

Abstract
Cell envelope lipids play an important role in the pathogenicity of mycobacteria, but the mechanisms by which they are transported to the outer membrane of these prokaryotes are largely unknown. Here, we provide evidence that LppX is a lipoprotein required for the translocation of complex lipids, the phthiocerol dimycocerosates (DIM), to the outer membrane of Mycobacterium tuberculosis. Abolition of DIM transport following disruption of the lppX gene is accompanied by an important attenuation of the virulence of the tubercle bacillus. The crystal structure of LppX unveils an U-shaped β-half-barrel dominated by a large hydrophobic cavity suitable to accommodate a single DIM molecule. LppX shares a similar fold with the periplasmic molecular chaperone LolA and the outer membrane lipoprotein LolB, which are involved in the localization of lipoproteins to the outer membrane of Gram-negative bacteria. Based on the structure and although an indirect participation of LppX in DIM transport cannot yet be ruled out, we propose LppX to be the first characterized member of a family of structurally related lipoproteins that carry lipophilic molecules across the mycobacterial cell envelope.
Keywords: crystallography, lipoprotein, Mycobacterium, phthiocerol dimycocerosates, tuberculosis
Introduction
Mycobacterium tuberculosis, the etiologic agent of tuberculosis (TB) in humans, is the second most deadly infectious agent in the world claiming about 2 million lives annually and the global number of TB cases is still rising at a rate of 2% per year (World Health Organization, 2004). A better understanding of the biology of the tubercle bacillus and of the molecular determinants underlying its pathogenicity are required to help the conception of new prophylactic and therapeutic strategies.
The complete sequence of the Mycobacterium tuberculosis genome (Cole et al, 1998) has revealed about one hundred lipoprotein genes representing roughly 2.5% of M. tuberculosis open-reading frames. Some of the putative lipoproteins they encode share sequence similarities with enzymes, components of transport systems or receptor domains of regulatory systems (Sutcliffe and Harrington, 2004). However, so far, studies on mycobacterial lipoproteins have only focused on a very limited set of proteins and have mainly addressed their potential implications in pathogenicity and immunogenicity (Young and Garbe, 1991; Bigi et al, 2004; Sander et al, 2004). As a result, only ∼10% of M. tuberculosis lipoproteins have been unambiguously associated with an enzyme activity or a function in the transport of inorganic phosphate, sulfur or potassium (Hackbarth et al, 1997; Lefèvre et al, 1997; D'Orazio et al, 2001; Torres et al, 2001; Wooff et al, 2002; Steyn et al, 2003; Sander et al, 2004; Sutcliffe and Harrington, 2004).
A possible explanation for this high number of putative lipoproteins is most probably linked to the distinctive architecture and composition of the mycobacterial cell envelope. Indeed, from the inside to the outside of the bacterium, the cell envelope can be schematically decomposed into four major structures: (i) the plasma membrane, (ii) the cell wall core that contains the peptidoglycan covalently linked to a highly branched heteropolysaccharide (arabinogalactan), which in turn is substituted at its nonreducing termini with very long-chain (C60–C90) fatty acids (mycolic acids), (iii) an outer lipid layer that is mainly composed of a large variety of noncovalently attached complex lipids and (iv) a capsule consisting of polysaccharides, proteins and lipids (Brennan and Nikaido, 1995; Daffé and Draper, 1998). It was proposed that the noncovalently bound lipids interact with the mycolic acids of the cell wall core to provide an asymmetric mycobacterial ‘outer membrane' system (Minnikin, 1982). Therefore, although mycobacteria are classified as Gram-positive bacteria, they possess an envelope that functionally resembles that of Gram-negative bacteria. This atypical organization associated with a large amount of lipids (lipids represent as much as 60% of the dry weight of the envelope) confers to the mycobacterial cell envelope an exceptionally low permeability, generally thought to be the major determinant of the intrinsic resistance of mycobacteria to antibiotics and chemotherapeutic agents (Brennan and Nikaido, 1995). Although such a unique architecture of the mycobacterial cell envelope implies the existence of general and specific pathways for export or secretion of macromolecules, to date, no transport mechanism allowing the translocation of (glyco)lipids and polysaccharides from their production site (generally cytosolic or membrane-associated) to the outer membrane has been yet described in mycobacteria. Based on what is currently known about the transport of lipoproteins and polysaccharides across the outer membrane of Gram-negative bacteria, such export routes could involve lipoproteins (Drummelsmith and Whitfield, 2000; Tokuda and Matsuyama, 2004).
In a search for novel M. tuberculosis virulence factors, we have isolated by Signature-tagged Transposon Mutagenesis (STM) an attenuated mutant carrying a transposon insertion in the putative lipoprotein gene lppX. Lipid analysis revealed that the lppX mutant fails to release complex lipids known as phthiocerol dimycocerosates (DIM) (Figure 1) into the culture medium, although it apparently retains the ability to translocate these lipids across the plasma membrane. This study describes the functional and structural analyses that were undertaken on the lppX mutant and on the LppX protein, respectively, to investigate the role of this lipoprotein in the transport of DIM to the cell surface of M. tuberculosis.
Figure 1.
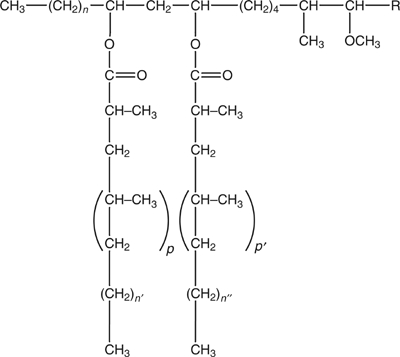
Structure of the phthiocerol dimycocerosates of M. tuberculosis. The long chain β-diol (phthiocerol moiety) is esterified with two mycocerosic acids; n=20–22; n′, n″=16, 18; p, p′=2–5; R=−CH2−CH3 or −CH3.
Results and discussion
Isolation and virulence attenuation of the lppX mutant of M. tuberculosis Mt103
In the context of our search for genes required for the multiplication and persistence of M. tuberculosis in vivo, ∼2000 new transposon mutants of M. tuberculosis Mt103 (Camacho et al, 1999) were screened by STM for attenuated virulence in the lungs of intranasally infected BALB/c mice 3 weeks postinfection. One of the mutants that displayed the greatest virulence attenuation carried a transposon inserted 187 bp downstream from the start codon of the lppX gene (Rv2945c).
This gene encodes a protein with a signal sequence and lipoprotein lipid attachment motif (PS00013) typically found in prokaryotic lipoproteins and thus presumably encodes a lipoprotein precursor. LppX, which was studied earlier for its immunogenic properties (Lefèvre et al, 2000; Al-Attiyah and Mustafa, 2004), was found in the membrane, cell wall, cell surface and culture medium of Mycobacterium bovis BCG and M. tuberculosis H37Rv (Lefèvre et al, 2000). Thus, as a potential lipoprotein, LppX seems not to remain anchored into the cytoplasmic membrane but, instead, to be exported to the outermost layers of the mycobacterial cell envelope.
lppX lies within a region of the M. tuberculosis genome dedicated to the synthesis and transport of DIM (Figure 1) (Camacho et al, 1999, 2001; Cox et al, 1999), complex lipids of the cell envelope that we have recently shown to play important roles in the modulation of the host immune response and in the resistance of M. tuberculosis to reactive nitrogen intermediates (Rousseau et al, 2004). Consistent with its potential role in the processing of DIM, lppX has orthologs in the DIM-producing species Mycobacteriumleprae and M. bovis, but not in Mycobacterium avium and Mycobacterium smegmatis, which are devoid of these lipids. lppX orthologs were also found in Mycobacterium marinum and Mycobacterium ulcerans, phthiocerol diphthioceranate (DIP)-producing species of mycobacteria in which phthioceranic acids instead of mycocerosic acids esterify the β-diol of phthiocerol (Daffé and Lanéelle, 1988).
The growth rate of the lppX mutant (strain 28AE5) in 7H9 broth was similar to that of wild-type M. tuberculosis Mt103, indicating that the disruption of lppX had no effect on growth in this medium (data not shown). However, when 103 CFUs of wild-type Mt103 or 28AE5 were used to infect BALB/c mice individually through the intranasal route, the attenuation phenotype of the mutant in vivo was obvious. Approximately 80-fold less mutant CFUs than Mt103 CFUs were recovered from the lungs of mice 3 weeks postinfection (data not shown). This important level of attenuation was comparable to that reported earlier for Mt103 mutants deficient in the synthesis or transport of DIM molecules (Camacho et al, 1999; Rousseau et al, 2004).
Functional characterization of LppX
Because of the chromosomal colocalization of lppX with genes dedicated to the synthesis and transport of DIM, we examined lipid extracts from wild-type Mt103 and the 28AE5 mutant following metabolic labeling of their methyl-branched fatty acids with [1-14C]propionate and compared the amounts and nature of DIM produced by the two strains. This experiment was performed on bacteria grown in 7H9 medium devoid of Tween 80 to minimize the amounts of lipids released by the bacterial cells in the culture medium. Thin-layer chromatography (TLC) analysis of bacteria-associated lipids in various solvent systems revealed that Mt103 and 28AE5 produced similar amounts of DIM (Figure 2A) and other methyl-branched fatty acid-containing lipids (data not shown).
Figure 2.
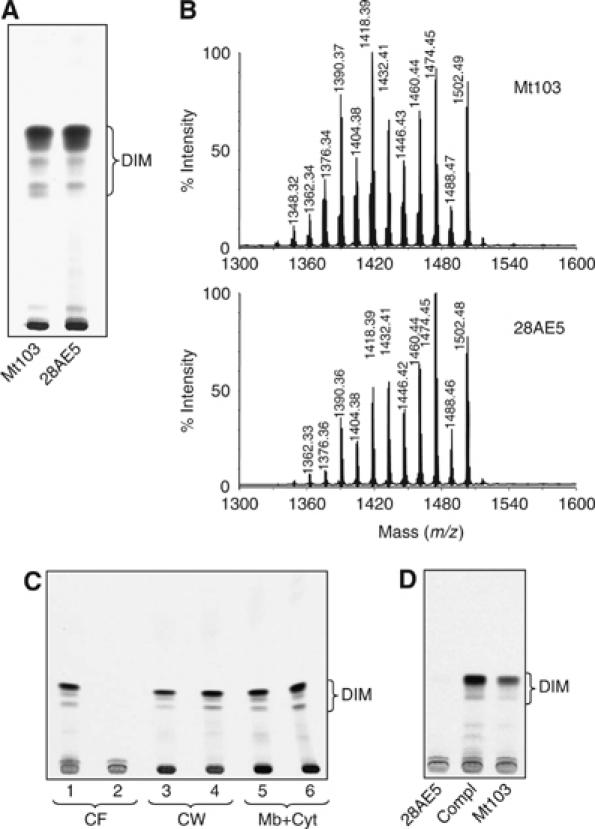
Production and distribution of DIM in the wild-type and lppX mutant strains of M. tuberculosis Mt103. (A) TLC analysis of [1-14C]propionate-derived lipids from wild-type Mt103 and 28AE5 grown in 7H9+ADC medium without Tween 80. Total lipids extracted from bacterial cells were subjected to TLC on silica gel G with 2% ethyl acetate in petroleum ether (60–80°C) as the solvent. (B) MALDI-TOF mass spectra of purified DIM molecules from wild-type Mt103 and 28AE5. (C) TLC analysis of [1-14C]propionate-derived lipids from bacteria grown in 7H9+ADC+Tween 80 and fractionated into culture filtrate (CF), cell wall (CW) and plasma membrane plus cytosol (Mb+Cyt). Mt103, lanes 1, 3 and 5; 28AE5, lanes 2, 4 and 6. (D) TLC analysis of [1-14C]propionate-derived lipids released in the culture medium of wild-type Mt103, 28AE5 and 28AE5/pVVlppX (Compl). In all TLCs, equal volumes of lipid preparations were loaded per lane. The solvent system used in (C) and (D) is the same as in (A).
To further compare the nature of the DIM produced by Mt103 and 28AE5, we fractionated the lipids from both strains on a silica gel column and analyzed the purified DIM-containing fractions by matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry (MS). The spectra from the two strains were almost superimposable (Figure 2B) and showed a series of intense pseudomolecular ion (M+Na)+ peaks differing from one another by 14 mass units. This difference corresponds to the mass of either a methylene unit or a methyl branch that is known to occur in the mycocerosic acids that esterify the β-diol of phthiocerol in DIM (Daffé and Lanéelle, 1988). We therefore concluded that wild-type Mt103 and its isogenic lppX mutant produced the same types and amounts of DIM molecules.
We next sought to compare the subcellular distribution of DIM in wild-type Mt103 and 28AE5. To this aim, 7H9-Tween 80 cultures of Mt103 and 28AE5 were metabolically labeled with [1-14C]propionate, fractionated into culture filtrate, cell wall and plasma membrane plus cytosol and each subcellular fraction was tested for the presence of DIM. In the presence of Tween 80, M. tuberculosis sheds some of its outermost components in the culture medium (Cox et al, 1999; Camacho et al, 2001) allowing an easier inventory of its outer membrane composition. A striking difference was observed between wild-type and mutant strains. While 36% of the DIM synthesized by wild-type Mt103 were found in the culture filtrate (with the remaining 27 and 37% being found in the cell wall and membrane plus cytosol fractions, respectively), no DIM could be detected in the culture filtrate of the mutant (Figure 2C). Instead, the DIM produced by 28AE5 remained associated with the cell wall (44%) and cytosol plus plasma membrane fractions (56%) (Figure 2C). Complementation of 28AE5 with a wild-type copy of the lppX gene carried on a multicopy shuttle plasmid restored the release of DIM into the culture medium (Figure 2D), clearly indicating that LppX is required for DIM to reach the cell surface of M. tuberculosis.
TLC analysis of the production and subcellular distribution of other lipids upon metabolic labeling with [1-14C]propionate or [1,2-14C]acetic acid revealed no other qualitative or quantitative difference between the two strains (data not shown), suggesting that LppX is only required for the translocation of DIM. Moreover, as the disruption of lppX did not result in the accumulation of DIM precursors, it is inferred that LppX is involved in the transport of full-size DIM molecules. Finally, given the facts that a significant portion of the DIM produced by 28AE5 were found in the cell wall fraction and that LppX was mainly detected in the cell wall and at the cell surface of M. tuberculosis and M. bovis BCG (Lefèvre et al, 2000), we propose that LppX is specifically required for the transport of DIM to the outer layers of the envelope after their translocation across the plasma membrane by a system involving DrrABC and MmpL7 (Cox et al, 1999; Camacho et al, 2001). The improper localization of DIM in 28AE5 is in good agreement with the virulence attenuation of this mutant, which was similar to that reported earlier for mmpL7 and drrC mutants of Mt103 (Camacho et al, 1999).
LppX is a lipoprotein
The lipoprotein character of LppX was assessed by applying Triton X-114 phase separation to the total proteins present in soluble extracts of the complemented mutant strain, 28AE5/pVVlppX. The resulting detergent and aqueous phases were then analyzed by SDS–PAGE and immunoblotting using a monoclonal anti-His antibody. As shown in Figure 3A, all of the recombinant LppX protein was found in the detergent phase, suggesting that LppX is acylated. This assumption was further supported by the demonstration that His-tagged LppX partially purified from 28AE5/pVVlppX using a cobalt-based metal affinity column incorporates [1-14C]palmitate (Figure 3B). The two bands of almost identical size (approximately 25 kDa) revealed by immunoblot and autoradiography (Figure 3A and B) are likely to correspond to the lipid-modified precursor form of LppX prior and after cleavage by signal peptidase II.
Figure 3.
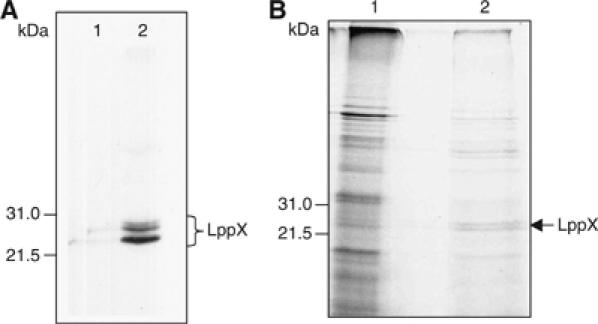
Triton X-114 phase partitioning and partial purification of the LppX protein. (A) Triton X-114 phase partitioning analysis of 28AE5/pVVlppX proteins. Total soluble proteins from 28AE5/pVVlppX were subjected to Triton X-114 phase partitioning and the resulting aqueous (lane 1) and detergent (lane 2) phases were then analyzed by SDS–PAGE and immunoblotting using a monoclonal anti-His antibody. (B) Partial purification of the LppX protein from [1-14C]palmitate-labeled 28AE5/pVVlppX. Lane 1: Total protein extract; lane 2: proteins eluted from the resin with 500 mM imidazole. Radiolabeled proteins were visualized by autoradiography. The elution profile reveals a significant enrichment of the total protein extract with two radiolabeled proteins of approximately 25 kDa that comigrate with the two bands of almost identical size revealed by immunoblotting with the anti-His antibody.
Structure quality and overall fold
Crystals of a truncated form of the mature LppX protein, devoid of its first nine amino acids but containing 21 additional N-terminal residues arising from cloning, were of poor diffraction quality. To overcome this problem, a TEV cleavage site was introduced upstream from the 10th amino acid of LppX (Glu10) and the resulting cleaved protein yielded crystals that diffract up to 2 Å resolution. The crystal structure of LppX was solved by the single-wavelength anomalous dispersion (SAD) method, using a mercury derivative, and refined at 2.15 Å resolution against a native data set. The final model, which has good stereochemistry (Table I), consists of residues Ser20–Val114 and Gly120–Asp207.
Table 1.
Data collection and refinement statistics
| Data collection | Hg-SAD | Native |
|---|---|---|
| Beamline | ESRF/ID23-1 | ESRF/ID29 |
| Wavelength (Å) | 1.00725 | 0.9792 |
| Space group | P6522 | |
| a=b, c (Å) | 54.20, 269.86 | 54.26, 269.98 |
| Resolution rangea | 50–2.3 (2.42–2.30) | 20–2.15 (2.27–2.15) |
| Rmergea,b | 0.041 (0.082) | 0.057 (0.121) |
| No. of observations | 366 260 | 115 100 |
| No. of unique reflections | 10 521 | 13 680 |
| Completenessa (%) | 91.8 (65.1) | 98.6 (91.8) |
| Redundancya | 34.8 (21.6) | 8.4 (5.2) |
| 〈I/σI〉a | 95.4 (25.4) | 24.0 (10.3) |
| B from Wilson statistics (Å2) | 31.93 | 28.34 |
| Refinement | ||
| Resolution (Å) | 20–2.15 | |
| No. of protein atoms | 1345 | |
| No. of water molecules/ ligand atoms | 127/92 | |
| Rcryst/Rfree (%) | 18.52/22.41 | |
| R.m.s. 1–2 bond distances (Å) | 0.008 | |
| R.m.s. 1–3 bond angles (deg) | 1.268 | |
| Average main/side chain B (Å2) | 41.85/42.98 | |
| Average B solvent/ligand (Å2) | 44.36/61.93 | |
| Average B fatty acids (Å2) | 63.9 | |
| Main chain ΔB, bonded atoms (Å2) | 1.34/1.75 | |
| Ramachadran plot | ||
| Residues in most favored regions | 92.4% | |
| Residues in additional allowed regions | 7.0% | |
| Residues in generously allowed regions | 0.6% | |
| aValues in parentheses are for the highest resolution shell. | ||
| bRmerge=∑hkl∑i∣Ihkl−〈Ihkli〉∣/∑hkl∑i〈Ihkli〉; Rcryst=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣; target mean values and standard deviations are given in parentheses. | ||
The structure of LppX reveals an α/β-fold consisting of an 11-stranded anti-parallel β-sheet (β1–11) and three α-helices (α1–3) attached to the concave face of the β-sheet, and has a globular shape with overall dimensions of 36 × 38 × 57 Å (Figure 5A). The β-sheet, which has strand order 789[10][11]123456, forms an U-shaped β-half-barrel with a helical twist. The three α-helices are located at the N-terminus (α1) and between the β-strands β6 and β7 (α2) and β8 and β9 (α3), respectively.
Figure 5.
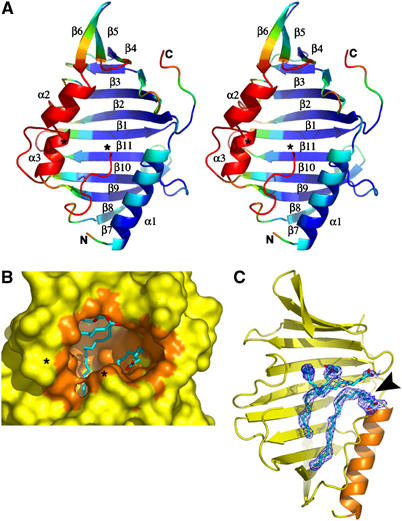
Overall view and cavity content of LppX. (A) Stereo view of the ribbon diagram of LppX with β-strands (β1–11) and α-helices (α1–3) colored as a rainbow gradient according to B-factor values (blue to red corresponds to B-factor values ranging from <30 to >55 Å2) viewed down the cavity entrance. (B) Molecular surface of LppX, oriented as in (A), surrounding the cavity (orange) partially occupied by three long-chain (C18–C22) fatty acids (blue carbon atoms with carboxylate in red). The invisible region (Leu115–Ala119) is symbolized by asterisks. (C) Sigma A-weighted difference electron density maps (contoured at 2.5σ, cyan) showing the location of the three fatty acids, C18:1 (top) and C22 (bottom), within the cavity. The maps were generated using an LppX model from the initial refinement stage devoid of fatty acids and solvent molecules. An arrow indicates the cavity entrance. For clarity, the α-helices α2 and α3 have been omitted.
The most striking feature of the LppX structure is a large hydrophobic cavity, with approximate dimensions of 25 × 12 × 12 Å, resulting in a surface area of ∼1375 Å2 and a volume of ∼2835 Å3 (Figure 5B). Its hydrophobic character arises from numerous small aliphatic and one Phe residue that line the pocket. Indeed, out of the 38 side chains that line the cavity, only three polar side chains (Arg113, Gln123 and Gln197) point toward the cavity. The cavity is partially filled with three long stretches of electron density (Figure 5C), which have been modeled as two cis-vaccenic acids (C18:1) and docosanoic acid (C22:0) (see Materials and methods). The presence of fatty acids in this cavity was corroborated by gas chromatography–MS (GC/MS) analyses of fatty acid methyl esters prepared from two independent batches of purified LppX used in the crystallography experiments. Gas chromatograms unambiguously revealed the presence of palmitic and cis-vaccenic acids in the preparations of LppX that were not found in the preparations of an unrelatedM. tuberculosis protein (Rv3849) purified from the same Escherichia coli strain under the same conditions (Figure 4).
Figure 4.
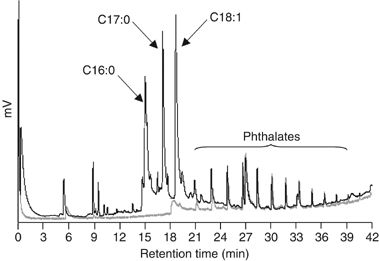
Gas chromatograms of fatty acid methyl esters recovered from purified protein preparations. Analyses of a preparation of LppX (black line) and Rv3849 (a M. tuberculosis 14.7 kDa cellular protein of unknown function) (gray line) are shown. Fatty acid methyl esters prepared from the equivalent of 50 μg of purified proteins were injected in both cases. Approximately 0.55 mol of fatty acids were recovered per mole of purified LppX. Peaks of similar intensities found in both protein preparations were identified by GC/MS as phthalates, likely to originate from plastic contamination. Heptadecanoic acid (C17:0) methyl ester (Sigma) was used as an internal standard for fatty acid quantification.
The concave face of the β-half-barrel forms the floor and the walls of the cavity, whereas the mouth is formed by the circular arrangement of the three α-helices, together with loop regions connecting β-strands and the highly mobile Gly120–127 loop region. This stretch of residues adopts a conformation reminiscent of an α-helix and, together with the preceding and invisible Leu115–Ala119 region, may restructure upon access of a ligand to the cavity. The rather short distance of 8.5 Å between the Cα atoms of Val114 and Gly120 residues suggests that formation of an additional α-helix could take place to accommodate the missing five amino acids upon binding of a large lipophilic ligand. Evidence for a high degree of dynamics for all structural elements forming the mouth of the cavity is given by the elevated thermal B-factor values of these elements compared to the rest of the three-dimensional structure (Figure 5A). In the absence of the Leu115–Ala119 residues, the cavity entrance has an oval shape of 7 × 15 Å, but motions of the structural elements lining the mouth are expected to alter its shape and dimension.
As mentioned above, PHI-PSI BLAST searches using the M. tuberculosis LppX sequence as a template identified top-ranked orthologs with e-values in the 10−60 range exclusively within the DIM- or DIP-producing mycobacterial species, M. bovis, M. leprae, M. ulcerans and M. marinum, resulting in sequence identities of 76–98%. LppX also shares 28% sequence identity with the M. tuberculosis lipoproteins LprA, LprF and LprG, suggesting that these proteins share a similar fold and may have evolved to recognize specific lipophilic ligands. Sequence alignment clearly indicates that all of the conserved aliphatic residues are confined within the cavity, consistent with our hypothesis that this large cavity is important for the biological functions of LppX and structurally related LprAFG proteins (see Supplementary data). With the exception of Lys36 near the cavity entrance, invariant polar residues are located on the protein surface and define a patch located on the opposite face to the cavity entrance. These residues may have a role during the translocation process or in targeting LppX to the outermost layers of the cell envelope.
The hydrophobic cavity of LppX could accommodate a DIM molecule
Given the fact that LppX appears to be directly or indirectly involved in DIM translocation, cocrystallization as well as crystal-soaking experiments with purified DIM molecules were performed in order to elucidate the putative mode of binding of these complex lipids. Unfortunately, all of our efforts to obtain LppX–DIM complexes were hindered by the extreme hydrophobic character of DIM. Preliminary crystallographic data collected with putative complexes (data not shown) did not allow the localization of the lipid and revealed only subtle conformational changes of the helical regions that line the cavity mouth, underlining their high mobility with respect to the β-sheet core, as evidenced by the B-factor distribution (Figure 5A). Modeling of the two mycocerosic acid chains and of the long-chain β-diol moiety of a DIM molecule into the cavity guided by the position of the three E. coli fatty acyl chains and followed by energy minimization and refinement against native X-ray data (data not shown), however, showed a nice fit of the two mycocerosate acyl chains and part of the phthiocerol moiety within the cavity, with only a small portion of the phthiocerol moiety emerging from the cavity entrance (Figure 6A and B). Small conformational changes within the α-helical region would be sufficient to envelop this latter portion and shield it from a hydrophilic environment. Although the network of aliphatic residues that line the cavity would be suitable to accommodate such a large lipophilic molecule as DIM (Figure 6C), given the relative small size of the cavity entrance, one should imagine major conformational changes to occur upon DIM uptake and release.
Figure 6.
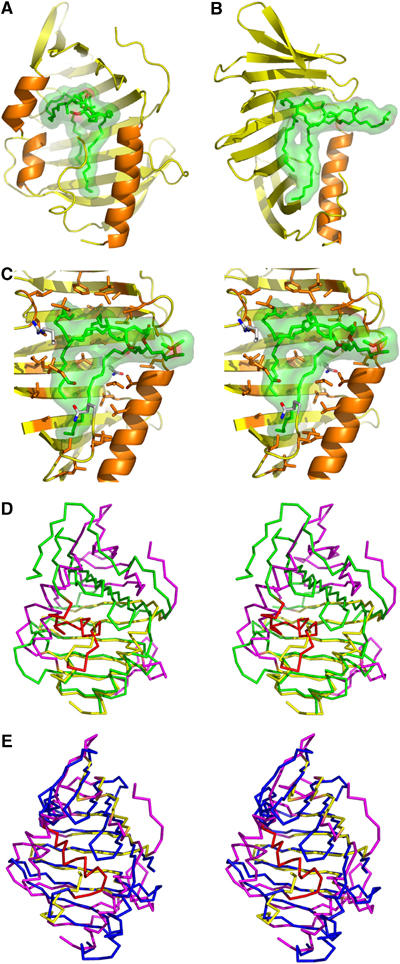
Hydrophobic cavity of LppX and structural comparison. A DIM molecule (green carbon atoms with carboxylate in red) is modeled into the cavity and is shown under a transparent surface, with LppX oriented as in Figure 5A (A) and rotated by 90° (B). (C) Close-up stereo view of the DIM molecule, oriented as in (B), showing the key aliphatic and polar residues (orange and with carbon atoms, respectively) that line the cavity. For clarity, the α-helices, α2 and α3, have been omitted in panels B and C. Overlay of LppX (yellow), oriented as in (A), and the crystal structure of (D) LolA (green, 1IWL) and (E) LolB (blue, 1IWN). Structural elements that significantly deviate in LppX are shown in magenta; those of LolA and LolB that differ from LppX cavity are in red.
Thus, our structural and functional data together with the demonstrated affinity of fatty acids for the hydrophobic cavity of LppX tend to suggest that LppX is a DIM carrier. Definitive proof of this assumption, however, awaits the direct biochemical demonstration of an interaction between LppX and DIM and the cocrystallization of LppX–DIM complexes.
Structural comparison
A DALI search for close structural homologs of LppX within the nonredundant set of protein structures from the PDB revealed top-ranked hits from the E. coli lipoprotein localization factors LolA (Z-score 5.8) and LolB (Z-score 5.8) (Takeda et al, 2003; Tokuda and Matsuyama, 2004), the autotransporter NalP from Neisseria meningitis (Z-score 5.8) (Oomen et al, 2004), the E. coli nucleoside transporter Tsx (Z-score 4.8) (Ye and van den Berg, 2004) and the integral membrane adhesin OpcA from N. meningitis (Z-score 4.3) (Prince et al, 2002). While NalP, Tsx and OpcA are integral outer membrane proteins from Gram-negative bacteria, forming closed β-barrels involved in either transport or adhesion, only LolA and LolB exhibit a similar β-half-barrel structure with the topology found in LppX. Other lipid-binding proteins exhibiting a β-half-barrel, such as the MLN64-START domain protein (Tsujishita and Hurley, 2000) and the phosphatidylinositol transfer protein (Yoder et al, 2001), present a different β-strand order from LppX and are hence not identified by DALI as structural homologs.
LolA plays the role of a periplasmic molecular chaperone, transferring outer-membrane-specific lipoproteins from the inner membrane to the outer membrane localization factor LolB. The three-dimensional structures of LolA and LolB, further supported by functional studies, suggest that the unbound closed form of LolA is more stable than the lipoprotein-bound form, whereas the opposite is true for LolB, in agreement with a vectorial transport of lipoproteins from low-affinity to high-affinity partners (Takeda et al, 2003). LppX shares the β-strand number and order found in LolB, whereas LolA is characterized by an additional β-strand at the C-terminal (see Supplementary data). The more extended shape of the β-half-barrel in LppX is more similar to that of LolB than to the curved shape β-half-barrel of LolA (Figure 6D and E). However, only the closed form of LolA has been observed and slight unfolding of the β-half-barrel towards a more extended conformation is expected to occur upon lipoprotein uptake. Such a variation in the shape of the β-sheet dramatically affects the shape of the cavity and thus may dictate ligand selectivity.
The most striking difference between LppX and LolA/B resides in the position of the α-helices forming the mouth and lid of the cavity. LppX helix α2 is replaced by a 310-helix and two α-helices in LolA and by two α-helices in LolB. LppX helix α3 is replaced by short loop regions both in LolA and LolB (Figure 6D and E). Altogether, the α-helices in LolA and LolB form a lid covering completely the concave face of the β-half-barrel, and an open channel between the β-half-barrel and the α-helical lid is only visible in LolB to access a rather small hydrophobic cavity of ∼300 Å3. Conversely, α-helices in LppX stay far apart, being aligned in a parallel manner, bordering a wide entrance to the large cavity facing the concave face of the β-half-barrel. These structural differences in the access and shape of the cavity between LppX and LolA/B might reflect the binding of different lipophilic molecules.
One of the crystal forms of LolB had a PEGMME2000 molecule bound within its cavity and it was suggested that this molecule mimicked one of the acyl chains of the N-acyl diacyl glycerol moiety of lipoproteins. Although LppX could easily bind the entire N-acyl diacyl glycerol moiety of lipoproteins, the large size of its cavity compared to that of LolB suggests that it is made to accommodate a bulkier group, such as a DIM molecule.
Conclusion
In both Gram-negative and Gram-positive bacteria, a substantial number of lipoproteins have been shown to be involved in the translocation of a wide range of substrates across the cell envelope (Tam and Saier, 1993; Sutcliffe and Russell, 1995; Drummelsmith and Whitfield, 2000; Tokuda and Matsuyama, 2004). In spite of the considerable interest the mycobacterial cell envelope has been stimulating for decades, little is known about the strategies used by mycobacteria to transport lipids, glycolipids, polysaccharides and lipoglycans from their site of production to their final extracytoplasmic location. In describing the first lipoprotein required for the transport of complex lipids to the outer layers of a bacterial envelope, this work supports the concept that lipoproteins are key players in the building of the outer membrane of mycobacteria and paves the way for future studies aimed at deciphering the biological role of this abundant class of proteins.
The precise role of LppX in DIM translocation now remains to be determined. Based on the restricted distribution of the lppX gene to DIM-producing species of mycobacteria, the binding of C16–C18 fatty acids to the hydrophobic cavity of LppX and our functional and structural data, it is tempting to speculate that LppX binds DIM directly to transport them from the outer leaflet of the plasma membrane to the outer membrane. DIM transport could then proceed associated with a chaperone analogous to the Lol-dependent transport mechanism of lipoproteins in Gram-negative bacteria. Until a direct biochemical evidence of an interaction between LppX and DIM is provided, however, other functions for LppX cannot be excluded. For instance, LppX might act as a receptor of DIM in the outer membrane stabilizing their surface localization, as is the case of LolB in the translocation of lipoproteins, or serve to transfer DIM to another lipoprotein located in the outer membrane as is the case of LolA. Alternatively, as its structural resemblance with the Lol system would initially suggest, LppX might participate in DIM translocation only indirectly transporting other (lipo)proteins involved in carrying DIM or stabilizing their localization at the cell surface. Further studies will be required to clarify this point.
Materials and methods
Bacterial strains and culture conditions
E. coli XL1-blue and BL21DE3pLysS were maintained in Luria Bertani (LB) broth (pH 7.5) (Becton Dickinson, Sparks, MD). Mt103, the clinical isolate of M. tuberculosis used in this study (Jackson et al, 1999), was grown in Middlebrook 7H9 medium supplemented with ADC (Difco) and 0.05% Tween 80 or on solid Middlebrook 7H11 medium supplemented with OADC (Difco). The antibiotics, kanamycin (Km) (25 μg/ml) and hygromycin (Hyg) (50 μg/ml) were added when appropriate.
STM and mice infection studies
Approximately 2000 new mutants (38 pools of 48 mutants) from the tagged Mt103 transposon mutant libraries described earlier (Camacho et al, 1999) were screened by STM in mice. The protocol used to screen the mutants was similar to the one described by Camacho et al (1999) with the following modification: 6- to 8-week-old female BALB/c mice purchased from CERJanvier were infected intranasally (Rousseau et al, 2004), instead of intravenously, with 103 CFU of the mutant pools. At 3 weeks postinfection, mice were killed, their lungs were removed aseptically and serial one in 10 dilutions of the lung homogenates were plated onto agar medium.
Mice were also infected individually through the intranasal route with 103 CFU of wild-type Mt103 and the Mt103 lppX mutant (strain 28AE5) and killed 3 weeks postinfection.
The transposon insertion site in 28AE5 was identified by ligation-mediated PCR as described (Prod'hom et al, 1998).
Complementation of the lppX mutant
Standard PCR strategies with Taq Gold DNA polymerase (Applied Biosystems, Roche) were used to amplify the M. tuberculosis Mt103 lppX gene. PCR amplification consisted of one denaturation step (95°C, 6 min) followed by 35 cycles of denaturation (95°C, 1 min), annealing (61°C, 1 min) and primer extension (72°C, 1 min), and a final extension step at 72°C for 10 min. The primers were Lppx.2 (5′-ggccgggcatatgaatgatggaaaacgggcgg-3′) and LppX.3 (5′-gcgaagcttgtcgacgttgacgggttcgttc-3′). The primers were designed to generate a PCR product corresponding to the entire lppX gene harboring NdeI and HindIII restriction sites (underlined in the primers sequences) to enable direct cloning into the pVV16 expression vector (Korduláková et al, 2002). The recombinant LppX protein produced with this system carries a six-histidine tag at its carboxyl-terminus. 28AE5 was transformed with the resulting expression vector, pVVlppX, and transformants were selected on 7H11-Km-Hyg plates. The production of recombinant LppX in 28AE5/pVVlppX was analyzed by immunoblotting with a mouse monoclonal anti-His antibody (Penta-His antibody; Qiagen) as described previously (Korduláková et al, 2002).
Whole-cell radiolabeling experiments
Radiolabeling of whole M. tuberculosis Mt103, 28AE5 and 28AE5/pVVlppX cells with [1,2-14C]acetic acid (specific activity, 55.3 Ci/mol; Perkin Elmer Life Sciences) and [1-14C]propionate (specific activity, 56.7 Ci/mol; ICN) was performed in 7H9 with or without Tween 80. Radiolabeling of whole 28AE5/pVVlppX cells with [1-14C]palmitic acid (specific activity, 50 Ci/mol; ICN) was performed in 7H9 medium without Tween 80. Radiolabeled precursors (0.5 μCi/ml for [1,2-14C]acetic acid and [1-14C]palmitate; 0.2 μCi/ml for [1-14C]propionate) were added to mid-log cultures and cultures were incubated at 37°C for a further 18 h with shaking.
Subcellular fractionation and analysis of lipids and fatty acids
Total lipids from cold or radiolabeled bacterial cells and culture media were extracted as described previously (Stadthagen et al, 2005). To analyze the subcellular distribution of DIM in wild-type Mt103 and 28AE5, [1-14C]propionate-labeled cells collected from 50 to 100 ml 7H9 cultures supplemented with Tween 80 were resuspended in 600 μl of water and broken with miniglass beads using a Bead Beater apparatus (Polylabo) for 8 min in the form of 8 × 60-s pulses with 1 min cooling intervals at 4°C between pulses. Beads and unbroken cells were removed by centrifugation at 3000 r.p.m. for 10 min and supernatants were recentrifuged for 30 min at 14 000 r.p.m. The 14 000 r.p.m. supernatants corresponded to cytosol and plasma membrane components, whereas the pellets consisted mainly of cell wall components. These pellets were checked for the absence of contamination with cytoplasmic compounds using the isocitrate dehydrogenase activity assay (Sigma). Lipids were analyzed by TLC on silica gel 60-precoated plates F254 (E Merck, Darmstadt, Germany) in various solvent systems as described (Rousseau et al, 2003). Radiolabeled lipids were visualized by exposure of TLC to Kodak BIOMAX MR films at −70°C. Fatty acid methyl esters from purified protein preparations were prepared by acid methanolysis using the Methanolic-HCl (3 N) kit from Supelco and analyzed by GC/MS. GC analyses were performed on a Girdel series 30 instrument equipped with a fused silica capillary column (25 m length × 0.22 mm inner diameter) containing WCOT OV-1 (0.3 μm film thickness spiral), using helium gas (0.7 bar) and a flame ionization detector at 310°C. A temperature gradient of 100–310°C with a programmed increase of 5°C/min was used.
Purification and structural analysis of DIM
DIM from wild-type Mt103 and 28AE5 were purified from total lipids extracts by chromatography on a silica gel (70–230 mesh) column. Lipid fractions were eluted with increasing concentrations of diethyl ether in petroleum ether (60–80°C) and the DIM content of each fraction was determined by TLC using petroleum ether/ethyl acetate (98:2 (v/v); three developments) as the eluent. MALDI-TOF MS detection of purified DIM samples in reflectron mode was performed as described previously (Camacho et al, 2001).
Purification of recombinant His-tagged LppX protein and Triton X-114 phase separation
For the purification of recombinant [1-14C]palmitate-labeled LppX, [1-14C]palmitate-labeled 28AE5/pVVlppX cells were washed and resuspended in buffer A (20 mM Tris–HCl buffer, pH 7.45, 500 mM NaCl, 10% glycerol) before being broken with miniglass beads as described above. The unbroken cells and bacterial debris were removed by centrifugation at 13 000 r.p.m. for 30 min and recombinant His-tagged LppX protein was purified from the supernatant using BD TALONspin columns™ (BD Biosciences Clontech, Palo Alto, CA) according to the supplier's recommendations. Unbound proteins were removed by washing the resin with buffer A. Proteins bound to the resin were then eluted twice with buffer A containing 500 mM imidazole. Radiolabeled proteins were visualized by exposure of dried SDS–PAGE gels to Kodak BIOMAX MR films at −70°C.
Extraction of proteins with Triton X-114 was performed as follows: total protein extracts from 28AE5/pVVlppX were obtained by breaking the cells with glass beads as described above. Unbroken cells and beads were removed by two consecutive centrifugations at 5000 r.p.m. for 5 min and the supernatant was recentrifuged at 14 000 r.p.m. for 30 min to separate the cell wall fraction (pellet) from the membrane plus cytosol fraction (supernatant). Triton X-114 (final concentration 2%) was added to the latter fraction and left overnight at 4°C. The temperature was then raised to 37°C for 10 min and the resulting detergent and aqueous phases were separated by centrifuging at 13 000 r.p.m. for 10 min at room temperature. Each phase was ‘back-washed' once and analyzed by SDS–PAGE and immunoblotting.
Cloning, expression and purification of the LppX protein used in crystallography studies
Hydrophobic cluster analysis permitted the identification of an unstructured and proline-rich region at the N-terminal region of mature LppX. Hence, a truncated form of LppX, starting at residue Glu10, was produced. The open-reading frame encoding LppX was amplified by PCR from the MTCY24G1 cosmid (Cole et al, 1998) using the forward primer 5′-cggaaaacctgtacttccagggtGAACAGGGTGTTCCCGTGAGC-3′ (TEV recognition site underlined) and the reverse primer 5′-CTAGTCGACGTTGACGGGTTC-3′ containing the attB1 and attB2 recombination sites at the 5′- and 3′-end, respectively. The TEV cleavage site was introduced between the 6xHis tag and the first LppX codon in order to allow release of LppX upon proteolysis. The PCR product was cloned into the expression vector pDEST 17 O/I by the Gateway technology (Gateway, Invitrogen). Expression of lppX was carried out using E. coli BL21 pLysS cells (Novagen) grown in LB broth containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol, and induced with 0.5 mM isopropyl β-D-thiogalactoside. Cells were harvested after 4 h and pellets were resuspended in lysis buffer containing 0.25 mg/ml lysozyme, 10 μg/ml DNAse and 20 mM MgSO4. Cells were disrupted by freeze thawing and ultrasonication and cell debris were removed by centrifugation. LppX was purified by Ni2+ affinity followed by size-exclusion chromatography in 5 mM Hepes (pH 7.5) and 150 mM NaCl. The 21 additional residues at the N-terminus of LppX, containing the 6xHis tag, were cleaved by TEV digestion and LppX was purified by exclusion on a Ni2+ affinity column. Purified LppX was further characterized by SDS–PAGE, MALDI-TOF mass spectroscopy, circular dichroism and dynamic light scattering. LppX was concentrated up to 11 mg/ml and stored at −80°C.
Crystallization and data collection
Sitting drop vapor diffusion crystallization screens were set up on customized microtiter plates using our automated crystallization facilities (Sulzenbacher et al, 2002) based on a TECAN Genesis and a Cartesian robot. Crystals appeared with condition 47 of the Wizard II crystallization kit (Emerald Biostructures) and were further optimized by mixing 1 μl of protein solution with 2 μl of precipitant solution consisting of 1.8 M NaCl, 0.1 M Zn acetate and 0.1 M imidazole/malate buffer (pH 8). Bipyramidal crystals grew within 2–3 days and were transferred to a cryoprotectant solution consisting of reservoir solution supplemented with 30% (v/v) glycerol, before flash freezing in a nitrogen gas stream at 100 K. Crystals belong to space group P6522 with cell dimensions: a=b=54.3 Å, c=270.0 Å and contain one molecule per asymmetric unit. Hg-derivatized crystals were prepared by overnight soaking of native crystals in mother liquor saturated with mercurous chloride, Hg2Cl2, followed by back soaking in mother liquor. A highly redundant data set for a Hg-derivatized crystal was collected at the mercury K edge, 12.231 KeV, up to 2.3 Å resolution on beam line ID23-1, and a native data set up to 2.15 Å resolution on beam line ID29 (European Synchrotron Radiation Facility; Grenoble). Data were indexed and integrated with MOSFLM (Leslie, 1992) and all further computing carried out with the CCP4 program suite (CCP4, 1994), unless otherwise stated. Data collection statistics are summarized in Table I.
Structure solution and refinement
Before phasing, data were processed with the program XPREP (Bruker Nonius, Madison, WI, USA). Two mercury and five zinc ions were located by the SAD method with the program SHELXD (Schneider and Sheldrick, 2002), taking advantage of data up to the highest resolution shell. The correlation coefficients from SHELXD for the correct solution were 0.43 and 0.24 for all and weak data, respectively. Phasing and density modification were carried out with the program SHELXE (Sheldrick, 2002). Starting from experimental phases, a model consisting of 143 residues, 120 of which docked into sequence, was automatically built by the program ARP/wARP (Perrakis et al, 1999). The program TURBO-FRODO (http://www.afmb.univ-mrs.fr/-TURBO-) was used to complete the model manually. The resulting model was refined against native data extending to 2.15 Å resolution with the program REFMAC (Murshudov et al, 1997), using the maximum-likelihood approach and incorporating bulk solvent corrections, anisotropic Fobs versus Fcalc scaling and TLS refinement. A random 10% (1356) of reflections were set aside for crossvalidation purposes. Automated solvent building was performed with the program ARP/wARP. The final model encompasses residues Ser20–Val114 and Gly120–Asp207, 130 water molecules, six Zn2+ ions, one acetate ion, two malate ions and three fatty acyl chains (two C18:1 and one C22:0). The large C22 fatty acid, which was not detected by GC/MS in the LppX samples (Figure 4) may reflect the alternate positions of smaller fatty acids, for example, C16:0, in crystalline LppX. The N-terminal residues Glu10–Ala19 and residues Leu115–Ala119 could not be built owing to either diffuse or totally absent electron density. It is worth mentioning that LppX crystallizes only in the presence of Zn2+ and that three out of the six Zn2+ ions present in the final model are coordinated by invariant residues (see Supplementary data). The positions of some of the Zn2+ ions are reminiscent of those of bivalent ions in the crystal structure of LolA. The final model has no residue in the disallowed region of the Ramachandran plot, as verified with PROCHECK (Laskowski et al, 1993). Cavity surfaces and volumes were calculated with the program CASTp (Liang et al, 1998). Coordinates have been deposited in the Protein Data Bank with accession reference number 2BYO. Figures 5 and 6 were generated with PyMOL (DeLano Scientific, San Carlos, CA, USA).
Supplementary Material
Supplementary Data
Acknowledgments
We thank Nadine Honoré for providing us with the M. tuberculosis cosmids and BACs libraries and Bernard Henrissat for bioinformatic analyses. This work was supported by the GPH-05 research program (Institut Pasteur), the CONACyT program from Mexico (to G Stadthagen), the European Commission contracts QLRT-2001-02018 (X-TB) and QLRT-1999-31761 (TB Prevention cluster) and by the Marseille–Nice Genopole. The European Synchrotron Radiation Facility (ESRF) is greatly acknowledged for beam time allocation.
References
- Al-Attiyah R, Mustafa AS (2004) Computer-assisted prediction of HLA-DR binding and experimental analysis for human promiscuous Th1-cell peptides in the 24 kDa secreted lipoprotein (LppX) of Mycobacterium tuberculosis. Scand J Immunol 59: 16–24 [DOI] [PubMed] [Google Scholar]
- Bigi F, Gioffre A, Klepp L, de la Paz Santangello M, Alito A, Caimi K, Meikle V, Zumarraga M, Taboga O, Romano MI, Cataldi A (2004) The knock-out of the LprG-Rv1410 operon produces strong attenuation of Mycobacterium tuberculosis. Microbes Infect 6: 182–187 [DOI] [PubMed] [Google Scholar]
- Brennan PJ, Nikaido H (1995) The envelope of mycobacteria. Annu Rev Biochem 64: 29–63 [DOI] [PubMed] [Google Scholar]
- Camacho LR, Constant P, Raynaud C, Lanéelle M-A, Triccas JA, Gicquel B, Daffé M, Guilhot C (2001) Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem 276: 19845–19854 [DOI] [PubMed] [Google Scholar]
- Camacho LR, Ensergueix D, Pérez E, Gicquel B, Guilhot C (1999) Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol 34: 257–267 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 [DOI] [PubMed] [Google Scholar]
- Cox JS, Chen B, McNeil M, Jacobs WR Jr (1999) Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402: 79–83 [DOI] [PubMed] [Google Scholar]
- D'Orazio M, Folcarelli S, Mariani F, Colizzi V, Rotilio G, Battistoni A (2001) Lipid modification of the Cu, Zn superoxide dismutase from Mycobacterium tuberculosis. Biochem J 359: 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffé M, Draper P (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol 39: 131–203 [DOI] [PubMed] [Google Scholar]
- Daffé M, Lanéelle M-A (1988) Distribution of phthiocerol diester, phenolic mycosides and related compounds in mycobacteria. J Gen Microbiol 134: 2049–2055 [DOI] [PubMed] [Google Scholar]
- Drummelsmith J, Whitfield C (2000) Translocation of group 1 capsular polysaccharide to the surface of Escherichia coli requires a multimeric complex in the outer membrane. EMBO J 19: 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackbarth CJ, Unsal I, Chambers HF (1997) Cloning and sequence analysis of a class A β-lactamase from Mycobacterium tuberculosis H37Ra. Antimicrob Agents Chemother 41: 1182–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Raynaud C, Lanéelle M-A, Guilhot C, Laurent-Winter C, Ensergueix D, Gicquel B, Daffé M (1999) Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol Microbiol 31: 1573–1587 [DOI] [PubMed] [Google Scholar]
- Korduláková J, Gilleron M, Mikusová K, Puzo G, Brennan PJ, Gicquel B, Jackson M (2002) Definition of the first mannosylation step in phosphatidylinositol synthesis: PimA is essential for growth of mycobacteria. J Biol Chem 277: 31335–31344 [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lefèvre P, Braibant M, De Wit L, Kalai M, Roeper D, Grotzinger J, Delville J-P, Peirs P, Ooms J, Huygen K, Content J (1997) Three different putative phosphate transport receptors are encoded by the Mycobacterium tuberculosis genome and are present at the surface of Mycobacterium bovis BCG. J Bacteriol 179: 2900–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefèvre P, Denis O, De Wit L, Tanghe A, Vandenbussche P, Content J, Huygen K (2000) Cloning of the gene encoding a 22-Kilodalton cell surface antigen of Mycobacterium bovis BCG and analysis of its potential for DNA vaccination against tuberculosis. Infect Immun 68: 1040–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. In Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography 26 [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C (1998) Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Science 7: 1884–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnikin DE (1982) Lipids: complex lipids, their chemistry, biosynthesis and roles. In The Biology of Mycobacteria, Ratledge C, Stanford J (eds), Vol. 1, pp 95–184. London: Academic Press [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Oomen CJ, van Ulsen P, van Gelder P, Feijen M, Tommassen J, Gros P (2004) Structure of the translocator domain of a bacterial autotransporter. EMBO J 23: 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Prince SM, Achtman M, Derrick JP (2002) Crystal structure of the OpcA integral membrane adhesin from Neisseria meningitidis. Proc Natl Acad Sci USA 99: 3417–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prod'hom G, Lagier B, Pelicic V, Hance AJ, Gicquel B, Guilhot C (1998) A reliable amplification technique for the characterization of genomic DNA sequences flanking insertion sequences. FEMS Microbiol Lett 158: 75–81 [DOI] [PubMed] [Google Scholar]
- Rousseau C, Sirakova TD, Dubey VS, Bordat Y, Kolattukudy PE, Gicquel B, Jackson M (2003) Virulence attenuation of two Mas-like polyketide synthase mutants of Mycobacterium tuberculosis. Microbiology 149: 1837–1847 [DOI] [PubMed] [Google Scholar]
- Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Avé P, Huerre M, Gicquel B, Jackson M (2004) Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol 6: 277–287 [DOI] [PubMed] [Google Scholar]
- Sander P, Rezwan M, Walker B, Rampini SK, Kroppenstedt RM, Ehlers S, Keller C, Keeble JR, Hagemeier M, Colston MJ, Springer B, Bottger EC (2004) Lipoprotein processing is required for virulence of Mycobacterium tuberculosis. Mol Microbiol 52: 1543–1552 [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM (2002) Substructure solution with SHELXD. Acta Crystallogr D 58: 1772–1779 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2002) Macromolecular phasing with SHELXE. Z Kristallografiya 217: 644–650 [Google Scholar]
- Stadthagen G, Korduláková J, Griffin R, Constant P, Bottová I, Barilone N, Gicquel B, Daffé M, Jackson M (2005) p-Hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J Biol Chem 280: 40699–40706 [DOI] [PubMed] [Google Scholar]
- Steyn AJC, Joseph J, Bloom BR (2003) Interaction of the sensor module of Mycobacterium tuberculosis H37Rv KdpD with members of the Lpr family. Mol Microbiol 47: 1075–1089 [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G, Gruez A, Roig-Zamboni V, Spinelli S, Valencia C, Pagot F, Vincentelli R, Bignon C, Salomoni A, Grisel S, Maurin D, Huyghe C, Johansson K, Grassick A, Roussel A, Bourne Y, Perrier S, Miallau L, Cantau P, Blanc E, Genevois M, Grossi A, Zenatti A, Campanacci V, Cambillau C (2002) A medium-throughput crystallization approach. Acta Crystallogr D 58: 2109–2115 [DOI] [PubMed] [Google Scholar]
- Sutcliffe IC, Harrington DJ (2004) Lipoproteins of Mycobacterium tuberculosis: an abundant and functionally diverse class of cell envelope components. FEMS Microbiol Rev 28: 645–659 [DOI] [PubMed] [Google Scholar]
- Sutcliffe IC, Russell RBB (1995) Lipoproteins of Gram-positive bacteria. J Bacteriol 177: 1123–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Miyatake H, Yokota N, Matsuyama S, Tokuda H, Miki K (2003) Crystal structures of bacterial lipoprotein localization factors, LolA and LolB. EMBO J 22: 3199–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam R, Saier MH Jr (1993) Structural, functional and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol Rev 57: 320–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda H, Matsuyama S (2004) Sorting the lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta 1693: 5–13 [DOI] [PubMed] [Google Scholar]
- Torres A, Juarez MD, Cervantes R, Espitia C (2001) Molecular analysis of Mycobacterium tuberculosis phosphate specific transport system in Mycobacterium smegmatis. Characterization of recombinant 38 kDa (PstS-1). Microb Pathogen 30: 289–297 [DOI] [PubMed] [Google Scholar]
- Tsujishita Y, Hurley JH (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol 7: 408–414 [DOI] [PubMed] [Google Scholar]
- WHO (2004) Tuberculosis http://www.who.int/mediacentre/fact sheets/fs104/en/print.html
- Wooff E, Michell SL, Gordon SV, Chambers MA, Bardarov S, Jacobs WR Jr, Hewinson RG, Wheeler PR (2002) Functional genomics reveals the sole sulphate transporter of the Mycobacterium tuberculosis complex and its relevance to the acquisition of sulphur in vivo. Mol Microbiol 43: 653–663 [DOI] [PubMed] [Google Scholar]
- Ye J, van den Berg B (2004) Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J 23: 3187–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MD, Thomas LM, Tremblay JM, Oliver RL, Yarbrough LR, Helmkamp GM Jr (2001) Structure of a multifunctional protein. Mammalian phosphatidylinositol transfer protein complexed with phosphatidylcholine. J Biol Chem 276: 9246–9252 [DOI] [PubMed] [Google Scholar]
- Young DB, Garbe TR (1991) Lipoprotein antigens of Mycobacterium tuberculosis. Res Microbiol 142: 55–65 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data