao. 2000. DNA microarray detection of metabolic responses to protein overproduction in Escherichia coli. Met. Eng. 2:201-209. [DOI] [PubMed] [Google Scholar]
- 37.Pease, A. J., and R. E. Wolf, Jr. 1994. Determination of the growth rate-regulated steps in expression of the Escherichia coli K-12 gnd gene. J. Bacteriol. 176:115-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Perez, J., G. Marquez, J.-L. Barbero, and J. Gutierez. 1994. Increasing the efficiency of protein export in Escherichia coli. Bio/Technology 12:178-180. [DOI] [PubMed] [Google Scholar]
- 39.Pogliano, J. A., and J. Beckwith. 1994. SecD and SecF facilitate protein export in Escherichia coli. EMBO J. 13:554-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roa, A., and J. L. Garcia. 1999. New insights into the regulation of the pac gene from Escherichia coli W ATCC 11105. FEMS Microbiol. Lett. 177:7-14. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt, M., E. Viaplana, F. Hoffmann, S. Martzen, A. Villaverde, and U. Rinas. 1999. Secretion-dependent proteolysis of heterologous protein by recombinant Escherichia coli is connected to an increased activity of the energy-generating dissimilatory pathway. Biotechnol. Bioeng. 66:61-67. [PubMed] [Google Scholar]
- 42.Schumacher, G., D. Sizmann, H. Hang, P. Buckel, and A. Böck. 1986. Penicillin acylase from E. coli: unique gene-protein relation. Nucleic Acids Res. 14:5713-5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobotkova, L., V. Stepanek, A. Plhackova, and P. Kyslik. 1996. Development of a high-expression system for penicillin G acylase based on the recombinant Escherichia coli strain RE3 (pKA18). Enzyme Microb. Technol. 19:389-397. [Google Scholar]
- 44.Suit, J. L., and S. E. Luria. 1988. Expression of the kil gene of the ColE1 plasmid in Escherichia coli Kilr mutants causes release of periplasmic enzymes and of colicin without cell death. J. Bacteriol. 170:4963-4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Mellaert, L., and J. Anne. 2001. Gram-positive bacteria as host cells for heterologous production of biopharmaceuticals, p. 277-300. In A. van Broekhoven et al. (ed.), Novel frontiers in the production of compounds for biomedical use. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 46.Weigert, T., H. Sahm, and G. A. Sprenger. 1997. Expression of the Zymomonas mobilis gfo gene for NADP-containing glucose: fructose oxidoreductase (GFOR) in Escherichia coli. Formation of enzymatically active preGFOR but lack of processing into a stable periplasmic protein. Eur. J. Biochem. 244:107-112. [DOI] [PubMed] [Google Scholar]
- 47.Wilms, B., A. Hauck, M. Reuss, C. Syldatk, R. Mettles, M. Siemann, and J. Altenbuchner. 2001. High-cell-density fermentation for production of l-N-carbomoylase by using an expression system based on the Escherichia coli rhaBAD promoter. Biotechnol. Bioeng. 73:95-103. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, Q., L. Zhang, H. Hau, and Y. Zhang. 1986. A method for screening penicillin G acylase-producing bacteria by means of 2-nitro-5-phenylacetamidobenzoic acid test paper. Anal. Biochem. 156:413-416. [DOI] [PubMed] [Google Scholar]