

Abstract
Arginine/lysine-rich motifs typically function as targeting signals for the translocation of proteins to the nucleus. Here, we demonstrate that such a motif consisting of four basic amino acids in the polyglutamine protein ataxin-3 (Atx-3) serves as a recognition site for the interaction with the molecular chaperone VCP. Through this interaction, VCP modulates the fibrillogenesis of pathogenic forms of Atx-3 in a concentration-dependent manner, with low concentrations of VCP stimulating fibrillogenesis and excess concentrations suppressing it. No such effect was observed with a mutant Atx-3 variant, which does not contain a functional VCP interaction motif. Strikingly, a stretch of four basic amino acids in the ubiquitin chain assembly factor E4B was also discovered to be critical for VCP binding, indicating that arginine/lysine-rich motifs might be generally utilized by VCP for the targeting of proteins. In vivo studies with Drosophila models confirmed that VCP selectively modulates aggregation and neurotoxicity induced by pathogenic Atx-3. Together, these results define the VCP–Atx-3 association as a potential target for therapeutic intervention and suggest that it might influence the progression of spinocerebellar ataxia type 3.
Keywords: ataxin-3, VCP, polyglutamine aggregation
Introduction
One of the most extensively studied AAA-ATPases is the mammalian valosin-containing protein VCP (also termed p97) and its yeast homolog Cdc48p (Wang et al, 2004). VCP is an essential, abundant protein involved in a variety of processes, including postmitotic homotypic membrane fusion of the endoplasmic reticulum and Golgi apparatus (Kondo et al, 1997), the degradation of soluble and membrane proteins by the ubiquitin–proteasome system (Ye et al, 2003), and the regulation of gene expression (Rape et al, 2001). VCP contains an N-terminal domain (N-domain) followed by two highly conserved AAA domains important for ATP binding and hydrolysis (D1 and D2). It forms a homohexamer with subunits arranged in a ring-shaped structure (Wang et al, 2003). The N-domain, which interacts with cofactors such as p47 or Ufd1-Npl4 (Meyer et al, 2000), is positioned at the outside of the hexamer (Zhang et al, 2000b; DeLaBarre and Brunger, 2003). Upon nucleotide binding and ATP hydrolysis, VCP undergoes structural rearrangements, including rotation of the AAA domains, closure and relaxation of the central cavity, and changes in the conformation and relative position of the N-domain. These movements are then thought to be transmitted to potential substrate proteins, exerting mechanical force (DeLaBarre and Brunger, 2005). Although different adaptors and cofactors have been defined (Wang et al, 2004), identification of additional VCP partners is critical for understanding its function in the different subcellular processes.
Recent studies revealed that VCP may be involved in the pathogenesis of protein misfolding diseases (Higashiyama et al, 2002). It colocalizes with abnormal inclusion bodies containing insoluble protein aggregates, which are characteristic features of many neurodegenerative diseases, including Alzheimer's, Parkinson's, and Huntington's disease (Hirabayashi et al, 2001; Mizuno et al, 2003). Furthermore, altered VCP function has been directly linked to disease. Missense mutations in VCP cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) (Watts et al, 2004; Schroder et al, 2005). IBMPFD is a rare dominant progressive disorder that appears at midlife and is characterized by VCP-containing protein deposits in neurons and muscle fibers. Mutations causing IBMPFD cluster in the N-domain of VCP, which is critical for its association with cofactors and ubiquitin (Wang et al, 2004). These findings suggest that the mutations either impair the binding of VCP to potential partners such as Ufd1-Npl4 or disrupt the interaction with polyubiquitinated substrates targeted by VCP for proteasomal degradation.
Ataxin-3 (Atx-3) is a polyubiquitin-binding protein with ubiquitin protease activity (Burnett et al, 2003; Donaldson et al, 2003). When it contains an expanded polyglutamine (polyQ) sequence, it causes the neurodegenerative disorder spinocerebellar ataxia type 3 (SCA3), also known as Machado Joseph disease (MJD) (Durr et al, 1996). The elongated polyQ sequence in Atx-3 induces misfolding and the formation of insoluble protein aggregates (Bevivino and Loll, 2001). This process is associated with neuronal dysfunction and cell loss in vivo (Goti et al, 2004). Factors that modulate aggregate formation and toxicity are therefore potential targets for therapeutic intervention. Experimental evidence suggests that VCP associates with soluble as well as aggregated forms of Atx-3 (Hirabayashi et al, 2001). Whether it directly binds to polyQ tracts in Atx-3 or requires other amino acids besides glutamine for its interaction is unknown. Also unclear is whether VCP can influence aggregation and/or neurodegeneration induced by the mutant Atx-3 protein. Given that altered Atx-3 and VCP activity are both associated with pathogenic processes, elucidation of the potential interactions between these proteins may provide insight into a variety of human disease mechanisms.
Here, we have identified VCP as a binding partner of Atx-3 in human brain. VCP binds directly to Atx-3 through an arginine/lysine-rich motif (termed VBM for VCP-binding motif), which is highly conserved among Atx-3 orthologs and has been previously identified as a potential nuclear localization signal. Using in vitro and in vivo model systems, we show that the VBM is critical for the VCP-mediated modulation of polyQ-induced Atx-3 aggregation and neurotoxicity. These studies define VCP as a specific interaction partner of Atx-3 that may significantly influence disease progression. Moreover, they indicate that arginine/lysine-rich motifs in proteins might be generally utilized as targeting signals for the molecular chaperone VCP. Modification of VCP activity, or potentially of VCP–Atx-3 interactions, may be of therapeutic benefit for SCA3.
Results
VCP associates directly with Atx-3 but not with huntingtin
In order to identify binding partners of Atx-3 which may play a role in SCA3, we performed pulldown assays with human brain cytosol and the polyQ-containing Atx-3 fusion proteins GST-Atx22Q and -Atx70Q (Figure 1A). These studies detected a protein selectively enriched by Atx-3 that migrated at 100 kDa; mass spectrometry (Figure 1B) and Western blotting identified the protein as VCP. VCP has been previously suggested to be a polyQ-binding protein (Hirabayashi et al, 2001). Our studies revealed that VCP was selectively enriched from brain extract by Atx-3, but not by the polyQ disease protein huntingtin (Htt) exon1 (GST-Htt51Q) or a control protein with no polyQ sequence (GST-Htt interacting protein (HIP)1; Figure 1A). Similar results were obtained when purified His-tagged VCP fusion protein was incubated with Atx-3 affinity matrices, indicating that VCP and Atx-3 interact directly with each other in vitro (Figure 1C). We did not observe an interaction of VCP with the Htt fusion proteins GST-Htt20Q and -Htt51Q (Figure 1C) or with the Parkinson's disease protein α-synuclein (A53T) (Figure 1D). In contrast, the well-characterized binding partner Ufd1 (Meyer et al, 2000) associated with VCP in the in vitro binding assay (Figure 1D). Taken together, these data indicate that VCP specifically and directly binds to Atx-3, but neither recognizes Htt exon 1 protein nor natively unfolded disease proteins, such as α-synuclein (A53T) in pulldown assays.
Figure 1.
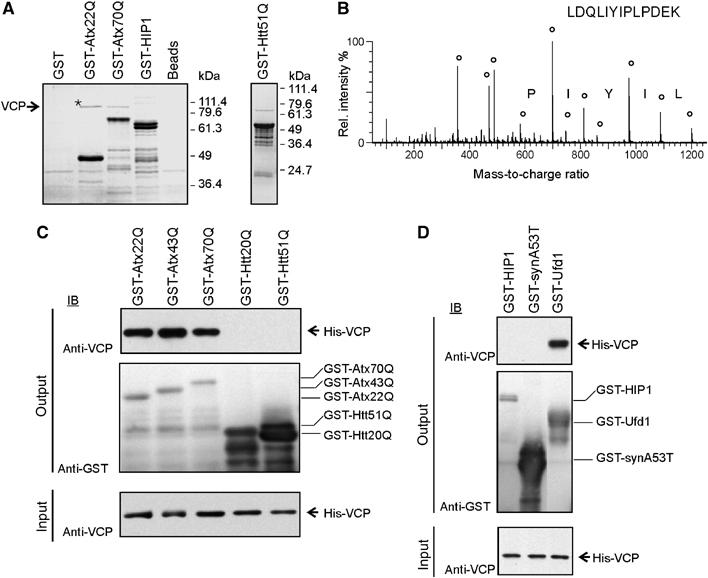
Identification and characterization of the VCP–Atx-3 protein interaction. (A) GST pulldown experiments with human brain extracts. GST fusion proteins used as baits are indicated on top of the gels. Bound proteins eluted from the affinity matrix were analysed by SDS–PAGE and Coomassie blue staining. An asterisk marks the band which was examined by mass spectrometry. (B) Fragmentation spectrum of a VCP peptide. Circles mark matches between peaks in the fragmentation spectrum and theoretical peptide masses for the identified VCP peptide. (C, D) GST pulldown experiments with purified His-VCP. Bound proteins were analyzed with an anti-VCP antibody (top panel) and bait proteins with an anti-GST antibody (middle panel). In all, 10% of the input binding mixture was also subjected to immunoblot analysis with anti-VCP antibody (bottom panel).
To confirm these findings, co-immunoprecipitation (IP) and colocalization studies were performed. Cell extracts were prepared from transiently transfected COS-1 cells and treated with antisera raised against Atx-3 or Htt. VCP was selectively precipitated by an anti-Atx-3, but not by an anti-Htt, antibody (Figure 2A and B). Interestingly, Atx-3 protein with a pathogenic length polyQ repeat (Atx70Q) co-immunoprecipitated a threefold larger amount of VCP than the wild-type protein Atx22Q (Figure 2A). This indicates that the polyQ expansion, which is not required for the interaction, significantly enhances the binding of VCP to Atx-3.
Figure 2.
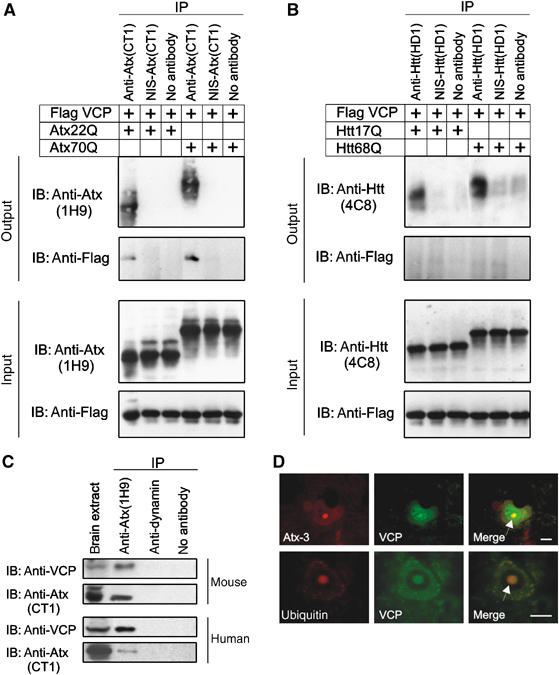
Analysis of the interaction between VCP and Atx-3. (A–C) Co-IP experiments with COS-1, mouse, and human brain cell extracts. Antisera used for IP and IB are indicated. The bottom panels in (A) and (B) show 10 μg of the input lysates. In (C), 50 μg of brain extracts were loaded as a control. NIS, nonimmune serum. (D) Co-immunofluorescence microscopy of SCA3 pons sections with anti-VCP (green), anti-Atx-3 (1H9, red) or anti-ubiquitin (red) antibodies. Neuronal intranuclear inclusions are indicated with an arrow. Scale bar: 10 μm (For color see web version).
The interaction between VCP and Atx-3 was also observed under physiological conditions. As shown in Figure 2C, VCP was co-immunoprecipitated from mouse and human brain extracts using an anti-Atx-3 antibody, but not by a control anti-dynamin antibody.
Finally, colocalization experiments were performed to examine the association of VCP with mutant Atx-3 in neurons. Immunohistochemical studies of brain sections prepared from SCA3 patients revealed that Atx-3, VCP and ubiquitin colocalize in neuronal nuclear inclusions (Figure 2D). This indicates that under pathogenic conditions VCP is present in inclusion bodies, which contain mutant Atx-3 protein aggregates, chaperones and ubiquitin (Chai et al, 1999). Together, these data indicate that both wild-type and mutant Atx-3 form protein complexes with VCP that can be detected by co-IP and immunohistochemistry using specific antibodies.
VCP interacts with Atx-3 in the presence and absence of ATP
As previous studies demonstrated that ATP binding and hydrolysis causes major conformational changes in the VCP hexamer (Rouiller et al, 2000; DeLaBarre and Brunger, 2005), we investigated whether the association of VCP with full-length Atx-3 or N-terminally truncated fragments is modulated by the addition of the nucleotides ATP, ATPγS, and ADP. As shown in Figure 3A, the GST-Atx22Q affinity matrix pulled down VCP from brain extracts in the presence and absence (apyrase treatment) of ATP. Moreover, the interaction was detected when the nonhydrolyzable ATP analog ATPγS was added to the binding reaction. In contrast, in the presence of ADP, VCP was not pulled down from the brain extract, suggesting that the addition of ADP changes the conformation of VCP and reduces the binding affinity for Atx-3. Similar results were obtained with reticulocyte lysate or when purified His-tagged VCP was used for in vitro binding assays (Figure 3A).
Figure 3.
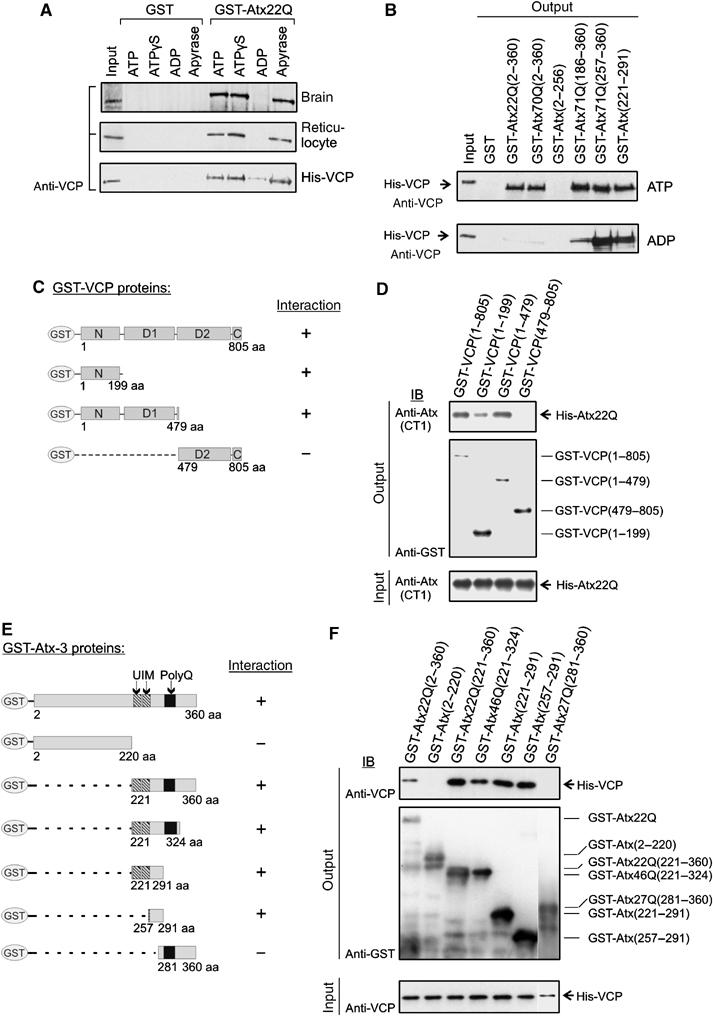
Analysis of the nucleotide dependence of the Atx-3–VCP interaction and mapping of the protein-binding sites. (A, B) GST fusion proteins were incubated with lysates containing endogenous VCP or purified recombinant His-VCP in the presence or absence of nucleotides (apyrase treatment), and protein complexes were enriched with affinity beads. After extensive washing of the beads, bound protein was detected by IB using an anti-VCP antibody. Input material: 10% of brain or reticulocyte lysates or 2% His-VCP. (C, E) Schematic representation of GST-VCP and GST–Atx-3 fusion proteins and summary of binding results. (D, F) GST pulldown experiments. Binding of recombinant His-Atx22Q and His-VCP was tested against various GST-VCP and GST–Atx-3 fusion proteins, respectively. Bound proteins (top panels) as well as immobilized bait proteins (middle panels) were detected by IB using specific antibodies. The bottom panels show 10% of the input mixtures.
Recent studies indicate that C-terminal Atx-3 fragments generated by proteolytic cleavage are critical for polyQ-mediated aggregation and neurodegeneration in transgenic mice (Goti et al, 2004). Therefore, we also examined whether the binding of VCP to C-terminal Atx-3 fragments is modulated by nucleotides (Figure 3B). Interestingly, we found that the N-terminally truncated Atx-3 proteins GST-Atx71Q(186–360), GST-Atx71Q(257–360), and GST-Atx(221–291) can bind VCP in the presence of ADP, although the full-length protein does not interact under this condition (Figure 3B). This suggests that ATP binding and hydrolysis influences the association of full-length Atx-3 with VCP in vitro, but not the interaction with N-terminally truncated Atx-3 fragments.
Mapping of the protein-binding regions in VCP and Atx-3
Using truncated versions of VCP and Atx-3, the protein-binding sites were determined using GST pulldown assays (for constructs, see Figure 3C and E). As shown in Figure 3D, an N-terminal VCP fragment (aa 1–199) is critical for the Atx-3 interaction, while the C-terminus containing the highly conserved ATP-binding domains D1 and D2 is not required. In Atx-3, a fragment of 34 amino acids (aa 257–291) located between the second ubiquitin-interacting motif (UIM) and the polyQ sequence was sufficient for the association with VCP (Figure 3E and F).
To determine the amino-acid residues in Atx-3 critical for the interaction with VCP, an array of synthetic peptides representing the C-terminus of Atx-3 with the potential binding region was prepared and probed for an interaction with His-VCP (Frank, 1992). Each synthetic peptide spotted onto the cellulose membrane was composed of 15 amino-acid residues, which overlapped in sequence with the next peptide by 12 residues. Peptides interacting with VCP were detected by overlay assays using a specific anti-VCP antibody. Figure 4A shows that binding peptide 43 (BP43; TSEELRKRREAYFEK, aa 277–291) interacted most strongly with the His-VCP protein, suggesting that it contains the amino acids crucial for the association with His-VCP.
Figure 4.
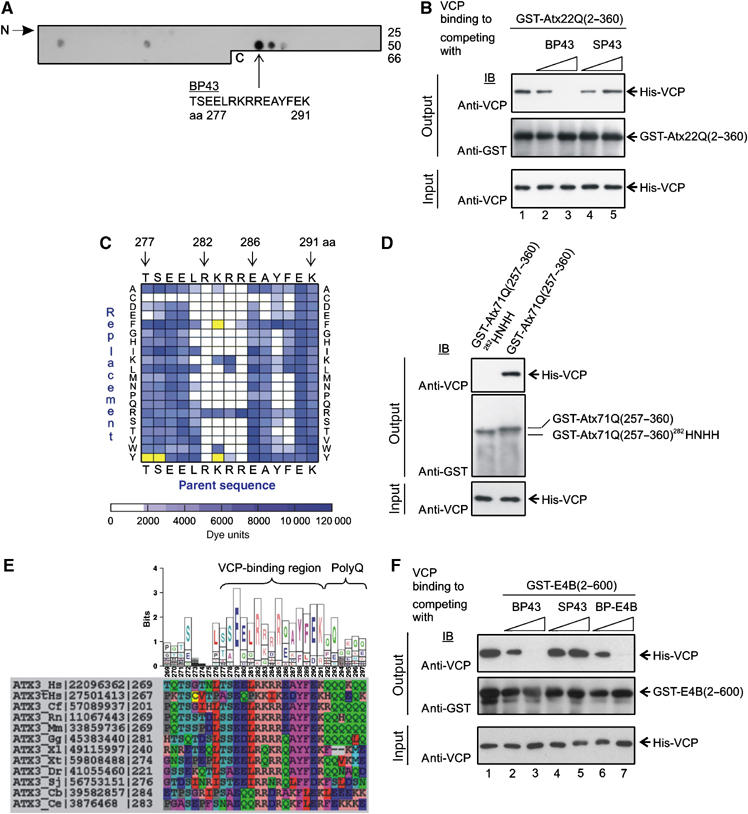
Identification of the amino-acid residues in Atx-3 responsible for VCP binding. (A) Detection of VCP-interacting Atx-3 peptides using overlay assays. An array with 66 overlapping 15mer peptides covering the C-terminus of Atx-3 (aa 151–360) was incubated with His-VCP, and binding peptides were identified by IB using an anti-VCP antibody. The Atx-3 binding peptide 43 (BP43) interacted most strongly with His-VCP. (B) BP43 at a concentration of 1 mM prevents the interaction between GST-Atx22Q(2–360) and His-VCP (lane 3). No competition of the protein–protein interaction was observed with the control peptide SP43. (C) Identification of the amino acids in BP43 required for the interaction with His-VCP. An array with 300 synthetic peptide analogs of BP43 with single amino-acid substitutions was incubated with His-VCP, and the amino acids critical for the peptide–protein interaction were determined by overlay assays. Signals were quantified and are represented as a spectral diagram. The BP43 parent sequence was displayed on the top and bottom of the matrix. The replaced amino acids are shown on the right and left sides. The bar below the matrix gives the signal intensity scale in arbitary dye units. As a control, anti-VCP antibody alone was incubated with the peptide array. BP43 derivatives, which were detected as false positives by the antibody alone, are indicated as yellow squares. (D) GST-pulldown experiments. The arginine/lysine-rich motif (282RKRR) in Atx-3 is critical for the interaction with His-VCP in vitro. Mutation of this sequence (282HNHH) prevents His-VCP binding. The bottom panel shows 10% of input mixture. (E) Conservation of the VCP-binding region in homologous Atx-3 sequences. The sequence logo (top) is derived from the sequence alignments of Atx-3 orthologs (bottom). Physicochemically similar amino acids are colored identical in the alignment. NCBI sequence accession numbers and the starting positions of the amino-acid sequences are indicated. Hs, Homo sapiens; tHs, Atx-3 paralog expressed in testis; Cf, Canis familiaris; Rn, Rattus norvegicus; Mm, Mus musculus; Gg, Gallus gallus; Xl/Xt, Xenopus laevis/tropicalis; Dr, Danio rerio; Sj, Schistosoma japonicum; Cb/Ce, Caenorhabditis briggsae/elegans. (F) The peptides BP43 and BP-E4B at a concentration of 1 mM prevent the interaction between GST-E4B(2–600) and His-VCP (lanes 3 and 7). The control peptide SP43 has no effect.
To test whether BP43 can specifically inhibit the VCP–Atx-3 interaction, competition assays were performed (Figure 4B). Indeed, the addition of soluble BP43 to binding reactions completely abolished the interaction between His-VCP and GST-Atx22Q (Figure 4B, lane 3), while the control peptide SP43 (a scrambled version of BP43) had no effect (Figure 4B, lane 5). Similar results were obtained when GST-Atx70Q was used for the competition assays (data not shown), indicating that BP43 can prevent the VCP interaction with both wild-type and pathogenic forms of Atx-3.
To identify the amino-acid residues in BP43 that are important for the interaction with VCP, a comprehensive replacement analysis with 300 synthetic peptide analogs derived from the BP43 sequence by single amino-acid substitutions was performed (Figure 4C). Data analysis revealed that amino acids R282 and R285 are essential, and that the amino acids L281, K283, R284, Y288 and F289 are critical for the interaction with VCP. For example, R282 and R285 cannot be replaced by any other of the 20 genetically coded amino acids, while K283 can be substituted by the amino acids R and W without significant loss of VCP binding (Figure 4C).
To determine whether the basic amino acids 282RKRR (arginine/lysine-rich motif) are crucial for the interaction with VCP in the context of the folded protein, we analyzed the binding of VCP to mutant Atx-3 (282RKRR to HNHH) using GST pulldown assays (Figure 4D). We found that His-VCP interacts with GST-Atx71Q(257–360), but not with the mutant protein GST-Atx71Q(257–360)282HNHH in vitro, confirming that the amino acids 282RKRR in Atx-3 are important for the association with VCP. The same result was also obtained when full-length Atx-3 proteins with a mutant arginine/lysine-rich motif (GST-Atx22Q(2–360)282HNHH and GST-Atx68Q(2–360)282HNHH) were used for the in vitro binding assay (Supplementary Figure S1).
VBM analysis
Based on the systematic mutational analysis, the following VBM can be postulated: 281(L/I/V/Y)-282R-283(K/R/W)- 284(R/K/L)-285R-286X-287X-288(Y/F)-289(F/K/L/Y). This bipartite motif consists mainly of four consecutive basic amino acids (282RKRR) and two hydrophobic residues (288YF). Strikingly, sequence comparison of homologous Atx-3 proteins from different species revealed that the VBM is highly conserved from human to nematode (Figure 4E). The only exceptions are the Atx-3 proteins from Cryptosporidium hominis/parvum, Plasmodium falciparum, and Theileria annulata, which might use a ubiquitin regulatory X (UBX) domain instead of the VBM for the association with VCP (Dreveny et al, 2004a).
Finally, we examined whether short stretches of four basic amino acids in other proteins are also critical for VCP binding. Using bioinformatic tools, we searched for such sequences in the amino-acid sequences of known VCP interaction partners (Dreveny et al, 2004b). This approach revealed that the ubiquitin chain assembly factor E4B (UFD2a) (Kaneko et al, 2003) contains an N-terminal stretch of four basic amino acids (10RRRR) that could be important for VCP binding. Competition assays with the peptides BP-E4B (SADEIRRRRLARLAG, aa 5–19) and BP43 (Figure 4A) were performed and the association between VCP and E4B was analyzed by immunoblotting (IB). As shown in Figure 4F, both peptides prevented the interaction between E4B and VCP in vitro, while the control peptide SP43 had no effect. In comparison, the binding of VCP to Ufd1, which does not contain a stretch of four basic amino acids, was not inhibited by the addition of BP43 (Supplementary Figure S2). Together, these results suggest that short stretches of arginine residues in other proteins also might be crucial for the association with VCP.
VCP modulates Atx-3 aggregation in vitro
Since VCP specifically interacts with Atx-3, which forms fibrillar structures (Bevivino and Loll, 2001), we examined whether VCP can modulate the self-assembly of insoluble protein aggregates in vitro. GST-Atx71Q(242–360) was incubated with purified His-VCP and PreScission protease (PP), and the formation of fibrillar aggregates was monitored by filter retardation assay (Wanker et al, 1999). PP removes the GST tag from the fusion protein and thereby stimulates the assembly of insoluble Atx71Q(242–360) protein aggregates. As shown in Figure 5A, VCP significantly enhanced the formation of SDS-insoluble protein aggregates in comparison to the control protein ovalbumin. Interestingly, the assembly of protein aggregates was significantly reduced when the protein GST-Atx68Q(242–360)282HNHH, which does not contain a functional VBM, was incubated with VCP and PP (Figure 5A). The addition of an ATP-regenerating system did not influence the formation of protein aggregates (data not shown), indicating that ATP binding and hydrolysis is not crucial for the VCP-mediated modulation of Atx71Q(242–360) aggregation in vitro.
Figure 5.
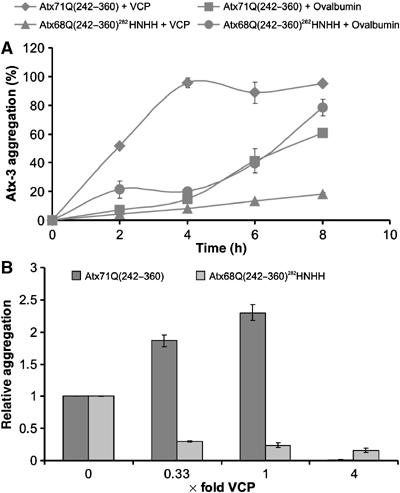
Binding of VCP to Atx-3 influences the assembly of polyQ-containing protein aggregates. (A) VCP stimulates the formation of Atx-3 aggregates in vitro. 3 μM GST-Atx71Q(242–360) or GST-Atx68Q(242–360)282HNHH was incubated with 1 μM VCP or 1 μM ovalbumin in the presence of PP. The assembly of SDS-stable Atx-3 protein aggregates was monitored by filtration using an anti-Atx (CT1) antibody. The sample with the highest signal intensity was arbitrarily set as 100%. (B) Effect of different VCP concentrations on Atx-3 aggregation in vitro. GST fusion proteins were incubated for 6 h with PP and different amounts of VCP. The assembly of SDS-insoluble Atx71Q(242–360) and Atx68Q(242–360)282HNHH aggregates was monitored by filtration using the CT1 antibody. Relative amounts of aggregates were quantified by phosphorimager densitometry. The X-axis of the diagram shows the ratio of VCP to Atx-3.
Next, we tested whether the effect of VCP on Atx-3 fibrillogenesis is concentration-dependent. We found that concentrations of VCP up to equimolarity enhanced Atx71Q(242–360) aggregation, while a fourfold molar excess of the chaperone completely suppressed it (Figure 5B). No such effect was observed with mutant GST-Atx68Q(242–360)282HNHH, which does not bind VCP in pulldown assays (Supplementary Figure S3). On the contrary, the increasing VCP concentrations caused a reduction of Atx68Q(242–360)282HNHH protein aggregates (Figure 5B). The aggregation of an Htt exon 1 protein with 51 glutamines, which does not contain a functional VBM, was not significantly altered by the addition of VCP (data not shown).
VCP mitigates toxicity in Drosophila models of SCA3
In order to examine the effect of VCP on Atx-3 toxicity in vivo, we used Drosophila models expressing full-length Atx-3 with an expanded polyQ tract (Atx3Q78 and Atx3Q84) in photoreceptor neurons. These proteins induce progressive degeneration of photoreceptors coupled with the formation of protein aggregates in the transgenic flies. However, when VCP was coexpressed, the loss of photoreceptor neurons and the accumulation of large inclusion bodies were significantly reduced (Figure 6A–D). The ratio of VCP to Atx-3 in these transgenic models was 1.42 × to 4.2 × depending upon the transgenic line (Supplementary Figure S4). In these experiments in vivo, added VCP never enhanced degeneration. These data indicate that high concentrations of VCP can suppress polyQ-mediated toxicity and reduce protein aggregation in vivo, consistent with the effect of VCP to mitigate aggregation of Atx3 at high concentrations in vitro.
Figure 6.
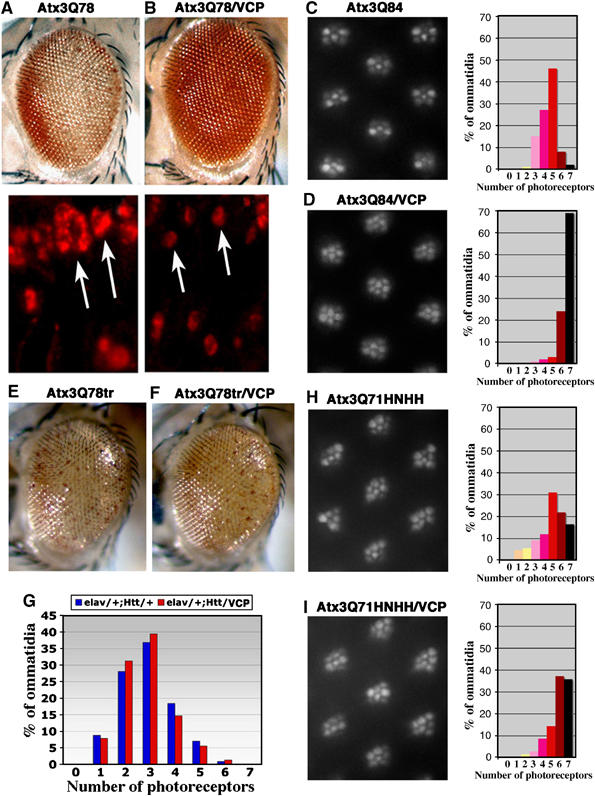
Human VCP suppresses Atx-3-induced neurodegeneration in Drosophila. (A) Flies expressing pathogenic Atx3Q78 protein alone (top is the eye, bottom are protein accumulations in adult retinal cryosections) or (B) together with VCP. Degeneration of the eye and size of protein aggregates are mitigated with added VCP activity. Arrows highlight nuclear inclusions. (C) Photographs of ommatidia from 2-day-old flies expressing Atx3Q84 alone or (D) with VCP, with distribution (in percentage) of photoreceptor neurons in ommatidia, with 7 being normal. Mean number of photoreceptor neurons per ommatidium is 4.5 in (C) and 6.6 in (D), indicating an improvement of 30% with added VCP. Genotypes: (A) gmr-Gal4;UAS-Atx3Q78#24.1/+, (B) gmr-Gal4;UAS-Atx3Q78#24.1/+;UAS-VCP/+, (C) elav-Gal4/+;UAS-Atx3Q84#16.1/+, (D) elav-Gal4/+;UAS-Atx3Q84#16.1/+;UAS-VCP/+. The VCP/Atx-3 ratio is ∼1.45 for Atx-3 line #24.1 and ∼4.2 for line #16.1. (E, F) Coexpression of VCP has little effect on degeneration induced by a truncated version of Atx-3 with a shortened VBM domain. Flies expressing Atx3Q78tr (E) alone or (F) with VCP. Genotypes: gmr-GAL4;UAS-Atx3Q78tr(S)/+ (E) or in trans to UAS-VCP (F). (G) Coexpression of VCP has little effect on degeneration induced by pathogenic Htt protein. Distribution in percentage of photoreceptor neurons in ommatidia of flies expressing HttQ120 alone or with VCP. Genotypes: elav-Gal4/+;gmr-HttQ120/+ (blue) and elav-Gal4/+;gmr-HttQ120/UAS-VCP (red). Photographs of ommatidia from 14-day-old flies expressing Atx3Q71HNHH (H) alone or with VCP (I), with distribution (in percentage) of photoreceptor neurons in ommatidia, with 7 being normal. Mean number of photoreceptors per ommatidium is 4.9 in (H) and 5.9 in (I), indicating an improvement of only 14% by VCP. Thus, photoreceptor neuron degeneration in flies expressing Atx3Q71HNHH is partially mitigated by VCP, but only to half the extent as degeneration by pathogenic Atx-3 with an intact VBM.
To test whether the effect on neurotoxicity is dependent on the interaction between VCP and Atx-3, we also overproduced VCP in flies which express a mutant N-terminally truncated Atx-3 fragment (Atx3Q78tr) or an Htt exon 1 fragment with a pathogenic polyQ sequence (Htt120Q). These proteins do not contain a functional VBM and therefore should not bind VCP with high affinity in vivo. We found that overexpression of VCP had little effect on degeneration induced by AtxQ78tr or Htt120Q (Figure 6E–G), indicating that VBM-mediated association of VCP with Atx-3 may be critical for modulation of protein aggregation and neurodegeneration in Drosophila. We then generated transgenic lines of Atx3Q71 with the mutated VCP-binding site (Atx3Q71HNHH). Coexpression of VCP had some effect to mitigate the toxicity of Atx3Q71HNHH, but the effect was less than that of the full-length protein with an intact VBM (Figure 6H and I). These data emphasize the importance of the VCP-binding domain.
Discussion
We isolated VCP as an in vivo binding partner of Atx-3 in human brain. Further analysis of this interaction revealed that the potential nuclear localization signal of Atx-3 is a highly conserved VBM. Moreover, our data demonstrated that VCP modulates the aggregation and in vivo neurotoxicity of pathogenic forms of Atx-3, which are associated with human neurodegeneration. These studies define Atx-3 as a novel partner protein of VCP, and indicate that a further investigation of the VCP–Atx-3 interaction may be of therapeutic benefit in human disease.
A highly conserved arginine/lysine-rich motif in Atx-3 is critical for VCP binding
Using cell-free and cell-based assays, we demonstrated that VCP is not a polyQ tract binding protein as suggested previously (Higashiyama et al, 2002). Rather, it directly associates with a conserved arginine/lysine-rich motif, termed VBM in the Atx-3 protein. This motif is located in close proximity to the polyQ sequence (see Figure 3E) and overlaps a potential nuclear localization signal (aa 282–285) (Albrecht et al, 2004). A critical implication of our findings is that predicted nuclear localization signals consisting of four basic amino acids might function as recognition signals for the promiscuous molecular chaperone VCP, which modulates, depending on its interaction partners, a vast array of fundamental subcellular processes (Wang et al, 2004).
These findings raise the question of whether other VCP interaction partners also utilize arginine/lysine-rich motifs for their association with VCP. Strikingly, we discovered that a motif of four consecutive basic amino acids (10RRRR) is critical for the interaction between the ubiquitination factor E4B and VCP. This indicates that short stretches of basic amino acids are used for the targeting of proteins to VCP more generally. Bioinformatic analysis revealed that stretches of arginines and lysines are also present in the known VCP-binding proteins SVIP (Nagahama et al, 2003), gp78 (Zhong et al, 2004) and BRCA1 (Zhang et al, 2000a). Whether these amino acids are indeed required for the association of the proteins with VCP needs to be investigated.
Nucleotide binding modulates VCP interactions with Atx-3
Previous studies have demonstrated that VCP hexamers have different conformations in the ATP-free and ATP-bound states (DeLaBarre and Brunger, 2005). We found that full-length Atx-3 interacts with VCP in the absence as well as in the presence of ATP or its nonhydrolyzable analog, ATP-γS. In striking contrast, no interaction was detected in the presence of ADP, suggesting that VCP might adopt an alternative conformation that is unable to bind Atx-3. Interestingly, the inhibitory effect of ADP was lost when N-terminally truncated Atx-3 fragments lacking the conserved Josephin domain were used for binding experiments (see Figure 3A and B). As Atx-3 is composed of a folded N-domain and most likely has an unstructured polyQ tail (Masino et al, 2003), we propose that the N-terminally truncated Atx-3 fragments are more flexible than the full-length protein and are therefore able to associate with the VCP-binding site in the ADP state.
However, our data also suggest that in vivo full-length Atx-3 binds to VCP in the ATP state, and is released after ATP hydrolysis in the ADP state. N-terminally truncated Atx-3 fragments with expanded polyQ sequences, which are generated under disease conditions (Goti et al, 2004), might lose their ability to dissociate from VCP in the ADP state. Thus, altered binding of truncated Atx-3 proteins to VCP in neurons could contribute critically to neuronal dysfunction in SCA3.
VCP modulates Atx-3 aggregation in a concentration-dependent manner
We tested whether the addition of the molecular chaperone VCP can modulate the formation of insoluble Atx-3 aggregates in vitro. Strikingly, we found that low or intermediate concentrations of VCP significantly stimulated Atx-3 aggregation, while a fourfold molar excess of the chaperone completely prevented fibrillogenesis. Currently, the molecular mechanism of this effect is unclear. We assume that at low or equimolar VCP concentrations the hexamers provide an optimal catalytic surface for the efficient conversion of bound Atx-3 molecules into fibrillar structures. At higher VCP concentrations (e.g. four hexamers with one Atx-3 molecule), however, the access to the aggregation-prone Atx-3 molecules exposed by the hexamers might be masked, because the chaperone molecules themselves start to form oligomeric structures (A Boeddrich and EE Wanker, unpublished results). Therefore, a fourfold molar excess of VCP prevents fibrillogenesis, potentially because the bound Atx-3 molecules cannot be delivered to the growing fibrils due to sterical hindrance. In Drosophila, we only saw evidence of suppression by added VCP. In vitro experiments do not have the vast complexity of the in vivo situation, where other pathways, other binding partners of VCP and of Atx3, and other VCP-like activities will be present. However, it is also possible that the lack of a concentration dependence may indicate that all situations of added transgenic VCP could be considered as ratios above equimolar.
We observed that ATP binding and hydrolysis are not required for the highly concentration-dependent modulation of Atx-3 aggregation mediated by VCP. This is in agreement with previous studies of the VCP yeast homolog Cdc48p, which equally does not require ATP hydrolysis for the binding of non-native proteins and the inhibition of protein aggregation (Thoms, 2002).
Our data indicating that VCP can enhance or slow down Atx-3 fibrillogenesis in a concentration-dependent manner are strikingly similar to results obtained with the chaperone Hsp104 and its interaction partner Sup35 (Chernoff et al, 1995). Hsp104 is a AAA ATPase with structural similarities to VCP; Sup35 is a translation termination factor, which forms self-propagating protein aggregates (prions) in yeast (Parsell et al, 1994; Serio and Lindquist, 2000). In vitro, intermediate concentrations of Hsp104 catalyze the self-assembly of Sup35 fibrillar structures, while high concentrations of the chaperone can dissemble fibrils (Shorter and Lindquist, 2004). The finding that the behaviour of Hsp104 is comparable to that of VCP suggests that both proteins modulate aggregation by similar mechanisms. This is supported by the fact that Hsp104, like VCP, does not require ATP hydrolysis for influencing Sup35 fibrillogenesis.
As expected, the concentration-dependent stimulation and suppression of Atx-3 fibrillogenesis in vitro was prevented when a mutated version of the protein [Atx68Q(242–360)282HNHH], which did not bind VCP in pulldown assays, was used for the aggregation reactions. This demonstrates that the high-affinity interaction between VCP and Atx-3 mediated by the VBM is critical for the stimulation/suppression effect in vitro. Interestingly, the addition of VCP significantly reduced the aggregation of the Atx68Q(242–360)282HNHH protein, indicating that the chaperone can associate with the Atx-3 protein even without an arginine/lysine-rich binding site. We suggest that a low-affinity, potentially unspecific, binding of VCP to Atx68Q(242–360)282HNHH is sufficient to mitigate the self-assembly of protein aggregates. For the efficient concentration-dependent assembly and disassembly of fibrils, however, a specific VBM-mediated interaction between VCP and Atx-3 is essential.
What is the functional relevance of the VCP–Atx-3 interaction?
Previous studies have demonstrated that cofactors such as p47 or the heterodimer Udf1/Npl4 bind with high affinity and independent of ATP to the N-domain of VCP (Meyer et al, 2000; Ye et al, 2003; Wang et al, 2004). The finding that Atx-3 also interacts very specifically with this domain suggests that it might also function as a VCP cofactor. Several lines of evidence indicate that the binding of mutually exclusive cofactors determines the specific activity of the chaperone in the cell. A complex of VCP and p47 plays an important role in homotypic membrane fusion events (Kondo et al, 1997), while a complex consisting of VCP, Ufd1 and Npl4 recognizes ubiquitinated proteins and mediates their degradation by the proteasome (Ye et al, 2003). We hypothesize that a protein complex formed of VCP, Atx-3 and other associated proteins could be involved in the ubiquitin-dependent protein degradation. Atx-3 binds to polyubiquitin chains and has ubiquitin protease activity (Burnett et al, 2003; Donaldson et al, 2003). Moreover, it associates with the proteins HHR23A and B, which play a functional role in the ubiquitin/proteasome pathway (Wang et al, 2000). Therefore, the association of Atx-3 might help VCP to present ubiquitinated proteins to the proteasome for degradation. Currently, it is thought that VCP functions as a disassembly factor which extracts ubiquitinated proteins from protein complexes or membranes for proteasomal degradation. Atx-3 with its deubiquitinating activity might contribute to this pathway, possibly by removing the polyubiquitin chains from substrates prior to digestion.
Besides functioning as a cofactor, Atx-3 could also be a substrate for VCP, which alters Atx-3 conformationally and functionally. The fact that different concentrations of VCP can modulate Atx-3 aggregation suggests that the assembly and disassembly of VCP–Atx-3 protein complexes might influence the cellular activity of Atx-3. Preliminary in vivo studies in mouse brain indicate that VCP is present in a 17-fold molar excess over the Atx-3 protein (A Boeddrich and EE Wanker, unpublished results). This suggests that, under physiological conditions, most, if not all, Atx-3 molecules are bound to VCP and the aggregation of the protein is prevented. An alteration of the VCP/Atx-3 ratio or a release of Atx-3, however, could lead to the formation of oligomers/aggregates, which might have novel functional properties. Such Atx-3 oligomers could be much more potent in cleaving polyubiquitin chains than soluble monomers or VCP–Atx-3 protein complexes. On the other hand, abnormal polyQ-mediated Atx-3 aggregation could be toxic and cause neurodegenerative disease. Notably, VCP also modulates the oligomerization of the cofactor p47, which, similar to Atx-3, has a high propensity to self-associate (Yuan et al, 2004).
VCP overexpression mitigates polyQ-induced neurotoxicity in Drosophila
Using Drosophila as an in vivo model system, we demonstrated that overproduction of VCP reduces toxicity of pathogenic full-length Atx-3 protein. Drosophila has been used successfully for defining pathways of relevance to human disease (Marsh and Thompson, 2004). The activity of VCP in vivo was selective for Atx-3, as it failed to modulate neurodegeneration induced by the polyQ-containing disease protein Htt. Suppression of neurodegeneration by VCP was in part dependent on an intact VBM domain in vivo, although it was still seen to some extent with a pathogenic Atx-3 with a mutant arginine/lysine-rich motif. This is consistent with the in vitro studies, where VCP reduced, but did not eliminate, aggregation of an Atx-3 protein with a nonfunctional VBM (see Figure 5A and B).
Taken together, these studies suggest that increasing the levels of VCP in SCA3 patients might have beneficial effects on disease pathogenesis. However, the molecular mechanism by which high VCP concentrations in photoreceptor neurons reduce toxicity is unclear. We suggest that VCP overexpression, similar to Hsp40/70 (Warrick et al, 1999), can protect neurons because it may reduce the amount of toxic protein aggregates (most likely oligomers or protofibrils) or mask the surface of toxic soluble Atx-3 molecules. However, in vivo, higher concentrations of VCP might also stimulate the selective degradation of misfolded Atx-3 molecules by the ubiquitin–proteasome system. These studies define VCP as an important interaction partner of Atx-3 that modulates the pathogenicity of the human mutant protein associated with disease. Further elucidation of the interactions between VCP and its partners promises to yield additional insights into human disease.
Materials and methods
Screening of membrane-bound peptide arrays
Peptides derived from the Atx-3 protein sequence were chemically synthesized as arrays on cellulose membranes (Frank and Overwin, 1996). After washing with ethanol and Tris-buffered saline (TBS), the peptide arrays were blocked with membrane-blocking solution (MBS) overnight at 4°C. MBS contains a casein-based blocking buffer concentrate (Genosys Biotechnologies, Cambridge) diluted 1:5 in T-TBS (TBS with 0.05% Tween-20) with 5% sucrose. After washing the membrane with T-TBS, 0.1 μM His-VCP in MBS was added and incubated for 3 h at room temperature. Then, the membrane was washed with T-TBS, followed by incubation with rabbit anti-VCP antibody and secondary peroxidase-conjugated anti-rabbit antibody. Immunoreactive protein was detected using enhanced chemiluminescence (ECL, Perkin-Elmer Life Sciences). For image quantification, the Phoretix Array analysis software 1.0 (NonLinear dynamics Ltd, Newcastle on Tyne) was used.
Aggregation assays
For in vitro aggregation studies, GST–Atx-3 fusion proteins were incubated with or without hexameric His-VCP in the presence of PP (Amersham Biosciences, Freiburg, Germany) at 30°C with 300 r.p.m. shaking for the indicated times. Reactions were stopped by adding an equal volume of 4% SDS/100 mM DTT and boiling for 5 min. Aliquots corresponding to 200–900 ng of GST-fusion protein were diluted into 0.2 ml 0.1% SDS and filtered through a 0.2 μm cellulose acetate membrane (Wanker et al, 1999). Captured SDS-resistant aggregates were detected by IB using the fluorescent substrate AttoPhos.
Drosophila strains and immunohistochemistry
Fly lines bearing Atx3Q78tr (previously named as MJDtr-Q78), Atx3Q84 (previously named as SCA3-Q84) and gmr-HttQ120 were used (Jackson et al, 1998; Warrick et al, 2005). UAS-VCP and UAS-Atx3Q71HNHH transgenics were generated by standard methods. Photoreceptors were counted (120–300 ommatidia/genotype) using a standard light optical method. Heads were embedded and cryosectioned following standard protocols, and immunostained with anti-c-Myc (9E10, diluted 1:50; Santa Cruz Biotechnology, Santa Cruz, CA) and secondary anti-mouse IgG Alexa Fluor 594 (diluted 1:50; Molecular Probes, Eugene, OR).
The experimental procedures for plasmid construction, protein expression, in vitro binding experiments, mass spectrometry analysis, antibody production, microscopic analysis, bioinformatics analysis, cell transfections and IPs are described in Supplementary data.
Supplementary Material
Supplementary Figure S1
{kind=link}
Supplementary Figure S2
{kind=link}
Supplementary Figure S3
{kind=link}
Supplementary Figure S4
{kind=link}
Supplemental Data
Acknowledgments
We thank S Schnögl, D Lessing, A Dröge and S Wälter for critical reading of the manuscript, G Jackson, SL Zipursky, D Ruden and R Pittman for fly lines and constructs, S Daenicke for expert help in SPOT synthesis, B Kornak for synthesis of soluble peptides and C Hänig for bioinformatics analysis. The project was funded by the DFG grants WA1151/4-3, WA1151/4-4, the NGFN grants 01GR471 and 01GR474, the EURO-SCA grant LSHM-CT-2004-503304, the Apopis grant LSHM-CT-2003-503330, the BioFuture grant 0311853, the NINDS and the Packard Foundation. NMB is an investigator of the Howard Hughes Medical Institute.
References
- Albrecht M, Golatta M, Wullner U, Lengauer T (2004) Structural and functional analysis of ataxin-2 and ataxin-3. Eur J Biochem 271: 3155–3170 [DOI] [PubMed] [Google Scholar]
- Bevivino AE, Loll PJ (2001) An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta-fibrils. Proc Natl Acad Sci USA 98: 11955–11960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett B, Li F, Pittman RN (2003) The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet 12: 3195–3205 [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL (1999) Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet 8: 673–682 [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268: 880–884 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2003) Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol 10: 856–863 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2005) Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol 347: 437–452 [DOI] [PubMed] [Google Scholar]
- Donaldson KM, Li W, Ching KA, Batalov S, Tsai CC, Joazeiro CA (2003) Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates. Proc Natl Acad Sci USA 100: 8892–8897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS (2004a) Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J 23: 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreveny I, Pye VE, Beuron F, Briggs LC, Isaacson RL, Matthews SJ, McKeown C, Yuan X, Zhang X, Freemont PS (2004b) p97 and close encounters of every kind: a brief review. Biochem Soc Trans 32: 715–720 [DOI] [PubMed] [Google Scholar]
- Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, Brice A (1996) Spinocerebellar ataxia 3 and Machado–Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 39: 490–499 [DOI] [PubMed] [Google Scholar]
- Frank R (1992) Spot-Synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48: 9217–9232 [Google Scholar]
- Frank R, Overwin H (1996) SPOT synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol Biol 66: 149–169 [DOI] [PubMed] [Google Scholar]
- Goti D, Katzen SM, Mez J, Kurtis N, Kiluk J, Ben-Haiem L, Jenkins NA, Copeland NG, Kakizuka A, Sharp AH, Ross CA, Mouton PR, Colomer V (2004) A mutant ataxin-3 putative-cleavage fragment in brains of Machado–Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci 24: 10266–10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, Kakizuka A (2002) Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ 9: 264–273 [DOI] [PubMed] [Google Scholar]
- Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A (2001) VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ 8: 977–984 [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL (1998) Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21: 633–642 [DOI] [PubMed] [Google Scholar]
- Kaneko C, Hatakeyama S, Matsumoto M, Yada M, Nakayama K, Nakayama KI (2003) Characterization of the mouse gene for the U-box-type ubiquitin ligase UFD2a. Biochem Biophys Res Commun 300: 297–304 [DOI] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388: 75–78 [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM (2004) Can flies help humans treat neurodegenerative diseases? BioEssays 26: 485–496 [DOI] [PubMed] [Google Scholar]
- Masino L, Musi V, Menon RP, Fusi P, Kelly G, Frenkiel TA, Trottier Y, Pastore A (2003) Domain architecture of the polyglutamine protein ataxin-3: a globular domain followed by a flexible tail. FEBS Lett 549: 21–25 [DOI] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19: 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Hori S, Kakizuka A, Okamoto K (2003) Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci Lett 343: 77–80 [DOI] [PubMed] [Google Scholar]
- Nagahama M, Suzuki M, Hamada Y, Hatsuzawa K, Tani K, Yamamoto A, Tagaya M (2003) SVIP is a novel VCP/p97-interacting protein whose expression causes cell vacuolation. Mol Biol Cell 14: 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Lindquist S (1994) Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem 269: 4480–4487 [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107: 667–677 [DOI] [PubMed] [Google Scholar]
- Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM (2000) A major conformational change in p97 AAA ATPase upon ATP binding. Mol Cell 6: 1485–1490 [DOI] [PubMed] [Google Scholar]
- Schroder R, Watts GD, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR (2005) Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol 57: 457–461 [DOI] [PubMed] [Google Scholar]
- Serio TR, Lindquist SL (2000) Protein-only inheritance in yeast: something to get [PSI+]-ched about. Trends Cell Biol 10: 98–105 [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304: 1793–1797 [DOI] [PubMed] [Google Scholar]
- Thoms S (2002) Cdc48 can distinguish between native and non-native proteins in the absence of cofactors. FEBS Lett 520: 107–110 [DOI] [PubMed] [Google Scholar]
- Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N (2000) Ataxin-3, the MJD1 gene product, interacts with the two human homologs of yeast DNA repair protein RAD23, HHR23A and HHR23B. Hum Mol Genet 9: 1795–1803 [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC (2003) Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun 300: 253–260 [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC (2004) Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol 146: 44–57 [DOI] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H (1999) Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol 309: 375–386 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23: 425–428 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, Bonini NM (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell 18: 37–48 [DOI] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36: 377–381 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Simpson P, McKeown C, Kondo H, Uchiyama K, Wallis R, Dreveny I, Keetch C, Zhang X, Robinson C, Freemont P, Matthews S (2004) Structure, dynamics and interactions of p47, a major adaptor of the AAA ATPase, p97. EMBO J 23: 1463–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang Q, Kajino K, Greene MI (2000a) VCP, a weak ATPase involved in multiple cellular events, interacts physically with BRCA1 in the nucleus of living cells. DNA Cell Biol 19: 253–263 [DOI] [PubMed] [Google Scholar]
- Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS (2000b) Structure of the AAA ATPase p97. Mol Cell 6: 1473–1484 [DOI] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279: 45676–45684 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplemental Data