

Abstract
Homeobox (Hox) gene mutations and their altered expressions are frequently linked to human leukemia. Here, we report that transforming growth factor β (TGFβ)/bone morphogenetic protein (BMP) inhibits the bone marrow transformation capability of Hoxa9 and Nup98-Hoxa9, the chimeric fusion form of Hoxa9 identified in human acute myeloid leukemia (AML), through Smad4, the common Smad (Co-Smad) in the TGFβ/BMP signaling pathway. Smad4 interacts directly with the homeodomain of Hoxa9 and blocks the ability of Nup98-Hoxa9 to bind DNA, thereby suppressing its ability to regulate downstream gene transcription. Mapping data revealed that the amino-terminus of Smad4 mediates this interaction and overexpression of the Hoxa9 interaction domain of Smad4 was sufficient to inhibit the enhanced serial replating ability of primary bone marrow cells induced by Nup98-Hoxa9. These studies establish a novel mechanism by which TGFβ/BMP regulates hematopoiesis and suggest that modification of Hox DNA-binding activity may serve as a novel therapeutic intervention for those leukemias that involve deregulation of Hox.
Keywords: Hox, leukemia, myeloid, Smad, TGFβ/BMP
Introduction
The transforming growth factor β (TGFβ) superfamily consists of TGFβs, bone morphogenetic proteins (BMPs), activins and related proteins. These secreted proteins regulate a broad range of cellular responses during hematopoiesis, including cell proliferation, differentiation and apoptosis (Bhatia et al, 1999; Fortunel et al, 2000). Smad proteins are intracellular signal transducers in the TGFβ/BMP signaling pathway. Upon ligand binding cooperatively to the type I and II transmembrane receptors, receptor-regulated Smads (R-Smads) are activated through phosphorylation (Derynck and Zhang, 2003). Specifically, Smad2 and Smad3 are phosphorylated by TGFβs, whereas phosphorylation of Smad1 is induced by BMPs. Consequently, phosphorylated R-Smads form heteromeric complexes with Smad4, the common Smad (Co-Smad) that is shared by both the TGFβ and BMP signaling pathways. Subsequently, these heteromeric complexes translocate to the nucleus (Attisano and Wrana, 2002), where they control target gene expression either by directly binding to the DNA or by interacting with other cofactors (Wotton et al, 1999; Hata, 2001).
Although the role of TGFβ/BMP in hematopoiesis is recognized increasingly, the mechanistic basis for their functions has not been well established. TGFβ is considered to be one of the most potent autocrine-negative regulators of hematopoiesis, and accumulated evidence indicates its role as a tumor suppressor in hematological malignancy (Sing et al, 1988; Tessier and Hoang, 1988). Abnormalities in the expression of TGFβ receptors have been described in proliferative syndromes, including both early myeloid (Bousse-Kerdiles et al, 1996; Rooke et al, 1999) and lymphoid leukemia (DeCoteau et al, 1997; Lagneaux et al, 1997). A missense mutation of the Smad4 gene in the MH1 domain (P102L) and a frameshift mutation resulting in termination in the MH2 domain (Δ (483–552)) have been identified in acute myelogenous leukemia (Imai et al, 2001). The products of these mutated Smad4 genes are susceptible to rapid degradation through the ubiquitin–proteosome pathway. In addition to disruptions of the components of the TGFβ signaling pathway, aberrant expression of oncoproteins that abrogate TGFβ responses also has been implicated in myeloid leukemia (Kurokawa et al, 1998; Lin et al, 2004). BMPs also inhibit proliferation and induce differentiation of highly purified human hematopoietic cells (Bhatia et al, 1999). Constitutive activation of BMPs causes an increase in commitment of hematopoietic progenitors to myeloid differentiation (Walters et al, 2002). Inhibition of Smad5 in human hematopoietic progenitors blocks erythroid differentiation induced by BMP-4 (Fuchs et al, 2002). In addition, loss of the Smad5 gene leads to enhanced proliferation of high proliferative potential precursors during embryonic hematopoiesis (Liu et al, 2003).
Homeobox (Hox) genes are also key regulators of hematopoiesis (Lawrence and Largman, 1992). In vertebrates, Hox genes are grouped into four clusters (Hox-A to Hox-D) on separate chromosomes (Sharkey et al, 1997). The expression of Hox genes during hematopoietic development is stage dependent and tightly controlled. Hoxa9, a member of the abdominal-B subclass of Hox genes, is one of the most studied Hox genes in hematopoiesis. Hoxa9 is expressed abundantly in early self-renewing CD34+ cells and is downregulated gradually as cells undergo maturation and terminal differentiation (Sauvageau et al, 1994; Lawrence et al, 1997). Hoxa9-deficient mice show defects in blood formation, whereas enforced expression of Hoxa9 immortalizes and blocks the differentiation of myeloid progenitors, eventually leading to acute myeloid leukemia (AML) in mice (Kroon et al, 1998). Hoxa9 is also upregulated in human AML (Golub et al, 1999). The detection of the Nup98-Hoxa9 fusion gene in cases of AML, with the amino-terminal portion of Nup98 fused to the Hoxa9 carboxyl-terminal DNA-binding homeodomain as a result of the t (7; 11) chromosomal translocation, further suggests a direct oncogenic effect of Hoxa9 in leukemia (Borrow et al, 1996; Nakamura et al, 1996). Overexpression of Nup98-Hoxa9 in murine bone marrow cells causes immortalization in vitro and induces chronic and AML in vivo (Kroon et al, 2001; Calvo et al, 2002). It is widely accepted that Nup98-Hoxa9 acts as an aberrant DNA-binding transcription factor and upregulates a broader range of genes than Hoxa9 (Kasper et al, 1999; Ghannam et al, 2004). Meis1 and PBX1a, members of the 3-amino-acid loop extension (TALE) homeodomain family, are cofactors of Hox (Moskow et al, 1995). Meis1 can collaborate with Nup98-Hoxa9 to accelerate the onset of leukemia. PBX1a has been shown to enhance the DNA-binding affinity of Nup98-Hoxa9 through heterodimerization with Nup98-Hoxa9 on the TGATTTA (C/T) consensus sequence (Kasper et al, 1999).
We have shown previously that Hox proteins are downstream transcription factors of TGFβ/BMP signaling pathways (Shi et al, 1999, 2001). Upon TGFβ/BMP stimulation, Smad4 and the BMP-specific R-Smad, Smad1, can interact directly with Hox proteins and block their DNA-binding activity. Besides our investigations of Smad/Hox interactions, other research has shown that Smad4 interacts with DLX1 at its homeodomain and blocks activin signaling in hematopoietic cells (Chiba et al, 2003). Here, we report that TGFβ/BMP inhibits the bone marrow transformation capability of Hoxa9 and Nup98-Hoxa9 through Smad4. Biochemical and cellular data demonstrate that the interaction between the amino-terminus of Smad4 and Nup98-Hoxa9 mediates this effect by inhibiting the DNA-binding ability of Nup98-Hoxa9. This study reveals a novel regulatory mechanism through which TGFβ/BMP regulates hematopoiesis and raises the possibility that Hox DNA-binding activity may serve as a potential therapeutic target in AML.
Results
TGF β /BMP inhibits bone marrow transformation capability of Hoxa9 and Nup98-Hoxa9
Previously, we have shown that Smad1 and Smad4 interact directly with Hox proteins such as Hoxc8 or Hoxa9 at their conserved homeodomains and inhibit their DNA-binding activities. This suggests that TGFβ/BMP may have an inhibitory effect on the bone marrow transformation capability of Hoxa9 or Hoxa9 fusion proteins by modulating their DNA-binding activities through Smads. To test this possibility, we first used a myeloid colony formation assay to analyze the effects of TGFβ/BMP on bone marrow cells overexpressing Hoxa9 or Nup98-Hoxa9. To achieve this goal, cDNAs encoding Hoxa9 or Nup98-Hoxa9 were cloned individually into the upstream of an internal ribosome entry site (IRES) linked with a blue-excited green fluorescent protein (GFP) variant (BEX) within murine stem cell virus (MSCV) (Figure 1A) (Anderson et al, 1996). Western blotting of extracts from transiently transfected BOCS23 retroviral packaging cells confirmed that both Hoxa9 and Nup98-Hoxa9 constructs were expressed efficiently (Figure 1C). Bone marrow cells infected with retrovirus bearing BEX, Hoxa9, or Nup98-Hoxa9 were then isolated by fluorescence-activated cell sorting (FACS) and cultured in methylcellulose for 7–10 days with or without TGFβ or BMP treatment (Figure 1B and D). Transduction efficiencies ranged from 5 to 20% for Hoxa9 and Nup98-Hoxa9 and from 35 to 45% for BEX (Figure 1D and data not shown).
Figure 1.
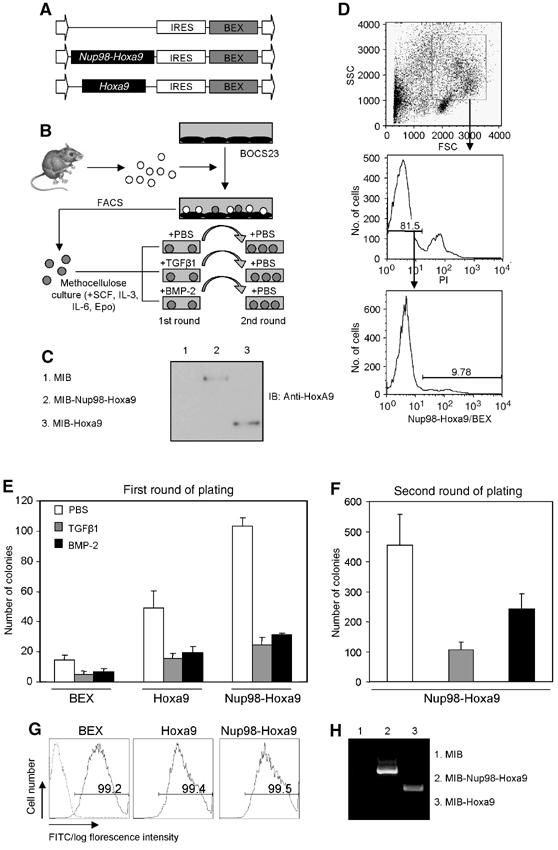
TGFβ/BMP inhibits bone marrow transformation capability of Hoxa9 or Nup98-Hoxa9. (A) Diagram of retroviral constructs expressing Hoxa9 and Nup98-Hoxa9 generated in MSCV. MSCV consists of long terminal repeat, IRES and BEX. (B) Schematic presentation of retroviral transduction procedures. Bone marrow cells were purified from 5-fluorouracil-injected C57BL/6-Ly5.2 mice and infected through cocultivation with transfected BOSC23 retroviral packaging cells for 24–48 h. BEX-positive cells were isolated by FACS and then grown in methylcellulose culture with various treatments as indicated. (C) Western blot analysis of BOSC23 cells transfected with MIB-Hoxa9 or MIB-Nup98-Hoxa9 as detected with an anti-Hoxa9 polyclonal antibody. (D) Bone marrows were gated on myeloid cells by forward scatter (FSC) and side scatter (SSC) and on propidium iodide (PI)-negative cells. Histograms indicate the percentage of BEX-positive cells that were isolated by FACS. (E) Colony numbers generated in the first plating of 2600 transduced bone marrow cells are shown. TGFβ1 (2 ng/ml) and BMP-2 (300 ng/ml) were used for treatment as indicated. Data presented are an average of at least three independent experiments with error bars. (F) Replating of 2600 transduced bone marrow cells harvested from first round of plating. Open, gray and black bars indicate treatment of PBS, TGFβ1 and BMP-2 in the first round of plating, respectively. Data presented are an average of at least three independent experiments with error bars. (G) FACS analysis of cells from second round of platings. Dash line represents nontransduced cells. (H) RT–PCR detection of the expression of the transduced genes in cells derived from the second round of platings.
Control BEX-expressing cells exhibited an average of 20 myeloid colonies of heterogeneous size and morphology, which was similar to the number and type of plating the same number of nontransduced bone marrow cells (Figure 1E and Supplementary data). Cells transduced with Hoxa9 or Nup98-Hoxa9 gave rise to large compact colonies, with an average of 50 and 100 myeloid colonies per 2600 plated cells, respectively (Figures 1E, 2A and C, upper panels). Treatment of TGFβ1 (2 ng/ml) reduced the number of colonies formed from Hoxa9- and Nup98-Hoxa9-transduced cells by 3.1- and four-fold, respectively (Figure 1E). BMP-2 (300 ng/ml) exhibited similar effects and reduced the number of colonies formed from Hoxa9- and Nup98-Hoxa9-transduced cells by 2.5- and 3.2-fold, respectively (Figure 1E). The inhibitory effects of TGFβ/BMP showed on BEX-transduced cells are likely owing to the expression of endogenous Hox genes in bone marrow progenitor cells (Figure 1E). All colonies were florescence positive, indicating that retroviral gene transductions were stable (Figure 2A and C, second rows).
Figure 2.
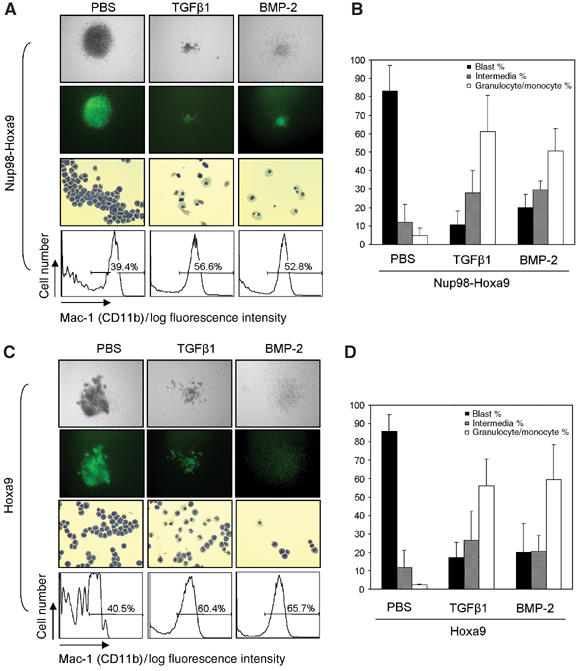
TGFβ/BMP-induced myeloid differentiation of bone marrow cells immortalized by Hoxa9 or Nup98-Hoxa9. (A, C, upper panel) Morphology of colonies formed in methylcellulose assays; original magnification × 5. (Second panel) Fluorescence photomicrographs of colonies. (Third panel) Wright–Giemsa-stained cytospin preparation of cells isolated from colonies derived from the second round of platings. Original magnification × 20. (Lower panel) Immunophenotyping of cells from pools of colonies transduced by Nup98-Hoxa9 or Hoxa9 as indicated. (B, D) The percentages of myeloblastic, intermediate and granulocytic/monocytic cells were obtained by counting a total of 500 cytospun cells in each experiment. Data are presented as mean±s.d. from three independent experiments.
To test the replating ability of cells within the primary cultures, primary colonies were harvested and replated without further treatment (Figure 1B). Although BEX-transduced cells isolated from untreated platings showed lowered replating efficiency owing to exhaustion of their proliferation capacity (data not shown), Nup98-Hoxa9-transduced cells, consistent with a previous report (Kroon et al, 2001), exhibited enhanced replating efficiency (Figure 1F). However, plating of an equivalent number of cells harvested from cultures previously treated with TGFβ1 or BMP-2 showed significantly lower replating efficiency than nontreated cells (Figure 1F), suggesting that TGFβ/BMP reduces the frequency of colony-forming cells in the first round of plating. FACS analysis of these cells showed that more than 99% of the cells were BEX positive (Figure 1G), indicating the stable expression of transduced genes. We further verified that both Hoxa9 and Nup98-Hoxa9 mRNA were expressed in cells derived from second-round colonies (Figure 1H).
Further examination of the morphology of cells within each colony showed that myeloid differentiation was partially blocked in untreated Hoxa9- and Nup98-Hoxa9-expressing colonies (Figure 2A and C, third rows). Strikingly, both TGFβ1 and BMP-2 were able to induce myeloid differentiation of Hoxa9- and Nup98-Hoxa9-expressing colonies into cells that exhibited monocytic or granulocytic cell morphology (Figure 2A and C). This observation was further supported by differential cell counts of cytospun colonies (Zhang et al, 2003), where TGFβ1- or BMP-2-treated cells expressing Hoxa9 or Nup98-Hoxa9 showed significant increases in the percentages of mature granulocytic or monocytic cells (Figure 2B and D). Immunophenotyping of these cells within the colonies revealed that Hoxa9- or Nup98-Hoxa9-transduced colonies had lower frequencies of cells expressing the myeloid cell marker Mac-1 (39.4 and 40.5%, respectively) (Figure 2A and C, bottom panels) than BEX-transduced colonies (around 75%, Supplementary data). Treatment with TGFβ1 or BMP-2 increased the frequency of Mac-1-positive cells to 56.6 and 52.8% for Nup98-Hoxa9-transduced cells, respectively, and 60.4 and 65.7% for Hoxa9-transduced cells, respectively (Figure 2A and C, bottom panels). In summary, these data indicate that TGFβ/BMP inhibits the bone marrow transformation capability of Hoxa9 and Nup98-Hoxa9 by induction of myeloid differentiation.
Smad4 inhibits binding of Nup98-Hoxa9 to DNA
To gain an understanding of the mechanism through which TGFβ/BMP inhibits the function of Nup98-Hoxa9, we examined the effects of TGFβ/BMP on Nup98-Hoxa9 downstream gene transcription. As the downstream transcriptional targets of Nup98-Hoxa9 are not clear, we selected Hoxa9 as a potential target gene because (a) Hoxa9 expression is induced in primary bone marrow cells transduced with Nup98-Hoxa9 (Calvo et al, 2002), (b) there are multiple Hox-binding sites (TTA(C/T)) in the Hoxa9 promoter (Patel et al, 1999), and (c) it has been determined that Hox genes are positively autoregulated by their own products or crossregulated by the products of other Hox genes (Gould et al, 1997). These findings suggest that Hoxa9 could be a direct transcription target of Nup98-Hoxa9. A Hoxa9 promoter luciferase reporter (Hoxa9-luc) was transfected into NIH/3T3 cells together with Nup98-Hoxa9 in combination with Smad1, Smad4 and treatment with BMP-2 (300 ng/ml), with or without PBX1a, the heterodimer partner of Nup98-Hoxa9 that is thought to stabilize the DNA-binding activity of Nup98-Hoxa9 (Kasper et al, 1999). Nup98-Hoxa9 alone significantly stimulated the transcription activity, which was further enhanced by coexpression of PBX1a (Figure 3A). Importantly, these transactivations were inhibited by Smad1, Smad4 or BMP-2 (Figure 3A). In Ba/F3 hematopoietic progenitor cells, Nup98-Hoxa9 was also able to transactivate the Hoxa9-luc by approximately 10-fold (Figure 3B). TGFβ1 or Smad4 inhibited Nup98-Hoxa9-induced transcription, but Smad2 had no such effect (Figure 3B). This result is consistent with our prior finding that TGFβ-specific R-Smads do not interact with Hox proteins (Shi et al, 1999). There were no significant effects of Smad2, Smad4, TGFβ or BMP-2 on the basal activity of the Hoxa9 promoter, indicating that the inhibitory effects were specific for the Nup98-Hoxa9-induced transcriptional activation (Figure 3A and data not shown). To verify the critical role of Smad4 in TGFβ/BMP-mediated inhibition, we eliminated endogenous Smad4 expression with a small interfering RNA against Smad4 (si-Smad4) (Wan et al, 2004) and repeated the transfection assay using NIH/3T3 cells. As Figure 3C shows, si-Smad4 completely abolished TGFβ/BMP-induced inhibition of Nup98-Hoxa9 transactivation and elevated Nup98-Hoxa9-induced transcription of Hoxa9-luc, whereas control si-GFP lacked such an effect. In summary, Nup98-Hoxa9 activates transcription of the Hoxa9 promoter, and this transcription activation is suppressed by TGFβ/BMP through Smad4.
Figure 3.
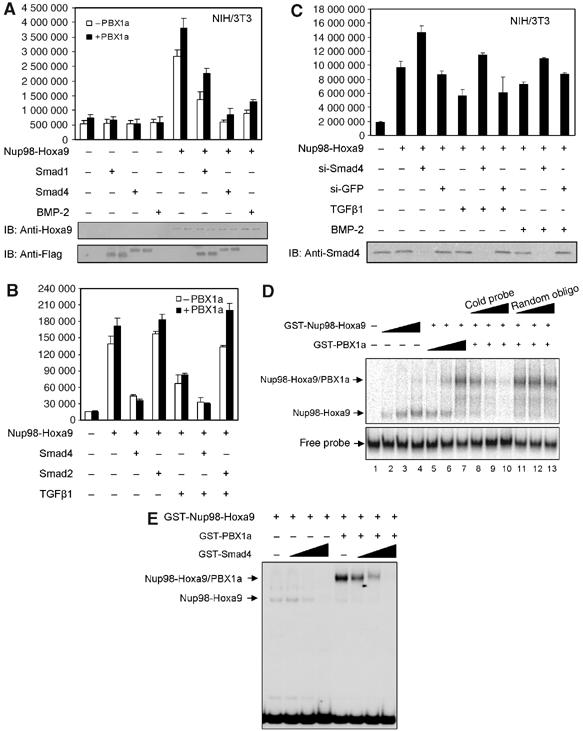
TGFβ/BMP represses Nup98-Hoxa9 transcription through Smad4-mediated inhibition of Nup98-Hoxa9 binding to DNA. (A) The luciferase activities under the control of Hoxa9 promoter were measured in NIH/3T3 cells expressing Nup98-Hoxa9, in the absence or presence of PBX1a, in combination with Smad1, Smad4 or BMP-2 as indicated. Each bar represents the mean and the standard deviations of at least three independent experiments. The expression level of Nup98-Hoxa9, Smad1 and Smad4 were shown in the lower panel by Western blot. (B) Effect of TGFβ on Nup98-Hoxa9-induced transcription, in the absence or presence of PBX1a, on Hoxa9 promoter measured by Hoxa9-luc transcription reporter assay in Ba/F3 cells. Each bar represents the mean and the standard deviations of at least three independent experiments. (C) Effect of loss of Smad4 by si-RNA on Nup98-Hoxa9 transcription on Hoxa9-luc. Western blot (bottom) confirms the absence of Smad4 in si-RNA Smad4-transfected cells. (D) EMSA. Nup98-Hoxa9 alone dose-dependently binds to Hoxa9 promoter (lanes 2–4). PBX1a supershifts this protein–DNA complex (lanes 5–7). Increasing amount of specific (lanes 8–10) but not of nonspecific cold competitor (lanes 11–13) at 0, 50 × and 100 × molar excess of radiolabeled probe inhibits binding of Nup98-Hoxa9 to Hoxa9 promoter probe. (E) EMSA. Smad4 dose-dependently inhibits the DNA-binding ability of Nup98-Hoxa9 to Hoxa9 promoter probe, whether in the absence or in the presence of PBX1a, at molar ratios of 0.2, 0.4 and 1 of Smad4 compared to Nup98-Hoxa9.
To further characterize the mechanism of Smad4-mediated inhibition, we examined whether Smad4 disrupts Nup98-Hoxa9 DNA binding in an electrophoretic mobility shift assay (EMSA). Purified glutathione S transferase (GST)-Nup98-Hoxa9 or GST-PBX1a or both were incubated with a 32P-labeled probe containing a Hox-PBX consensus DNA-binding site (TGATTTAC) from the Hoxa9 promoter (Shen et al, 1999). GST-Nup98-Hoxa9 migrated as a retarded band in a dose-dependent manner (Figure 3D, lanes 2–4), and the band shifted when PBX1a protein was added (Figure 3D, lanes 5–7), indicating that Nup98-Hoxa9 directly binds to the Hoxa9 promoter and that Nup98-Hoxa9 can heterodimerize with PBX1a. The specificity of the binding was demonstrated by a dose-dependent competition of Nup98-Hoxa9 binding with an excess of unlabeled Hoxa9 DNA probe (Figure 3D, lanes 8–10) but not with an excess of unlabeled random oligonucleotides (Figure 3D, lanes 11–13). Most importantly, Smad4 effectively inhibited the binding of Nup98-Hoxa9 to the DNA element in a concentration-dependent manner, whether or not PBX1a was present (Figure 3E). These results suggest that TGFβ/BMP inhibits Nup98-Hoxa9-induced transcription activity by disrupting the DNA-binding activity of Nup98-Hoxa9 through Smad4.
The Smad4 MH1 domain contributes to the interaction with Nup98-Hoxa9
To map the domain(s) of Smad4 that interacts with Hoxa9 protein, we generated a series of GST-Smad4-truncated fusion proteins as shown in Figure 4A. In the absence of Hoxa9, none of the GST-Smad4 or truncated GST-Smad4 fusion proteins bound to the probe (Figure 4B, lanes 3–8). Full-length Smad4, its MH1 domain and its MH1 domain with the linker region all inhibited Hoxa9 DNA-binding activity (Figure 4B, lanes 10, 13 and 15). Moreover, this effect was not observed when the MH2 domain, linker region and MH2 domain with the linker region of Smad4 were used (Figure 4B). These results indicate clearly that the amino-terminus of Smad4 mediates the interaction of Smad4 with Hoxa9. To localize the interaction region within the amino-terminus of Smad4, we generated four smaller fragments as shown in Figure 4C. The fragments containing amino acids 52–148 or amino acids 101–148 of the MH1 domain inhibited the binding of Hoxa9 to DNA (Figure 4D), whereas the fragments containing amino acids 1–51 or amino acids 52–101 did not (Figure 4D). These results suggest that residues 101–148 of Smad4 are crucial to its direct interaction with Hoxa9 and inhibition of Hoxa9 DNA-binding activity. We then tested whether the mapped domains also inhibited the DNA-binding activity of Hoxa9 and Nup98-Hoxa9 in the presence of PBX1a. Both Smad4D (MH1 domain plus linker region) (Figure 4E, lanes 4 and 10) and Smad4.4 (fragment containing amino acids 101–148) (Figure 4E, lanes 5 and 11) inhibited Hoxa9/PBX1a or Nup98-Hoxa9/PBX1a binding to DNA, whereas Smad4E (MH2 domain plus linker) had no such effect (Figure 4E, lanes 6 and 12).
Figure 4.
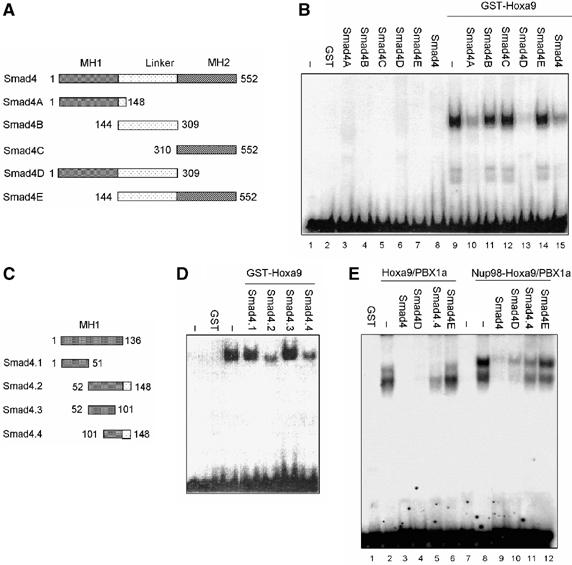
Amino-terminal domain of Smad4 interacts with Hoxa9. (A) Schematic representation of Smad4 deletion constructs. (B) EMSA was performed by using purified GST fusion proteins illustrated in panel A and 32P-labeled probe. (C) Schematic representation of Smad4 MH1 plus partial linker region deletion constructs. (D) EMSA was performed by using purified GST fusion proteins illustrated in panel B and 32P-labeled probe. (E) EMSA was performed by using purified GST fusion proteins as indicated and 32P-labeled probe.
Smad4 inhibits endogenous Hoxa9 expression induced by Nup98-Hoxa9
Having shown that Smad4 inhibits Nup98-Hoxa9 transactivation by blocking its DNA-binding activity, we then attempted to examine whether Smad4 inhibits Hoxa9 endogenous gene transcription induced by Nup98-Hoxa9. Cells were infected with retrovirus-bearing HA-tagged Nup98-Hoxa9 or Nup98b-Hoxa9, which is an alternatively spliced form of Nup98-Hoxa9 (Kasper et al, 1999). Expression of each construct was confirmed by RT–PCR and Western blot analysis (Figure 5B and C). Consistent with an earlier report (Calvo et al, 2002), both Nup98-Hoxa9 and Nup98b-Hoxa9 enhanced the levels of endogenous Hoxa9 mRNA (Figure 5B) and protein expression (Figure 5C) and enforced expression of Smad4 inhibited the induction of Hoxa9 expression (Figure 5B and C), suggesting that Smad4 suppresses Nup98-Hoxa9-induced gene transcription in these cells.
Figure 5.
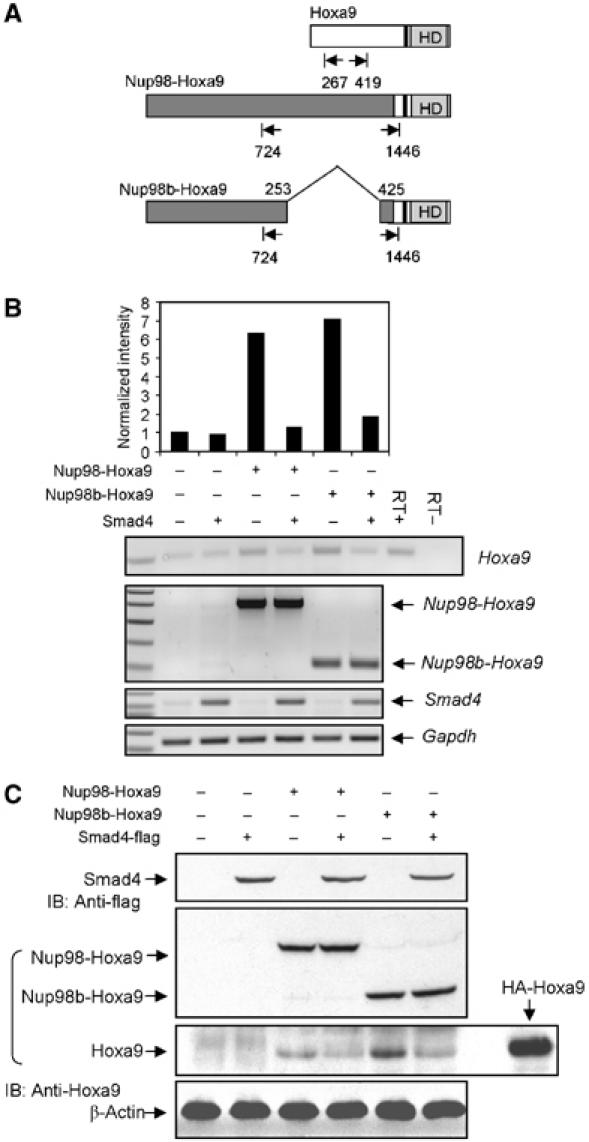
Smad4 inhibits endogenous Hoxa9 expression induced by Nup98-Hoxa9. (A) Schematic representation of primers used for RT–PCR analysis. (B) RT–PCR analysis of Hoxa9 gene expression in NIH/3T3 cells. NIH/3T3 cells were infected with retroviral vectors encoding GFP, Nup98-Hoxa9 or Nup98b-Hoxa9 and transfected with expression plasmids for Smad4-flag or empty vector (EV) as control where Smad4-flag was not used. Total cellular RNA was isolated from transfected cells after 48 h and RT–PCR was performed with specific primers as indicated. A 1-kb DNA ladder was used for size markers (Fisher Scientific). The density of the bands was quantitated with Amersham Pharmacia Biotech Storm System and image analysis software. (C) Western blot analysis of endogenous Hoxa9 expression. Overexpressed HA-tagged Hoxa9 was used as positive control to indicate the size of Hoxa9.
Smad4 inhibits binding of Nup98-Hoxa9 to DNA in cells
To determine whether TGFβ/BMP regulates Nup98-Hoxa9 downstream gene transcription in cells by blocking Nup98-Hoxa9 DNA-binding activity, we investigated the binding of Nup98-Hoxa9 to the chromatin-associated Hoxa9 promoter and analyzed the effect of TGFβ/BMP on this binding using a chromatin immunoprecipitation (ChIP) assay. Retrovirally transduced NIH/3T3 cells were treated with TGFβ1 or BMP-2 for 2–4 h and then with formaldehyde to cross-link DNA–protein complexes. After sonication, chromatin fragments were immunoprecipitated with an HA antibody specific for HA-Nup98-Hoxa9 and then analyzed by PCR to amplify Hoxa9 promoter DNA (between −284 and −91) containing the Hox-PBX consensus-binding element TGATTTAC.
As illustrated in Figure 6A, the Hoxa9 promoter element co-immunoprecipitated with HA-Nup98-Hoxa9 from NIH/3T3 cells. Furthermore, both TGFβ1 and BMP-2 inhibited co-immunoprecipitation of Nup98-Hoxa9 with Hox-binding DNA element. These results indicate specific association of Nup98-Hoxa9 with the proximal Hoxa9 promoter and that TGFβ and BMP-2 suppress the association. We have shown previously that Smad4 mediates TGFβ/BMP inhibition of Hoxa9 DNA binding and that Smad2 does not interact with Hoxa9 (Shi et al, 1999). Consequently, we determined whether Smad4 or Smad2 affects the association of Nup98-Hoxa9 with the proximal Hoxa9 promoter. In a ChIP assay similar to that described above, NIH/3T3 cells infected with retrovirus-expressing HA-Nup98-Hoxa9 or GFP were transfected transiently with expression plasmids for Smad4 or Smad2 or with empty vector as a control. Consistent with the results obtained using the EMSA assay, Smad4 specifically inhibited binding of both Nup98-Hoxa9 and Nup98b-Hoxa9 to DNA, whereas Smad2 did not exhibit such an effect (Figure 6B).
Figure 6.
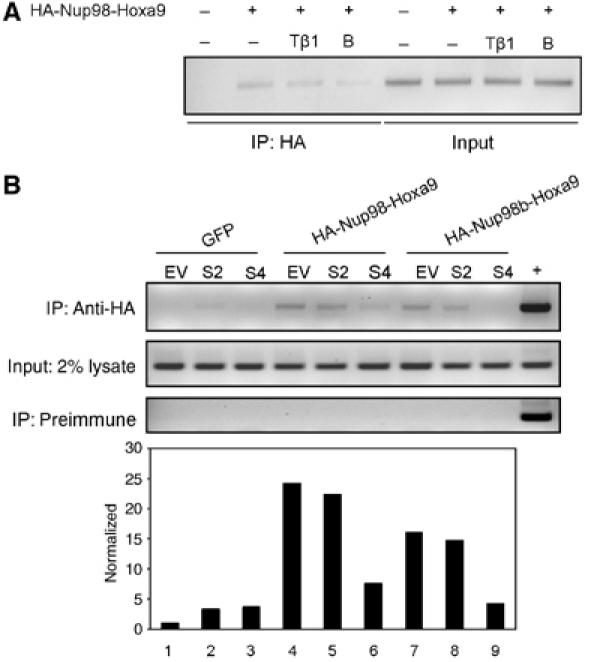
TGFβ/BMP inhibits binding of Nup98-Hoxa9 to DNA in vivo. (A) NIH/3T3 cells were treated with or without TGFβ1 (Tβ1) or BMP-2 (B) for 2 h, and ChIP assays were performed with the indicated antibody and PCR primers. (B) ChIP assays. Overexpression of Smad4 but not Smad2 inhibited the binding of Nup98-Hoxa9 to Hoxa9 promoter. The density of the bands was quantitated by using Amersham Pharmacia Biotech Storm System and image analysis software.
Smad4 inhibits Nup98-Hoxa9-induced bone marrow transformation
Our results suggest that TGFβ/BMP inhibits the function of Nup98-Hoxa9 through Smad4. To examine whether Smad4 alone can inhibit the bone marrow transformation capability of Hoxa9 or Nup98-Hoxa9, cDNAs of Smad4 or Smad4FG, which encodes the Smad4/Hoxa9 interaction domain of Smad4 (amino acids 52–148) fused with a nuclear localization signal (NLS), were individually cloned into the MSCV retroviral vector, which carries a spectrally distinct GFP variant, violet-excited GFP (VEX) (Figure 7A). The expression of BEX and VEX constructs can be detected simultaneously in a single cell owing to their differential excitation properties (Anderson et al, 1996) (Figure 7B). We also generated a chimeric construct, Smad2/4, in which the Smad4/Hoxa9 interaction MH1 domain of Smad4 was replaced by the MH1 domain of Smad2. Equivalent numbers of double-transduced cells were then isolated by FACS and cultured in methylcellulose for 7–10 days. Expression of Smad4, Smad4FG or Smad2/4 alone resulted in a reduced number of colonies in serial platings approximating the numbers of colonies generated by cells transduced with the control vector (data not shown). Cells transduced with Nup98-Hoxa9/VEX gave rise to large compact colonies with enhanced replating ability (Figure 7D). Coexpression of Smad4 or Smad4FG together with Nup98-Hoxa9 not only inhibited the colony numbers in the first round of plating but also significantly impaired the enhanced serial replating ability of Nup98-Hoxa9-transduced cells. Examination of colony morphology revealed that the colony sizes were smaller in the cultures of cells expressing Smad4 or Smad4FG than those that expressed Nup98-Hoxa9 alone (Figure 7E). Wright–Giemsa staining confirmed that the expression of Smad4 or Smad4FG induced myeloid differentiation into monocytic or granulocytic cells (Figure 7E). In contrast, coexpression of Smad2/4 with Nup98-Hoxa9 resulted in only a slight increase in the colony-forming potential of bone marrow cells and was unable to release the differentiation block imposed by Nup98-Hoxa9 (Figure 7D and E).
Figure 7.
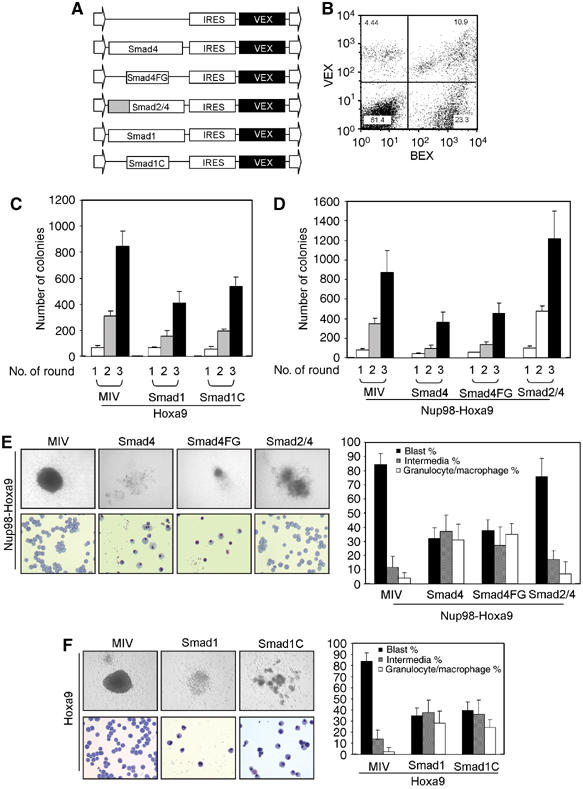
Smad1 and Smad4 inhibit the serial replating ability of Hoxa9- or Nup98-Hoxa9-transduced cells. (A) Schematic representation of retroviral constructs generated in MSCV. (B) Representative FACS plot indicates the BEX and VEX double-positive cell population. (C) Smad1 inhibits the replating ability of bone marrow cells by Hoxa9. Colony numbers generated in the first, second and third round of plating of 3000 double-transduced bone marrow cells isolated from FACS are shown. Data presented are an average of at least two independent experiments with error bars. (D) Smad4 inhibits serial replating ability of bone marrow cells by Nup98-Hoxa9. Colony numbers generated in the first, second and third round of plating of 3000 double-transduced bone marrow cells isolated by FACS are shown. Data presented are an average of at least two independent experiments with error bars. (E, F) Morphology of colonies formed in methylcellulose assays (upper left panels). Wright–Giemsa-stained cytospin preparation of cells from methylcellulose colonies (lower left panels). Differential cell counts of the representative cytospun colonies are indicated on the right. The percentages of myeloblastic, intermediate and granulocytic/monocytic cells were obtained by counting a total of 500 cells in each experiment and data are presented as mean±s.d. from three independent experiments.
On the basis of our previous finding that Smad1 interacts with Hox proteins at their homeodomain and blocks their DNA-binding activity (Shi et al, 1999), we tested Smad1 for its ability to inhibit the Hoxa9-induced enhancement of serial replating ability of murine primary bone marrow cells. Toward this end, cDNAs of Smad1 and its Hox interaction domain, Smad1C, fused with an NLS (Yang et al, 2000), were cloned into the MSCV-IRES-VEX retroviral vector and coexpressed with Hoxa9 in primary bone marrow cells (Figure 7A). Both Smad1 and Smad1C expression were shown to inhibit the sustained replating ability of Hoxa9-transduced bone marrow cells (Figure 7C). In addition, myeloid differentiation into monocytic and granulocytic cells was induced in Smad1- and Smad1C-expressing colonies (Figure 7F).
Coexpression of VEX alone with Hoxa9 or Nup98-Hoxa9 resulted in largely undifferentiated mononucleated blast (∼85%) or intermediate stages (∼12%). Granulocytic or monocytic differentiation blocked by Nup98-Hoxa9 or Hoxa9 was enhanced by coexpressing Smad4, Smad4FG, Smad1 or Smad1C, with significantly higher levels of terminally differentiated populations (31% for Smad4, 35% for Smad4FG, 28% for Smad1, 24% for Smad1C) compared with the VEX-expressing colonies (4% for Nup98-Hoxa9, 2% for Hoxa9) (Figure 7E and F, right panels). Taken together, these data indicate that the interaction of Smad4 with Hoxa9 inhibits the ability of Hoxa9 to block myeloid differentiation at an immature stage.
Discussion
In the present study, we characterized a novel mechanism that mediates the inhibitory effect of TGFβ/BMP on the transformation of murine primary bone marrow cells by Hoxa9 or Nup98-Hoxa9, the chimeric fusion form of Hoxa9 identified in a subset of human AML. Biochemical studies demonstrate that Smad4 inhibits the DNA-binding activity of Nup98-Hoxa9 through its amino-terminus and suppresses its downstream gene transcription via a TGFβ/BMP-mediated mechanism. Furthermore, enforced expression of either Smad4 or Smad4FG, a 98-amino-acid interaction domain of Smad4 with Hoxa9, was sufficient to inhibit Nup98-Hoxa9-induced transformation of primary bone marrow cells. These results reveal a critical antagonistic role for TGFβ/BMP in controlling Hox activity and provide a model of regulatory orchestration between external cytokines and intrinsic transcription factors during hematopoiesis as shown in Figure 8A: Hoxa9 maintains hematopoietic stem cells and progenitor cells in an undifferentiated stage. Upon stimulation of TGFβ/BMP, Smads interact with Hoxa9 and remove it from its DNA target, resulting in myeloid differentiation (Figure 8). In pathological situations, however, mutations causing overexpression of Hoxa9 break the regulatory balance between Smads and Hoxa9, resulting in constitutive activation of Hoxa9 function and leading to leukemia (Figure 8).
Figure 8.
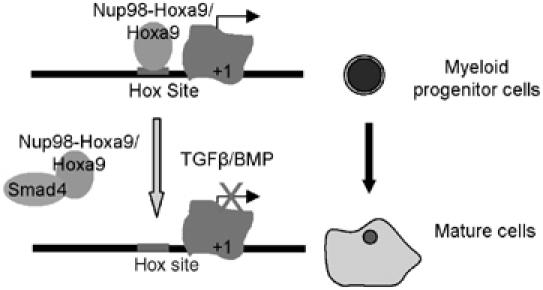
Model illustrates the regulation of Hoxa9 activity by TGFβ/BMP through Smad4. In primitive myeloid blast, Hoxa9 occupies target gene promoter and regulates its transcription. Upon TGFβ/BMP stimulation, Smad4 translocates into nucleus, where they interact with Hoxa9 or its fusion proteins and inhibit their DNA-binding activity, resulting in myeloid differentiation.
TGFβ signaling plays an important role in hematopoiesis by regulating the differentiation and proliferation of hematopoietic cells; however, the mechanism by whhich TGFβ/BMP regulates hematopoiesis is not well characterized. TGFβ is one of the most potent negative regulators of cell cycle progression in general, and a line of evidence indicates that TGFβ maintains quiescence of hematopoietic stem cells through autocrine secretion of this cytokine (Ploemacher et al, 1993). In addition, a number of related mechanisms can mediate cell cycle inhibition. These mechanisms are involved primarily in transcriptional regulation of cell cycle regulators, including downregulation of cyclin-dependent kinases and cyclins during the G1 and G2 phases of the cell cycle or upregulation of the cell cycle inhibitors, such as p27, p21 and p15 (Kim and Letterio, 2003). TGFβ inhibition of cell cycle progression in hematopoietic cells also has been shown to occur independently of p21 or p27 (Cheng et al, 1998). Here, we showed that TGFβ/BMP-induced interaction of Smad4 with Hoxa9 negatively regulates Hoxa9 activity by inhibiting Hoxa9 DNA-binding activity in murine primary bone marrow cells. This may represent a major mechanism through which TGFβ/BMP controls hematopoietic cell proliferation and differentiation as at least 21 of the 39 Hox genes are expressed in the hematopoietic system and, in general, maintain hematopoietic cells in an undifferentiated state (Sauvageau et al, 1994).
TGFβ/BMP can elicit a broad range of intracellular responses. In order to confirm that the interaction between Smad4 and Hoxa9 has a regulatory role in hematopoiesis, we overexpressed Smad4FG or Smad1C, which are the domains from full-length Smads that associated with Hoxa9 directly. Enforced expression of Smad4FG or Smad1C was sufficient to inhibit the enhanced serial replating ability of Nup98-Hoxa9- or Hoxa9-transformed bone marrow cells. To further demonstrate this point, we generated a chimeric fusion construct, in which the MH1 domain of Smad4 was substituted by the corresponding sequence of Smad2. As expected, Smad2/4 lost the inhibitory effect. Taken together, these data suggest that the interaction of Smad4/Hoxa9 plays a major role in how TGFβ/BMP modulates myeloid development.
Both TGFβ and BMP are able to induce the phosphorylation of their R-Smads and subsequent translocation of these R-Smads into nucleus together with Smad4. We have shown that Hoxa9 is able to interact with Smad4 and BMP-specific Smad1 but not TGFβ-specific Smad2. Thus, BMP has both Smad1 and Smad4 available as effectors to inhibit the ability of Hoxa9 to bind DNA and antagonize its function, whereas TGFβ has only Smad4. This suggests that BMP may have more regulatory influence on Hox activity than TGFβ. In addition, there are two inhibitory Smads, Smad6, which preferentially inhibits BMP signaling (Hata et al, 1998), and Smad7, which has a broader inhibition profile (Itoh et al, 2000). Interestingly, Smad6 forms heterodimers with Hox transcription factors when binding to DNA as a negative feedback loop in the nucleus, but Smad7 does not interact with Hox proteins (Bai et al, 2000). Once the Smad6/Hox heterodimer is formed, neither Smad1 nor Smad4 is able to regulate its DNA-binding and transcription activity. Smad4 and the BMP-specific Smad1 and Smad6 have been observed to interact with Hox transcription factors from each of the 13 paralogs in vertebrate animals (Li et al, 2006). It is likely that Smad1, Smad4 and Smad6 interact with all 39 Hox proteins depending on the individual expression pattern of the Hox protein, the promoter context and the cell type. BMP employs the interaction of Smads with Hox in the regulation of hematopoiesis. Our understanding of the function of BMP in hematopoiesis is still in its infancy, but it is known that BMPs are involved in hematopoietic development and that Hox also plays a critical role in this process. If the numerous BMP ligands, multiple BMP receptors and R-Smads and the complex pattern of Hox gene expression during hematopoiesis are taken into consideration, it seems highly likely that interactions between Hox- and BMP-regulated Smads generate intricate signals that negatively modulate Hox transcription activity during hematopoietic cell lineage commitment and maturation. Both TGFβ and BMP can regulate this composite transcription network in hematopoiesis, but many of the fine details of the mechanisms through which these factors regulate this mechanism have yet to be described.
Elevated expression of Hox genes is found frequently in leukemias, particularly in AML (Golub et al, 1999). Animal models, as well as retrovirus-mediated overexpression of individual Hox genes, indicate that many Hox genes can cause leukemia. In addition to acting as a fusion partner with Hoxa9, Nup98 has been identified as a fusion partner with other Hox genes in leukemia, including Hoxa11, HoxC13 and HoxD13 (Moore, 2005). In all cases, the amino-terminus of Nup98 is fused with the carboxyl-terminus of the Hox protein, which contains the complete DNA-binding homeodomain. However, how the function of Hox proteins is regulated remains poorly described. Hox proteins function as transcription factors that are capable of regulating the expressions of a wide variety of gene families (Dorsam et al, 2004; Ghannam et al, 2004). The DNA-binding activity of the Hox proteins is required for their function. For example, protein kinase C has been reported to phosphorylate Hoxa9 at S204 and to impair the DNA-binding activity of Hoxa9, thus inducing myeloid differentiation of Hoxa9-immortalized murine bone marrow cells (Vijapurkar et al, 2004). Mutations causing loss of Nup98-Hoxa9 DNA-binding activity abolished the ability of Nup98-Hoxa9 to transform NIH/3T3 cells (Kasper et al, 1999). Our data suggest that Smad4-mediated inhibition of Hoxa9 DNA-binding activity will trigger myeloid differentiation of Hoxa9-immortalized primary bone marrow cells, as Hoxa9-immortalized primary murine myeloid progenitors retain the capacity to differentiate into macrophages (Calvo et al, 2000).
Here, we report that TGFβ/BMP negatively regulates Hox DNA-binding activity through a Smad4-mediated mechanism. Although mutations and abnormal expression of Hox genes are frequently observed in leukemias, particularly in AML, mutations of components of the TGFβ/BMP signaling pathway are not common in leukemias. Two distinct Smad4 mutations, as well as enhanced proteolytic degradation of the Smad4 protein, have been described in human myeloid leukemia (Imai et al, 2001; Wierenga et al, 2002). As Smad4 is a potent inhibitor of Hox DNA-binding activity, mutations or loss of Smad4 may elevate Hoxa9 activity and promote the development of leukemias. Thus, inhibition of Hox binding to DNA by Smads may represent a potential therapeutic intervention for those leukemias that involve deregulation of Hox expression.
Materials and methods
Bone marrow culture
See Supplementary data for detailed procedures of bone marrow harvesting, retrovirus production and transduction and methylcellulose colony-forming assay.
GST fusion proteins and EMSA
GST fusion constructs were generated by using pGEX5-N1 vector and were transformed into the BL21 strain of Escherichi coli. GST proteins were purified as described previously (Shi et al, 1999). DNA probes (50 000 c.p.m./binding reaction) consisted of PCR-generated, gel-purified, end-labeled oligonucleotides. EMSA was performed as described (Chang et al, 1995). Briefly, DNA-binding reactions were performed at 4°C for 30 min and contained purified GST fusion proteins in a total volume of 20 μl containing 2 μg poly(d(I–C)), 75 mM NaCl, 1 mM EDTA, 1 mM DTT, 10 mM Tris–HCl (pH 7.5), 6% glycerol, 2 μg BSA and 50 000 c.p.m. of DNA probe. Binding reactions were subject to nondenaturing electrophoresis on a 6% polyacrylamide gel (acrylamide:bis-acrylamide, 29:1) in 0.25 × TBE buffer at 10 mA per gel. Dried gels were autoradiographed at −80°C overnight.
Plasmid and constructs
Nup98-Hoxa9 cDNA was a generous gift from T Nakamura. Nup98b-Hoxa9 cDNA was generated by PCR mutagenesis. Both Nup98-Hoxa9 chimeras were cloned into pcDNA3 vector tagged with HA. Different cDNAs as indicated in the figures were also cloned into parental vectors MSCV-IRES-BEX and MSCV-IRES-VEX. All genes were cloned into the EcoRI and XhoI sites upstream of the IRES sequence. si-RNA-GFP and si-RNA-Smad4 have been described elsewhere (Wan et al, 2004).
Cell culture and transfection
Ba/F3 cells were cultured in RPMI-1640 containing 10% FBS and 1 ng/ml recombinant murine IL-3 (Sigma, I4144). NIH/3T3 cells were cultured in DMEM containing 10% FCS and 4 mM L-glutamine. Ba/F3 cells were transfected by electroporation. Briefly, 107 cells in RPMI-1640 were mixed with 20–30 μg of DNA (typically 5 μg reporter, 2 μg prl-0 to normalize, expression constructs and pcDNA3 empty vectors to an equivalent total) and electroporated in a 4-mM gap cuvette (Bio-Rad) at 350 V, 950 μf. NIH/3T3 cells were transfected by Lipofectamine according to the manufacturer's suggestions.
Western-blotting analysis and RT–PCR
Western-blotting analysis of cell lysates was performed as described previously (Wan et al, 2004). All blots were developed by the enhanced chemiluminescence technique (Amersham, Little Chalfont, UK). In RT–PCR analysis, total RNA was isolated from cultured cells by using RNA STAT-60 (Tel-test Inc.). Total RNA (1 μg) was used for the synthesis of first-strand cDNA by using the Superscript pre-amplification system (Life Technologies). Primers used were listed in Supplementary data.
Luciferase assays and statistical analysis
Luciferase activities were assayed with the Dual-Luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer's directions. Luciferase values shown in the figures are representative of transfection experiments performed in triplicate in at least three independent experiments.
ChIP assay
ChIP assay was performed as described previously (Hussein et al, 2003). See Supplementary data for detailed description.
Supplementary Material
Supplementary Materials
Acknowledgments
We are indebted to AD Friedman and DG Gilliland for cell lines, T Nakamura for Nup98-Hoxa9 cDNA, MP Kamps for constructs and advice on primary bone marrow cell culture and PE DiCorleto for Hoxa9-luc reporter construct. Our special thanks go to ML Spell at the UAB Center for AIDS Research flow cytometry core for his expertise. We thank AB Friedman for polishing the manuscript. This work was supported by National Institutes of Health (NIH) Grant DK60913 (to X Cao).
References
- Anderson MT, Tjioe IM, Lorincz MC, Parks DR, Herzenberg LA, Nolan GP, Herzenberg LA (1996) Simultaneous fluorescence-activated cell sorter analysis of two distinct transcriptional elements within a single cell using engineered green fluorescent proteins. Proc Natl Acad Sci USA 93: 8508–8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L, Wrana JL (2002) Signal transduction by the TGF-β superfamily. Science 296: 1646–1647 [DOI] [PubMed] [Google Scholar]
- Bai S, Shi X, Yang X, Cao X (2000) Smad6 as a transcriptional corepressor. J Biol Chem 275: 8267–8270 [DOI] [PubMed] [Google Scholar]
- Bhatia M, Bonnet D, Wu DM, Murdoch B, Wrana J, Gallacher L, Dick JE (1999) Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. J Exp Med 189: 1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow J, Shearman AM, Stanton VP, Becher R, Collins T, Williams AJ, Dube I, Katz F, Kwong YL, Morris C, Ohyashiki K, Toyama K, Rowley J, Housman DE (1996) The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 12: 159–167 [DOI] [PubMed] [Google Scholar]
- Bousse-Kerdiles MC, Chevillard S, Charpentier A, Romquin N, Clay D, Smadja-Joffe F, Praloran V, Dupriez B, Demory JL, Jasmin C, Martyre MC (1996) Differential expression of transforming growth factor-β, basic fibroblast growth factor, and their receptors in CD34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood 88: 4534–4546 [PubMed] [Google Scholar]
- Calvo KR, Sykes DB, Pasillas M, Kamps MP (2000) Hoxa9 immortalizes a granulocyte–macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol 20: 3274–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo KR, Sykes DB, Pasillas MP, Kamps MP (2002) Nup98-HoxA9 immortalizes myeloid progenitors, enforces expression of Hoxa9, Hoxa7 and Meis1, and alters cytokine-specific responses in a manner similar to that induced by retroviral co-expression of Hoxa9 and Meis1. Oncogene 21: 4247–4256 [DOI] [PubMed] [Google Scholar]
- Chang CP, Shen WF, Rozenfeld S, Lawrence HJ, Largman C, Cleary ML (1995) Pbx proteins display hexapeptide-dependent cooperative DNA-binding with a subset of Hox proteins. Genes Dev 9: 663–674 [DOI] [PubMed] [Google Scholar]
- Cheng T, Shen H, Rodrigues N, Scadden DT (1998) TGF-β1 induced cell cycle arrest of hematopoietic cells is independent of p21. Blood 92: 62A. [DOI] [PubMed] [Google Scholar]
- Chiba S, Takeshita K, Imai Y, Kumano K, Kurokawa M, Masuda S, Shimizu K, Nakamura S, Ruddle FH, Hirai H (2003) Homeoprotein DLX-1 interacts with Smad4 and blocks a signaling pathway from activin A in hematopoietic cells. Proc Natl Acad Sci USA 100: 15577–15582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoteau JF, Knaus PI, Yankelev H, Reis MD, Lowsky R, Lodish HF, Kadin ME (1997) Loss of functional cell surface transforming growth factor β (TGF-β) type 1 receptor correlates with insensitivity to TGF-β in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 94: 5877–5881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425: 577–584 [DOI] [PubMed] [Google Scholar]
- Dorsam ST, Ferrell CM, Dorsam GP, Derynck MK, Vijapurkar U, Khodabakhsh D, Pau B, Bernstein H, Haqq CM, Largman C, Lawrence HJ (2004) The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood 103: 1676–1684 [DOI] [PubMed] [Google Scholar]
- Fortunel NO, Hatzfeld A, Hatzfeld JA (2000) Transforming growth factor-β: pleiotropic role in the regulation of hematopoiesis. Blood 96: 2022–2036 [PubMed] [Google Scholar]
- Fuchs O, Simakova O, Klener P, Cmejlova J, Zivny J, Zavadil J, Stopka T (2002) Inhibition of Smad5 in human hematopoietic progenitors blocks erythroid differentiation induced by BMP4. Blood Cells Mol Dis 28: 221–233 [DOI] [PubMed] [Google Scholar]
- Ghannam G, Takeda A, Camarata T, Moore MA, Viale A, Yaseen NR (2004) The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J Biol Chem 279: 866–875 [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286: 531–537 [DOI] [PubMed] [Google Scholar]
- Gould A, Morrison A, Sproat G, White RA, Krumlauf R (1997) Positive cross-regulation and enhancer sharing: two mechanisms for specifying overlapping Hox expression patterns. Genes Dev 11: 900–913 [DOI] [PubMed] [Google Scholar]
- Hata A (2001) TGF β signaling and cancer. Exp Res 264: 111–116 [DOI] [PubMed] [Google Scholar]
- Hata A, Lagna G, Massague J, Hemmati-Brivanlou A (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev 12: 186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Duff EK, Sirard C (2003) Smad4 and beta-catenin co-activators functionally interact with lymphoid-enhancing factor to regulate graded expression of Msx2. J Biol Chem 278: 48805–48814 [DOI] [PubMed] [Google Scholar]
- Imai Y, Kurokawa M, Izutsu K, Hangaishi A, Maki K, Ogawa S, Chiba S, Mitani K, Hirai H (2001) Mutations of the Smad4 gene in acute myelogenous leukemia and their functional implications in leukemogenesis. Oncogene 20: 88–96 [DOI] [PubMed] [Google Scholar]
- Itoh S, Itoh F, Goumans MJ, Ten Dijke P (2000) Signaling of transforming growth factor-β family members through Smad proteins. Eur J Biochem 267: 6954–6967 [DOI] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK, Schnabel CA, Pritchard CEJ, Cleary ML, van Deursen JMA (1999) CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol 19: 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Letterio J (2003) Transforming growth factor-β signaling in normal and malignant hematopoiesis. Leukemia 17: 1731–1737 [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G (1998) Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 17: 3714–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G (2001) NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J 20: 350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H (1998) The oncoprotein Evi-1 represses TGF-β signalling by inhibiting Smad3. Nature 394: 92–96 [DOI] [PubMed] [Google Scholar]
- Lagneaux L, Delforge A, Bron D, Massy M, Bernier M, Stryckmans P (1997) Heterogenous response of B lymphocytes to transforming growth factor-β in B-cell chronic lymphocytic leukaemia: correlation with the expression of TGF-β receptors. Br J Haematol 97: 612–620 [DOI] [PubMed] [Google Scholar]
- Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK, Largman C (1997) Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood 89: 1922–1930 [PubMed] [Google Scholar]
- Lawrence HJ, Largman C (1992) Homeobox genes in normal hematopoiesis and leukemia. Blood 80: 2445–2453 [PubMed] [Google Scholar]
- Li X, Nie S, Chang C, Qiu T, Cao X (2006) Smads oppose Hox transcriptional activities. Exp Cell Res (in press) [DOI] [PubMed] [Google Scholar]
- Lin HK, Bergmann S, Pandolfi PP (2004) Cytoplasmic PML function in TGF-β signaling. Blood 104: 140A–141A [DOI] [PubMed] [Google Scholar]
- Liu B, Sun YX, Jiang FZ, Zhang SX, Wu Y, Lan Y, Yang X, Mao N (2003) Disruption of Smad5 gene leads to enhanced proliferation of high-proliferative potential precursors during embryonic hematopoiesis. Blood 101: 124–133 [DOI] [PubMed] [Google Scholar]
- Moore MA (2005) Converging pathways in leukemogenesis and stem cell self-renewal. Exp Hematol 33: 719–737 [DOI] [PubMed] [Google Scholar]
- Moskow JJ, Bullrich F, Huebner K, Daar IO, Buchberg AM (1995) Meis1, a PBX1-related homeobox gene involved in myeloid leukemia in BXH-2 mice. Mol Cell Biol 15: 5434–5443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, Chen SJ, Willman CL, Chen IM, Feinberg AP, Jenkins NA, Copeland NG, Shaughnessy JD (1996) Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nature Genetics 12: 154–158 [DOI] [PubMed] [Google Scholar]
- Patel CV, Sharangpani R, Bandyopadhyay S, DiCorleto PE (1999) Endothelial cells express a novel, tumor necrosis factor-alpha-regulated variant of HOXA9. J Biol Chem 274: 1415–1422 [DOI] [PubMed] [Google Scholar]
- Ploemacher RE, van Soest PL, Boudewijn A (1993) Autocrine transforming growth factor β 1 blocks colony formation and progenitor cell generation by hemopoietic stem cells stimulated with steel factor. Stem Cells 11: 336–347 [DOI] [PubMed] [Google Scholar]
- Rooke HM, Vitas MR, Crosier PS, Crosier KE (1999) The TGF-β type II receptor in chronic myeloid leukemia: analysis of microsatellite regions and gene expression. Leukemia 13: 535–541 [DOI] [PubMed] [Google Scholar]
- Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, Humphries RK (1994) Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci USA 91: 12223–12227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey M, Graba Y, Scott MP (1997) Hox genes in evolution: protein surfaces and paralog groups. Trends Genet 13: 145–151 [DOI] [PubMed] [Google Scholar]
- Shen WF, Rozenfeld S, Kwong A, Kom ves LG, Lawrence HJ, Largman C (1999) HOXA9 forms triple complexes with PBX2 and MEIS1 in myeloid cells. Mol Cell Biol 19: 3051–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Bai S, Li L, Cao X (2001) Hoxa-9 represses transforming growth factor-β-induced osteopontin gene transcription. J Biol Chem 276: 850–855 [DOI] [PubMed] [Google Scholar]
- Shi X, Yang X, Chen D, Chang Z, Cao X (1999) Smad1 interacts with homeobox DNA-binding proteins in bone morphogenetic protein signaling. J Biol Chem 274: 13711–13717 [DOI] [PubMed] [Google Scholar]
- Sing GK, Keller JR, Ellingsworth LR, Ruscetti FW (1988) Transforming growth factor β selectively inhibits normal and leukemic human bone marrow cell growth in vitro. Blood 72: 1504–1511 [PubMed] [Google Scholar]
- Tessier N, Hoang T (1988) Transforming growth factor β inhibits the proliferation of the blast cells of acute myeloblastic leukemia. Blood 72: 159–164 [PubMed] [Google Scholar]
- Vijapurkar U, Fischbach N, Shen W, Brandts C, Stokoe D, Lawrence HJ, Largman C (2004) Protein kinase C-mediated phosphorylation of the leukemia-associated HOXA9 protein impairs its DNA-binding ability and induces myeloid differentiation. Mol Cell Biol 24: 3827–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters MJ, Wayman GA, Notis JC, Goodman RH, Soderling TR, Christian JL (2002) Calmodulin-dependent protein kinase IV mediated antagonism of BMP signaling regulates lineage and survival of hematopoietic progenitors. Development 129: 1455–1466 [DOI] [PubMed] [Google Scholar]
- Wan M, Tang Y, Tytler EM, Lu C, Jin B, Vickers SM, Yang L, Shi X, Cao X (2004) Smad4 protein stability is regulated by ubiquitin ligase SCF β-TrCP1. J Biol Chem 279: 14484–14487 [DOI] [PubMed] [Google Scholar]
- Wierenga ATJ, Eggen BJL, Kruijer W, Vellenga E (2002) Proteolytic degradation of Smad4 in extracts of AML blasts. Leukemia Res 26: 1105–1111 [DOI] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massague J (1999) A Smad transcriptional corepressor. Cell 97: 29–39 [DOI] [PubMed] [Google Scholar]
- Yang X, Ji X, Shi X, Cao X (2000) Smad1 domains interacting with Hoxc-8 induce osteoblast differentiation. J Biol Chem 275: 1065–1072 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Morrone G, Zhang J, Chen X, Lu X, Ma L, Moore M, Zhou P (2003) CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J 22: 6057–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials