

Abstract
The AAA+ protein ClpC is not only involved in the removal of misfolded and aggregated proteins but also controls, through regulated proteolysis, key steps of several developmental processes in the Gram-positive bacterium Bacillus subtilis. In contrast to other AAA+ proteins, ClpC is unable to mediate these processes without an adaptor protein like MecA. Here, we demonstrate that the general activation of ClpC is based upon the ability of MecA to participate in the assembly of an active and substrate-recognizing higher oligomer consisting of ClpC and the adaptor protein, which is a prerequisite for all activities of this AAA+ protein. Using hybrid proteins of ClpA and ClpC, we identified the N-terminal and the Linker domain of the first AAA+ domain of ClpC as the essential MecA interaction sites. This new adaptor-mediated mechanism adds another layer of control to the regulation of the biological activity of AAA+ proteins.
Keywords: AAA+, adaptorprotein, chaperones, HSP100/Clp, proteolysis
Introduction
HSP100/Clp proteins are ring-forming oligomeric proteins and members of the AAA+ superfamily, which use ATP-dependent conformational changes within the oligomer to drive various cellular processes (Ogura and Wilkinson, 2001; Sauer et al, 2004).
ClpC of the Gram-positive bacterium Bacillus subtilis is an HSP100/Clp protein with unique properties. Through regulated proteolysis, ClpC controls not only key steps of developmental processes, like competence (Turgay et al, 1998), sporulation (Pan et al, 2001) and regulation of stress response (Krüger et al, 2001; Kirstein et al, 2005), but also participates in protein quality control (Krüger et al, 2000; Schlothauer et al, 2003).
Like most members of the HSP100/Clp family, including ClpA and ClpX from Escherichia coli, ClpC associates with the oligomeric peptidase ClpP to form an ATP-dependent protease, similar to the eukaryotic proteasome (Turgay et al, 1998; Gerth et al, 2004). These HSP100/Clp proteins recognize, unfold and translocate substrate proteins for degradation by ClpP (Horwich et al, 1999). Another important activity of HSP100/Clp family proteins is the disaggregation and refolding of previously aggregated proteins, exemplified in E. coli by ClpB (Weibezahn et al, 2004).
ClpC consists of two AAA+ domains, of which the first AAA+ domain (D1) contains an additional N-domain, homologous to the N-domains of ClpA or ClpB, and a Linker domain homologous to, but half the size of the middle domain of ClpB (Figure 1A). The Linker domain consists of a coiled-coil structure, which is inserted into the smaller C-terminal subdomain of D1 (Lee et al, 2003).
Figure 1.
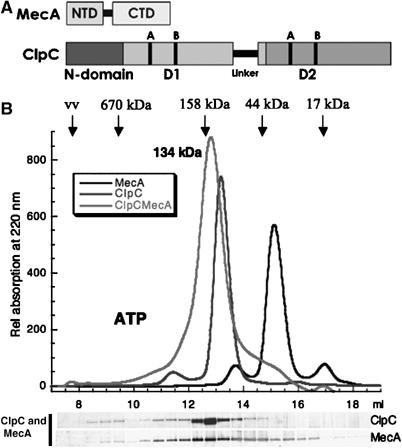
(A) Domain organization of MecA and ClpC. The relative size and location of the NTD and CTD of MecA are depicted. For ClpC, the location of the NTD (N-domain), the position of the AAA+ domains with the Walker A and B sites and the additional Linker domain are indicated. (B) A stable heterodimer of ClpC and MecA is detected by size-exclusion chromatography. ClpC (10 μM) and/or MecA (10 μM) (as indicated), preincubated with ATP (5 mM) and 0.5 mM ATP in running buffer. The elution profile of the protein detected at 220 nm is depicted in the graph. The positions of standard proteins with different molecular weights and the void volume (vv) are indicated. The size of the heterodimer (134±0.6 kDa) determined by multiangle light scattering is indicated. The eluted fractions of the gelfiltration run ClpC and MecA were analyzed by SDS–PAGE with subsequent colloidal Coomassie stain and depicted below the elution profile.
Several adaptor proteins for bacterial HSP100/Clp proteins were recently identified and characterized. They expand, change or enhance the substrate recognition of the respective partner AAA+ protein (Dougan et al, 2002a). For example, the adaptor proteins SspB (Levchenko et al, 2000) and RssB (Zhou et al, 2001; Stüdemann et al, 2003) target specific substrates to ClpX, forming a substrate delivery complex by interacting with the N-domain of ClpX and the substrate (Dougan et al, 2003; Levchenko et al, 2003; Song and Eck, 2003; Bolon et al, 2004). Another adaptor protein, ClpS, changes the substrate specificity of ClpA by interacting with the N-domain of ClpA (Dougan et al, 2002b; Guo et al, 2002a; Zeth et al, 2002). However, all these adaptor proteins are only modulators and not necessary for the basic functions of their partner proteins, which are also fully active in their absence. In contrast, all the general activities of ClpC of B. subtilis, including chaperone activities and degradation of general substrates like casein, depend on the presence of the adaptor protein MecA or its YpbH paralog, which are unrelated to the adaptor proteins identified in E. coli (Schlothauer et al, 2003).
In fact, MecA has been initially discovered together with ClpC in the same genetic screen for repressors of competence development (Dubnau and Roggiani, 1990). Subsequently, it was demonstrated that MecA is essential for ClpCP-mediated regulated degradation of the transcription factor ComK, which controls competence development (Turgay et al, 1997, 1998).
In vitro experiments demonstrated that either MecA or YpbH were essential for the general chaperone activity of ClpC, which included disaggregation and refolding or together with ClpP degradation of previously aggregated proteins. MecA consists of two domains. The N-terminal domain (NTD) of MecA is necessary for the recognition and targeting of substrate proteins (Persuh et al, 1999; Schlothauer et al, 2003), whereas the C-terminal domain (CTD) of MecA (Figure 1A) is responsible for the interaction with ClpC and the induction of the ClpC ATPase activity.
We were interested in understanding why an adaptor protein like MecA is necessary for virtually all the activities of ClpC. Here, we demonstrate that MecA regulates the activity of ClpC by facilitating the oligomerization of ClpC. In the presence of ATP, this results in the formation of an active substrate-recognizing higher oligomer consisting of ClpC and MecA. Our results also demonstrated that MecA interacts with both the N-domain and the Linker domain of ClpC and that this interaction is essential for the oligomerization. The adaptor-controlled oligomerization of ClpC, mediated by the N-domain and the Linker domain of ClpC, represents a new mechanism of regulating AAA+ protein activity.
Results
MecA and ClpC form a heterodimeric complex
Previously, it has been demonstrated that the induction of ATPase activity correlates with the increase of the chaperone activity of ClpC with MecA (Schlothauer et al, 2003). However, the molecular mechanism behind this activation of the HSP100/Clp protein by an adaptor protein remained elusive. A correlation between the ATPase activity, chaperone activity and the oligomeric ring formation of HSP100/Clp proteins has been observed for other homologs of ClpC, like ClpA, ClpB and HSP104 (Singh and Maurizi, 1994; Hattendorf and Lindquist, 2002; Mogk et al, 2003b). Therefore, we set out to examine the oligomeric state of ClpC and its dependence on MecA.
We used size-exclusion chromatography with different gelfiltration columns and varying buffer conditions either in the presence or in the absence of ATP, ADP or ATPγS to analyze the oligomeric state of ClpC and MecA. At concentrations ranging from 1 to 10 μM of ClpC, we could not detect higher oligomeric forms of ClpC (Figure 1B and data not shown), unlike previously demonstrated, for example, ClpA or ClpB. When we added MecA to ClpC, a peak, shifted to higher molecular weight of 134 kDa, as determined by multiangle light scattering (Mogk et al, 2003a), was detected in the elution profile (Figure 1B and data not shown). Analysis of the fractions by SDS–PAGE with colloidal Coomassie stain, silver stain or Western blot analysis confirmed the presence of both ClpC and MecA in this peak (Figure 1B and data not shown).
These results suggested that ClpC and MecA form a heterodimeric complex independent of the presence of ATP, ATPγS or ADP. However, the experiments do not rule out that a higher oligomeric complex might exist under equilibrium conditions, but is kinetically not stable enough to be detected by the gelfiltration experiments. To further analyze the oligomeric state of ClpC and MecA, we used analytical ultracentrifugation or chemical crosslinking with glutaraldehyde (GA) (Mogk et al, 2003b) with subsequent size-exclusion chromatography.
ClpC and MecA form a distinct higher oligomeric complex in the presence of ATP or ADP
We used analytical ultracentrifugation to probe the complex formation under equilibrium conditions. When ClpC and MecA were incubated simultaneously in equimolar concentrations, the analysis of the equilibrium sedimentation experiments demonstrated that a stably associated higher oligomeric complex appeared in the presence of ADP or ATP. The radial distribution of the protein upon equilibrium sedimentation could be described as a single species with an apparent molecular weight of 747±19 kDa (Figure 2A). When the relative amount of MecA was raised, the size of this complex did not increase, instead an additional species appeared, whose size corresponded to an MecA dimer (data not shown). These results are consistent with the formation of a hexamer of ClpC interacting with six MecA monomers.
Figure 2.
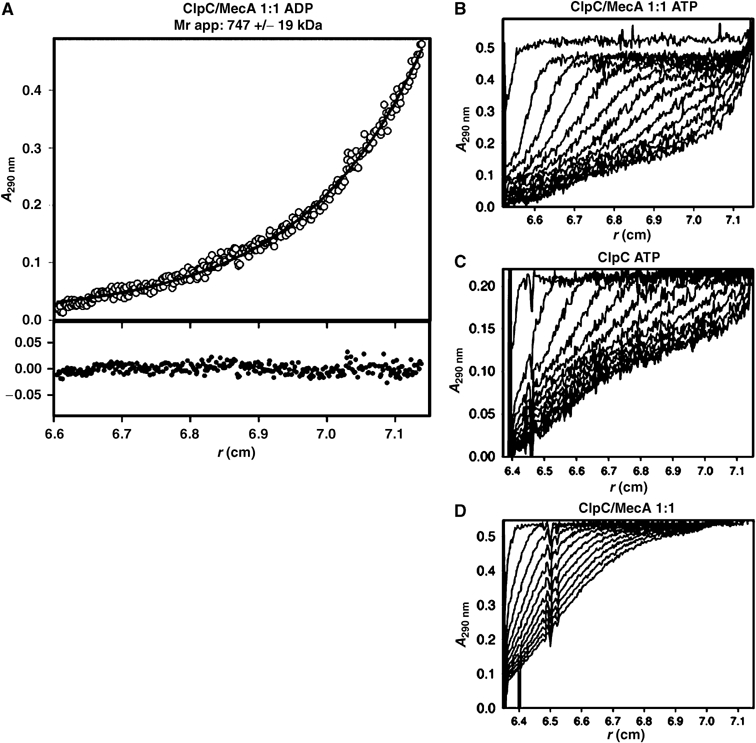
Complex formation of ClpC and MecA analyzed by analytical ultracentrifugation. (A) Sedimentation equilibrium of ClpC (4 μM) together with MecA (4 μM) in the presence of 1 mM ADP. A fit of the absorption data to a single species yielded an Mr of 747±19 kDa. Sedimentation velocity experiments with (B) 10 μM ClpC, 10 μM MecA, 1 mM ADP; (C) 10 μM ClpC, 1 mM ADP; and (D) 10 μM ClpC, 10 μM MecA without nucleotides.
Experiments with ClpC and MecA alone without nucleotides demonstrated that ClpC sediments as observed in the gelfiltration experiments as a monomer of 96 kDa and MecA in a monomer (28 kDa) to dimer (56 kDa) equilibrium (data not shown).
To compare directly the effect of the presence of MecA and ATP on the oligomerization state of ClpC, sedimentation velocity experiments performed with ClpC in the presence or absence of MecA and/or ATP are presented in Figure 2B–D. ClpC and MecA in the presence of ATP associated to a homogeneous particle with a sedimentation rate of 12.5S. The fast sedimenting species (80% of the signal) could be described by an apparent value of 12.5S, the slow sedimenting species (20% of the signal) corresponds to nonassociated MecA (Figure 2B). In contrast, in the absence of MecA, half of the ClpC population displayed heterogeneous association with a higher apparent sedimentation rate (19.2S) in the presence of ATP. The remaining ClpC did not associate at all (Figure 2C). Under the same conditions, ClpC and MecA without ATP displayed no apparent association to higher oligomeric structures (Figure 2D).
We also used a chemical crosslink approach with subsequent gelfiltration to examine the oligomeric state of ClpC and the influence of MecA. The results of these experiments (Supplementary Figure 1a) demonstrated that in the presence of ADP, ATP or ATPγS, a higher oligomeric complex larger than 670 kDa formed only in the presence of ClpC and MecA.
Using negative staining electron micrograph (EM), we obtained EMs of the crosslinked higher oligomeric complex of ClpC and MecA with ATPγS. The results depicted in Supplementary Figure 1b demonstrated that a homogeneous population of structured particles with distinct size and form was detected.
Consistent with the ultracentrifugation experiments (Figure 2C), we observed in the absence of MecA and the presence of ATP, ADP or ATPγS a distinct crosslinked high molecular weight complex of ClpC, which eluted at a significant higher molecular size of more than 1000 kDa compared to the complex formed in the presence of MecA. An inspection of EMs (Supplementary Figure 1b), of these crosslinked complexes, revealed a heterogeneous population of diffuse structures with not well-defined boundaries, which were generally about two-third larger in size compared to the structured particles stemming from the ClpC and MecA crosslinked in the presence of nucleotides. The heterogeneous higher oligomer formed by ClpC in the presence of ATP could be fully reactivated by the addition of MecA even after a prolonged incubation time (data not shown).
These results demonstrate that under equilibrium conditions, MecA facilitates the formation of a functional homogeneous higher oligomeric complex containing a ClpC hexamer and six MecA in the presence of nucleotides.
ClpC-DWB forms a distinct stable higher oligomeric complex with MecA in the presence of ATP
It has been shown for other AAA+ proteins that a mutation of the Walker B motif, which prevents hydrolysis but not binding of ATP, could result in stronger substrate interaction state and helped to stabilize the higher oligomeric form of these proteins (Babst et al, 1998; Hartman and Vale, 1999; Weibezahn et al, 2003; Bösl et al, 2005; Hersch et al, 2005). To further analyze the oligomeric state of ClpC and the influence of MecA, we used a mutated variant of ClpC in which the conserved glutamate residue within the Walker B motif in both ATPase domains was changed to alanine (ClpC E280A/E618A referred to as ClpC-DWB).
We examined the oligomeric state of the ClpC-DWB variant and could not detect higher oligomeric forms of ClpC-DWB by size-exclusion chromatography with or without ATP or ADP (Figure 3A and data not shown). We detected a ClpC-DWB MecA heterodimer peak, as demonstrated for MecA and ClpC (Figure 1B), in the absence of ATP. When we used ClpC-DWB in the presence of MecA and ATP, we observed the formation of a defined higher oligomeric complex, in addition to the already observed heterodimer complex (Figure 3A and data not shown). Using multiangle light scattering, we determined the size of this complex to be 769 kDa, which was consistent with the size determined by sedimentation equilibrium ultracentrifugation of the ClpC-DWB/MecA complex in the presence of ATP (data not shown). It eluted at the same volume as the crosslinked wild-type (wt) ClpC with MecA incubated with ATPγS (Supplementary Figure 1a).
Figure 3.
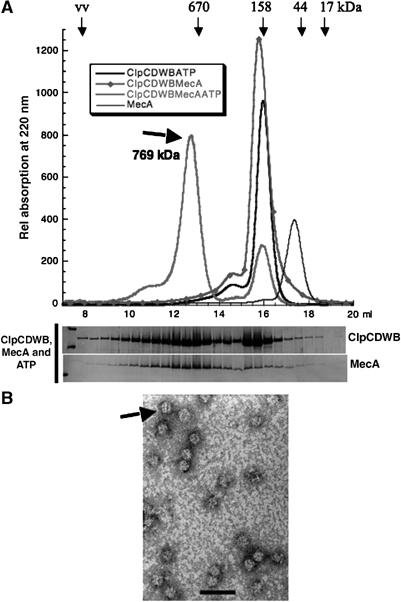
(A) A stable higher oligomeric complex containing both ClpC-DWB and MecA was detected by gelfiltration in the presence of ATP. The elution profile of size-exclusion chromatography experiments with ClpC-DWB (10 μM) with or without MecA (10 μM), preincubated with ATP (5 mM) and 0.5 mM ATP in the running buffer, as indicated. The relative position and molecular weights of standard proteins are indicated above the elution profile. The size of the higher oligomeric complex (769±3 kDa) determined by multiangle light scattering is indicated. The fractions of the experiment with ClpC-DWB and MecA in the presence of ATP were analyzed as described in Figure 1 and depicted below the elution profile. (B) The EM (76 000 × enlarged) of negatively stained (0.5% uranyl acetate) particles of ClpC-DWB and MecA (10 μM) preincubated with ATP (2 mM) is depicted. The length of the bar is 50 nm.
These results demonstrated that ClpC-DWB formed with MecA a complex similar to the one already detected for ClpC and MecA with the ultracentrifugation experiments (Figure 2). In addition, this complex formed by the ATP binding but not hydrolyzing variant of ClpC, interacting with MecA, was kinetically stable enough to be detected by gelfiltration (Figure 3A).
We further characterized this higher oligomeric complex by negative staining EM. The results depicted in Figure 3B revealed distinct particles, which are similar in size and form to the particles observed for the higher oligomers of crosslinked ClpC and MecA preincubated with ATPγS (see Supplementary Figure 1a). They appear even more structured, sometimes striated, resembling EMs of other HSP100/Clp proteins like ClpA (Beuron et al, 1998; Grimaud et al, 1998; Ishikawa et al, 2001). The CTD of MecA is sufficient to induce the ATPase of ClpC (Persuh et al, 1999; Schlothauer et al, 2003). If the induction of the ATPase activity correlates with ClpC oligomerization, then the addition of the CTD of MecA should also be sufficient for the oligomerization of ClpC. We tested this hypothesis and could demonstrate that in the presence of ATP, the CTD of MecA formed with ClpC-DWB the higher oligomeric complex (Supplementary Figure 2).
These results suggested that the interaction of MecA with ClpC in the nucleotide-bound state is a prerequisite for the formation of a stable higher oligomeric complex, possibly via the heterodimer intermediate.
Macromolecular crowding
We also confirmed in vitro that macromolecular crowding (van den Berg et al, 1999) did not influence the activity and the oligomerization of ClpC and MecA (see Supplementary data and Supplementary Figure 2b).
The higher oligomer complex of ClpC-DWB with MecA constitutes the active substrate-binding species of this chaperone system
As we were able to monitor the formation of the higher oligomer complex of ClpC-DWB and MecA, we were interested whether a substrate of MecA and ClpC, like ComK, could stably associate with this complex (Turgay et al, 1997). The result depicted in Figure 4A demonstrated that a stable distinct high oligomeric complex containing ClpC-DWB, MecA and ComK-MBP, which was shifted to a higher molecular size of 896 kDa (measured by multiangle light scattering) compared to the ClpC-DWB complex (769 kDa), was formed in the presence of ATP. Similar results were obtained for the alternative substrate α-casein (data not shown). This also demonstrates that substrate proteins like α-casein and ComK are not involved in oligomerization or activation of ClpC (Figure 4A and Turgay et al (1997), Persuh et al (1999) and Schlothauer et al (2003)).
Figure 4.
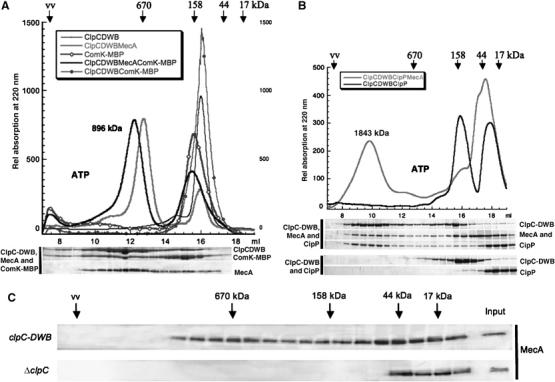
(A) The higher oligomeric complex of ClpC-DWB and MecA can bind substrate protein. The elution profile of size-exclusion chromatography experiments with ClpC-DWB (10 μM), MecA (10 μM), ComK-MBP (10 μM) (preincubated with ATP (5 mM) and 0.5 mM ATP in the running buffer) as indicated in the legend. The relative position and molecular weights of standard proteins are indicated above the elution profile. The size of the higher oligomeric complex consisting of ClpC-DWB, MecA and ComK-MBP (896±22 kDa) determined by multiangle light scattering is indicated. The fractions were analyzed as described in Figure 1 and depicted below the elution profile. (B) The ClpCP protease complex cannot assemble in the absence of MecA. The elution profile of size-exclusion chromatography experiments with ClpC-DWB (10 μM), MecA (10 μM), ClpP (10 μM) (preincubated with ATP (5 mM) and 0.5 mM ATP in the running buffer) as indicated in the legend. The relative position and molecular weights of standard proteins are indicated above the elution profile. The size of the higher oligomeric complex consisting of ClpC-DWB, MecA and ClpP (1843±10 kDa) determined by multiangle light scattering is indicated. The fractions were analyzed as described above and depicted below the elution profile. (C) Comparison of the size distribution of MecA in lysates prepared from clpC-DWB and ΔclpC B. subtilis strains. The soluble fractions of lysates of clpC-DWB and ΔclpC strains were separated by size-exclusion chromatography. The subsequent analysis by SDS–PAGE and Western blot with Anti MecA antibodies is depicted. The relative position and molecular weights of standard proteins are indicated (input is a 1:10 dilution of the lysate).
A complex of the same size was detected when ClpC, MecA and ComK-MBP were incubated and subsequently chemically crosslinked in the presence of ATPγS (data not shown). In contrast, a shift to a higher molecular weight was not observed when only ClpC and ComK-MBP were incubated and crosslinked in the presence of nucleotides, suggesting that the transient higher oligomer of ClpC, formed in the presence of nucleotides, could not interact with substrate (data not shown).
Substrate proteins, like ComK and α-casein, did not coelute with the higher oligomeric complex of ClpC-DWB, when the CTD of MecA, lacking the N-terminal substrate-binding domain, instead of full-length MecA was used (Turgay et al, 1997; Persuh et al, 1999; Schlothauer et al, 2003; data not shown).
Taken together, these results demonstrated that the higher oligomeric complex of ClpC with full-length MecA constitutes the active substrate-binding species of this chaperone system.
MecA is necessary for the formation of the ClpCP protease complex
It was demonstrated previously with in vitro and in vivo protein degradation and stability experiments that ClpC can form with ClpP analogous to ClpAP or ClpXP the ClpCP protease (Turgay et al, 1998; Schlothauer et al, 2003; Gerth et al, 2004). In addition, the sequence of ClpC contains the loop necessary for the interaction with ClpP (Kim et al, 2001; Singh et al, 2001). The E. coli ClpP can form a stable double-heptameric ring (Wang et al, 1997; Maurizi et al, 1998). It could be hypothesized that such a barrel-like structure might also act as platform to support the oligomerization of ClpC. To test this hypothesis, we performed the gelfiltration experiment with ClpP and ClpC-DWB in the presence of ATP and compared it to the experiment where we also added MecA (Figure 4B). In the absence of MecA and in the presence of ATP, both ClpC-DWB and ClpP appear to elute as monomers. This is in contrast to results obtained for ClpAP or ClpXP from E. coli (Kessel et al, 1995; Maurizi et al, 1998). Our observation that ClpP from B. subtilis, unlike ClpP from E. coli, behaved as a monomer was confirmed, when we compared the sedimentation behavior of ClpP from B. subtilis and E. coli by analytical ultracentrifugation (Supplementary Figure 2c).
When MecA was added, a new peak appeared, which contains ClpC-DWB, ClpP and MecA. The peak had a molecular weight of 1840 kDa and appeared homogeneous as determined by multiangle light scattering. This size is consistent with two ClpC hexamers, each interacting with six MecA's stacked on both sides of the double heptameric ClpP barrel. Such complexes are functional and had been observed before for ClpXP and ClpAP from E. coli (Kessel et al, 1995; Beuron et al, 1998; Maurizi et al, 1998; Ortega et al, 2000, 2002). These results again demonstrate that MecA is necessary for the formation of an active ClpC hexamer and in addition suggest that in B. subtilis the formation of this complex might have to precede the final assembly of ClpP barrel structure to create the functional ClpCP complex.
A high molecular weight complex of MecA and ClpC can be detected in vivo
A connection between the presence of MecA and the formation of higher oligomers of ClpC is difficult to prove in vivo, partially because at least two proteins were identified as adaptor proteins for ClpC (Persuh et al, 2002; Schlothauer et al, 2003) and the presence of not yet known adaptor proteins has been proposed (Pan and Losick, 2003; Kock et al, 2004). We prepared lysates from the exponential growth phase of the respective B. subtilis strains grown in full medium at 37°C. Subsequently, these lysates were separated by size-exclusion chromatography and analyzed by Western blot with the respective antibodies. In lysates prepared from a clpC deletion strain, MecA was detected only in a lower molecular weight range up to 44 kDa. But in wt and clpC-DWB strains, MecA was in addition also detected at a size consistent with the higher oligomer of ClpC and MecA (Figure 4C and data not shown), suggesting that the presence of ClpC was necessary to detect MecA molecules at a size consistent with the size of the higher oligomeric complex of ClpC and MecA.
Both the N-domain and the Linker domain of ClpC are essential for the interaction with MecA
We examined the effect of MecA on ClpC protein variants in which only the first or the second Walker B motif (ClpC E280A referred to as ClpC-WB1 and ClpC E618A referred to as ClpC-WB2) was modified. In the presence of ATP, only ClpC-WB1 and not ClpC-WB2 could form the stable higher oligomeric complex with MecA as detected by size-exclusion chromatography (data not shown). This indicated that a conformational change driven by ATP hydrolysis in the first AAA+ domain (D1) prevented a stable association of MecA, suggesting that the interaction site with MecA could be located in, or closely associated with D1 of ClpC. Interestingly, two accessory domains, the N-domain and the Linker domain, are directly associated with the D1 of ClpC (Figure 1A). Recently, it was shown that the N-domain of ClpA (Dougan et al, 2002a; Guo et al, 2002a; Zeth et al, 2002) and ClpX (Dougan et al, 2003; Wah et al, 2003) were responsible for interaction with specific adaptors. The Linker domain is unique to the ClpC subfamily (Schirmer et al, 1996) and is therefore an alternative candidate for the interaction with the ClpC-specific adaptor protein MecA (Schlothauer et al, 2003).
To determine if these domains are responsible for the MecA and ClpC interaction, we initially constructed variants of ClpC lacking the N-domain (ClpC-ΔN) or the Linker domain (ClpC-ΔLi) (see Figure 5A). Judged by circular dichroism spectroscopy measurements, both ClpC variants appeared to be properly folded (data not shown). They displayed the same very low basal ATPase rate as full-length ClpC (0.24 min−1/ClpC) (Schlothauer et al, 2003), but the basal ATPase of ClpC-ΔN and ClpC-ΔLi was no longer inducible by MecA. Furthermore, all MecA-dependent chaperone activity was lost (data not shown), suggesting that MecA could no longer interact with these ClpC variants. Judged by gelfiltration experiments, a ClpC/MecA heterodimer was no longer detectable for both deletion variants (data not shown).
Figure 5.
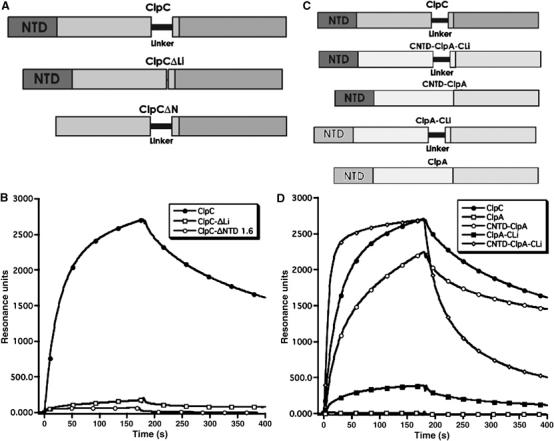
(A) A schematic drawing of the used deletion constructs of ClpC. (B) Interaction of ClpC, ClpC-ΔN and ClpC-ΔLi with the CTD of MecA analyzed by SPR. The binding response is measured in resonance units (RU). The CTD of MecA was immobilized on a CM5-chip (Biacore) and ClpC, ClpC-ΔN or ClpC-ΔLi was passed over the chip surface. (C) A schematic drawing of the primary structures of ClpA, ClpC and the constructed ClpA/ClpC hybrid proteins. (D) Interaction of ClpC, ClpA and ClpA-ClpC hybrid proteins (CNTD-ClpA, ClpA-CLi, CNTD-ClpA-CLi) with MecA, analyzed by SPR. The used proteins and hybrid constructs are schematically depicted. ClpC, CNTD-ClpA, ClpA-CLi, CNTD-ClpA-CLi or ClpA proteins were passed over the same surface used for the experiment depicted in (B).
To further understand the interaction between ClpC and MecA, we used surface plasmon resonance (SPR), as described previously (Persuh et al, 1999; Turgay et al, 2001). Here, we attached the CTD of MecA to the sensor surface and followed the formation of the heterodimer with ClpC, ClpC-ΔLi and ClpC-ΔN in the absence of ATP.
Removal of the Linker domain strongly reduced the interaction between ClpC-ΔLi and the CTD of MecA. The interaction of ClpC-ΔN with MecA, measured by SPR on the same surface, was detectable but weaker compared to ClpC-ΔLi (Figure 5B).
Owing to the deletion of the N-domain or the Linker domain, MecA could not form the stable heterodimer with ClpC. Consistent with this finding, MecA did not assist the oligomerization of ClpC-ΔN or ClpC-ΔLi as demonstrated by size-exclusion chromatography and GA crosslink experiments (data not shown). We conclude that the presence of both the NTD and the Linker domain of ClpC are necessary for the ATP-independent interaction with MecA and the subsequent formation of the higher oligomer consisting of ClpC and MecA.
The N-domain of ClpC and the Linker domain of ClpC interact with the CTD of MecA
To confirm the role of the N-domain and Linker of ClpC in the interaction with MecA, we used ClpA from E. coli as scaffold to construct several different hybrid proteins. In the first hybrid protein, we inserted the Linker domain of ClpC (ClpA-CLi). In the second hybrid, we exchanged the N-domain of ClpA and ClpC (CNTD-ClpA). Finally, a third ClpA double hybrid protein was constructed in which both the Linker domain and N-domain were introduced into ClpA (CNTD-ClpA-CLi) (see Figure 5C).
All purified hybrid proteins displayed the same basal ATPase activity as ClpA (42 min−1/ClpA), suggesting that they were not structurally perturbed. The addition of MecA stimulated the ATPase of all the hybrid proteins (approximately a 30% increase (54 min−1/CNTD-ClpA or ClpA-CLi) for each of the single domain hybrid proteins and 70% higher activity (72 min−1/CNTD-ClpA-CLi) for the double hybrid). Importantly, the ATPase of ClpA was not stimulated by MecA, which suggested that MecA specifically interacted with the hybrid proteins.
To study directly the interaction of the hybrid proteins with MecA, we examined the protein–protein interaction between the CTD of MecA and the hybrid proteins as described above by SPR. The results displayed in Figure 5D demonstrated that ClpC interacted with MecA, whereas it was not possible to measure an interaction with ClpA. In contrast to ClpA, ClpA-CLi exhibited a weak interaction with MecA, whereas the CNTD-ClpA hybrid interacted more strongly and the ClpA double hybrid protein interacted as strong as wt ClpC with MecA.
These results clearly demonstrate that the presence of the N-domain of ClpC and the Linker domain are not only necessary but also sufficient for the interaction with MecA.
As a next step, we wanted to examine whether this was a functional interaction. To do so, we tested the ability of the double hybrid CNTD-ClpA-CLi to degrade MecA in the presence of E. coli ClpP. As a control, we used ClpA and the general substrate α-casein. The in vitro degradation experiment illustrated in Figure 6A demonstrated that ClpAP could degrade α-casein but not MecA. In contrast, the double hybrid protein was able to degrade α-casein and MecA in the presence of ClpP, suggesting that CNTD-ClpA-CLi could form together with ClpP an active protease complex. Hence, the addition of both domains did not interfere with the ability of ClpA to recognize and degrade α-casein. Moreover, these domains in the framework of ClpA enabled the recognition and subsequent degradation of MecA. These results clearly demonstrate that the N-domain with the Linker domain of ClpC was sufficient not only for the recognition but also for the targeting and subsequent degradation of MecA.
Figure 6.

(A) The double hybrid ClpA and ClpP can degrade MecA. The Coomassie-stained SDS–PAGE of a degradation assay with E. coli ClpA or CNTD-ClpA-CLi together with ClpP. (B) MecA can target ComK for degradation by the CNTD-ClpA-CLi and ClpP. The Coomassie stained SDS–PAGE of a degradation assay with B. subtilis ClpCP or E. coli CNTD-ClpA-CLi with ClpP showing the degradation of ComK-MBP in the presence or absence of MecA. All proteins were used at a final concentration of 1 μM. The incubation time is indicated below the lanes.
A further question was whether the interaction of MecA with CNTD-ClpA-CLi could also support the targeting of a MecA-specific substrate like ComK. The results shown in Figure 6B demonstrate that in the presence of ComK-MBP, the MecA degradation was reduced, and ComK-MBP was concurrently degraded, albeit not as fast and as efficient as the MecA-mediated degradation of ComK by ClpCP. In the absence of MecA, no degradation of ComK-MBP took place. This indicated that the interaction of MecA with CNTD-ClpA-CLi was not only sufficient to degrade MecA but also enabled the targeting of ComK-MBP for degradation.
Discussion
We could demonstrate that MecA activated ClpC by participating in the assembly of the higher oligomeric complex of ClpC and MecA in the presence of nucleotides (Figure 7). All until now characterized adaptor proteins, like SspB for ClpX or ClpS for ClpA, were only expanding, modulating or improving the substrate recognition abilities of their cognate HSP100/Clp proteins, but were not necessary to activate them. And in contrast to E. coli, where, for example, the deletion of clpA or clpP revealed no strong phenotype, the tight control of ClpCP activity in B. subtilis appears to be very important.
Figure 7.
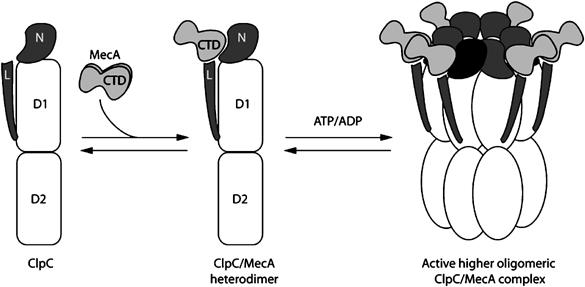
A model summarizing the mechanism of the activation by oligomerization of the AAA+ protein ClpC by the adaptor protein MecA.
To activate ClpC by facilitating its oligomerization is a very effective and new control mechanism. The pre-existing ClpC will be rendered inactive, unless activated by an adaptor protein. Thereby, ClpC is immediately available on demand and the adaptor MecA, when not delivering substrate protein, becomes itself a target for degradation by ClpCP (Schlothauer et al, 2003). In addition, the considerable consumption of ATP by hexameric HSP100/Clp proteins as demonstrated for ClpX (Sauer et al, 2004) is more stringently controlled by this molecular mechanism. We could also demonstrate that the B. subtilis ClpP double-heptamer complex only formed when ClpC was activated and oligomerized (Figure 5C). This indicated a more dynamic ClpP oligomerization in B. subtilis compared to E. coli, whose oligomerization would depend on the availability of activated, hexameric ClpC, ClpX and ClpE. Interestingly, it has been recently demonstrated that human mitochondrial ClpP is a stable heptamer, which only formed the double heptameric structure in the presence of ClpX (Kang et al, 2005). The dynamic state of the B. subtilis ClpP oligomer requires further investigation.
Other adaptor proteins are also available for the activation of ClpC. The MecA paralog YpbH, which is also an adaptor protein and can activate ClpC like MecA (Schlothauer et al, 2003), assisted the oligomerization of ClpC (data not shown). We identified and are currently analyzing a third possible adaptor protein McsB, which displayed similar adaptor and activator abilities for ClpC, including the assistance of ClpC oligomerization (J Kirsten, T Schlothauer, U Gerth, M Hecker and K Turgay, in preparation). We already established that McsB and MecA could compete for interaction with ClpC (Kirstein et al, 2005). Studies on the role of ClpC in sporulation (Pan and Losick, 2003) and on the identification of further ClpP substrates (Kock et al, 2004) suggested that even more than these three adaptor proteins for ClpCP could exist in B. subtilis. Taken together, a more than sufficient amount of adaptor proteins should be available in vivo to interact with and activate ClpC (Turgay et al, 1998). This suggests that our in vitro observation of a heterogeneous, unstructured species of higher oligomeric ClpC forming in the presence of ATP or ADP (Figure 2C, Supplementary Figure 1c and d) is not relevant for the in vivo situation in B. subtilis.
In vivo, different adaptor proteins with, for example, different substrate affinities, targeting abilities or localization could activate ClpC, thereby providing oligomeric ClpC complexes with distinct substrate specificities for different physiological or regulatory purposes at various times, conditions and cellular locations.
The necessity of the presence of nucleotides for the formation of the higher oligomeric complexes of ClpC is not unexpected, because an analysis of the crystal structure of various hexameric AAA+ proteins revealed that ATP binding always occurs at the interface of two neighboring AAA+ domains (Ogura and Wilkinson, 2001; Guo et al, 2002b). For different hexameric AAA+ proteins, it was observed that the binding and hydrolysis of ATP resulted in conformational changes (Rouiller et al, 2002) and it was demonstrated that the different nucleotide states correlated with different interaction strength within the oligomer (Gai et al, 2004). The results of a recent study on the nucleotide binding of a Walker B mutant variant of ClpX demonstrated that the active ClpX hexamer contains three to four ATP or ADP in its six ATP binding sites. This and other experiments suggested that a concerted ATPase mechanism, where all six bound ATP molecules would be simultaneously hydrolyzed and released, could be ruled out (Hersch et al, 2005). These results would suggest that due to the presence of the three to four bound ATP and ADP, the ClpC/MecA complex could remain assembled during the ATPase cycles.
The size of the complex of ClpC and MecA in the ATP- (769 kDa DWB, MecA and ATP; Figure 3A) or ADP- (747±19 kDa; Figure 2A) bound state was consistent with a hexameric ring of ClpC interacting with six MecA molecules, a ratio where the disaggregation and refolding activity of ClpC and MecA was at its maximum (Schlothauer et al, 2003). Analysis of EMs demonstrated that ClpC and MecA in the presence of ATP constituted distinct particles, which were in size and form similar to EMs taken from other ring-forming HSP100/Clp proteins like ClpA (Figure 3A).
Our experiments with the N-domain and Linker deletions as well as the ClpA–ClpC hybrid proteins demonstrate that the ClpC domains necessary for the interaction with MecA are the N-domain and the Linker domain. The ClpA–ClpC hybrid proteins displayed, unlike the ClpC deletion constructs, an additive effect in their ability to interact with MecA (compare Figure 5B and D). This suggests that additional contributions by ClpC, which are not present in ClpA, could also be necessary for the more complex interaction of ClpC with its adaptor MecA. This is consistent with the different off and on kinetics of ClpC and CNTD-ClpA-CLi interacting with MecA in the Biacore experiment (Figure 5D) and the observation that CNTD-ClpA-CLi could support the MecA-dependent degradation of ComK-MBP, but not as efficient and fast as ClpC (Figure 6B).
The structure of ClpB from Thermus thermophilus has recently been determined (Lee et al, 2003). A structural model of ClpC built upon the ClpB structure, which took the shorter Linker domain of ClpC into account, demonstrated that the NTD of ClpC could be positioned proximal to the Linker domain and a concurrent contact of MecA with the mobile ClpC Linker domain and the mobile NTD would be feasible (data not shown). This would fix the position of the Linker and could concomitantly also move the NTD in a position, which may allow better access of the substrate to the pore of the hexameric ClpC. A recent study demonstrated that the movement and the position of the middle domain of ClpB, which is homologous to the Linker domain, influences the ability of ClpB to form hexamers (Watanabe et al, 2005), suggesting that MecA interacting with the ClpC Linker might also influence the assembly of the ClpC hexamer.
Whether the same activation mechanism is also in place for other ClpC homologs is not known yet. It has been recently demonstrated that cyanobacterial ClpC displayed, unlike B. subtilis ClpC, a high basal ATPase activity and could form a hexameric complex. Nevertheless, the same study also demonstrated that MecA from B. subtilis interacted with this cyanobacterial ClpC and enabled its chaperone activities (Andersson et al, 2006). We plan to investigate the biochemical properties of different ClpC homologs from high GC Gram-positive organisms like, for example, Mycobacterium, which do not encode known adaptor homologs.
It has been previously observed that an adaptor protein could negatively regulate the activity of an AAA+ protein. PspA is an inhibitor of PspF, which is a bacterial enhancer protein of the AAA+ family (Chaney et al, 2001; Zhang et al, 2002). As PspA inhibits the ATPase activity of PspF, it has been suggested that it could act by inhibiting the oligomer formation of this AAA+ protein (Dworkin et al, 2000; Elderkin et al, 2002). Such a mechanism has also been suggested for the interaction of plant CDC48 with PUX1 (Rancour et al, 2004).
Our results for ClpC and MecA demonstrate that controlling the ability of an AAA+ protein to form an active substrate accepting higher oligomeric complex is an important functional aspect and a mechanism by which the activity of this protein family can be regulated. This notion of such a controllable dynamic oligomerization is a concept, which could also apply to the understanding of the function and regulation of other AAA+ proteins, involved in various and diverse fundamental cellular functions.
Materials and methods
Proteins
Monomer protein concentrations were determined by using the Bio-Rad Bradford assay with BSA as a standard. The ClpC-N-domain, ClpA, CNTD-ClpA, ClpA-CLi, CNTD-ClpA-CLi, E. coli and B. subtilis ClpP, MecA and the CTD of MecA were purified as described previously (Turgay et al, 1998; Persuh et al, 1999; Dougan et al, 2002b). ClpC variants and ClpC and ComK-MBP were purified as described previously (Turgay et al, 1997; Schlothauer et al, 2003). Pyruvate kinase was purchased from Sigma, α-casein from Fluka and luciferase from Sigma.
Supplementary Material
Supplementary Results
Acknowledgments
We thank C Escher (Universität Freiburg) and F Kaulbars (FU Berlin) for technical assistance. KT thanks D Dubnau (PHRI, Newark), in whose Lab this project started, for discussion, B Beatrix (FU Berlin) for suggestions and discussion, R Hengge (FU Berlin) for support, LW Hamoen (Oxford U), E Deuerling and C Schlieker (ZMBH) for discussion and critical reading of the manuscript. AM, BB and KT were supported by the Deutsche Forschungsgemeinschaft.
References
- Andersson FI, Blakytny R, Kirstein J, Turgay K, Bukau B, Mogk A, Clarke AK (2006) Cyanobacterial ClpC/HSP100 protein displays intrinsic chaperone activity. J Biol Chem 281: 5468–5475 [DOI] [PubMed] [Google Scholar]
- Babst M, Wendland B, Estepa EJ, Emr SD (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J 17: 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuron F, Maurizi MR, Belnap DM, Kocsis E, Booy FP, Kessel M, Steven AC (1998) At sixes and sevens: characterization of the symmetry mismatch of the ClpAP chaperone-assisted protease. J Struct Biol 123: 248–259 [DOI] [PubMed] [Google Scholar]
- Bolon DN, Grant RA, Baker TA, Sauer RT (2004) Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell 16: 343–350 [DOI] [PubMed] [Google Scholar]
- Bösl B, Grimminger V, Walter S (2005) Substrate binding to the molecular chaperone Hsp104 and its regulation by nucleotides. J Biol Chem 280: 38170–38176 [DOI] [PubMed] [Google Scholar]
- Chaney M, Grande R, Wigneshweraraj SR, Cannon W, Casaz P, Gallegos MT, Schumacher J, Jones S, Elderkin S, Dago AE, Morett E, Buck M (2001) Binding of transcriptional activators to sigma 54 in the presence of the transition state analog ADP-aluminum fluoride: insights into activator mechanochemical action. Genes Dev 15: 2282–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan D, Mogk A, Zeth K, Turgay K, Bukau B (2002a) AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett 529: 6. [DOI] [PubMed] [Google Scholar]
- Dougan DA, Reid BG, Horwich AL, Bukau B (2002b) ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9: 673–683 [DOI] [PubMed] [Google Scholar]
- Dougan DA, Weber-Ban E, Bukau B (2003) Targeted delivery of an ssrA-tagged substrate by its adaptor protein SspB to its cognate AAA+ protein ClpX. Mol Cell 12: 373–380 [DOI] [PubMed] [Google Scholar]
- Dubnau D, Roggiani M (1990) Growth medium-independent genetic competence mutants of Bacillus subtilis. J Bacteriol 172: 4048–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin J, Jovanovic G, Model P (2000) The PspA protein of Escherichia coli is a negative regulator of sigma(54)-dependent transcription. J Bacteriol 182: 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elderkin S, Jones S, Schumacher J, Studholme D, Buck M (2002) Mechanism of action of the Escherichia coli phage shock protein PspA in repression of the AAA family transcription factor PspF. J Mol Biol 320: 23–37 [DOI] [PubMed] [Google Scholar]
- Gai D, Zhao R, Li D, Finkielstein CV, Chen XS (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119: 47–60 [DOI] [PubMed] [Google Scholar]
- Gerth U, Kirstein J, Mostertz J, Waldminghaus T, Miethke M, Kock H, Hecker M (2004) Fine-tuning in regulation of Clp protein content in Bacillus subtilis. J Bacteriol 186: 179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR (1998) Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J Biol Chem 273: 12476–12481 [DOI] [PubMed] [Google Scholar]
- Guo F, Esser L, Singh SK, Maurizi MR, Xia D (2002a) Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone, ClpA. J Biol Chem 277: 46753–46762 [DOI] [PubMed] [Google Scholar]
- Guo F, Maurizi MR, Esser L, Xia D (2002b) Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem 277: 46743–46752 [DOI] [PubMed] [Google Scholar]
- Hartman JJ, Vale RD (1999) Microtubule disassembly by ATP-dependent oligomerization of the AAA enzyme katanin. Science 286: 782–785 [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, Lindquist SL (2002) Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J 21: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT (2005) Asymmetric interactions of ATP with the AAA+ ClpX(6) unfoldase: allosteric control of a protein machine. Cell 121: 1017–1027 [DOI] [PubMed] [Google Scholar]
- Horwich AL, Weber-Ban EU, Finley D (1999) Chaperone rings in protein folding and degradation. Proc Natl Acad Sci USA 96: 11033–11040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Beuron F, Kessel M, Wickner S, Maurizi MR, Steven AC (2001) Translocation pathway of protein substrates in ClpAP protease. Proc Natl Acad Sci USA 98: 4328–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Dimitrova MN, Ortega J, Ginsburg A, Maurizi MR (2005) Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J Biol Chem 280: 35424–35432 [DOI] [PubMed] [Google Scholar]
- Kessel M, Maurizi MR, Kim B, Kocsis E, Trus BL, Singh SK, Steven AC (1995) Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26 S proteasome. J Mol Biol 250: 587–594 [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA (2001) Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol 8: 230–233 [DOI] [PubMed] [Google Scholar]
- Kirstein J, Zuhlke D, Gerth U, Turgay K, Hecker M (2005) A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. EMBO J 24: 3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock H, Gerth U, Hecker M (2004) MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol Microbiol 51: 1087–1102 [DOI] [PubMed] [Google Scholar]
- Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M (2000) The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol 182: 3259–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Zühlke D, Witt E, Ludwig H, Hecker M (2001) Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J 20: 852–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe Y, Sigler PB, Chiu W, Yoshida M, Tsai FTF (2003) The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115: 229–240 [DOI] [PubMed] [Google Scholar]
- Levchenko I, Grant RA, Wah DA, Sauer RT, Baker TA (2003) Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol Cell 12: 365–372 [DOI] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA (2000) A specificity-enhancing factor for the ClpXP degradation machine. Science 289: 2354–2356 [DOI] [PubMed] [Google Scholar]
- Maurizi MR, Singh SK, Thompson MW, Kessel M, Ginsburg A (1998) Molecular properties of ClpAP protease of Escherichia coli: ATP-dependent association of ClpA and clpP. Biochemistry 37: 7778–7786 [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B (2003a) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278: 31033–31042 [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, Bukau B (2003b) Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J Biol Chem 278: 17615–17624 [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Ortega J, Lee HS, Maurizi MR, Steven AC (2002) Alternating translocation of protein substrates from both ends of ClpXP protease. EMBO J 21: 4938–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J, Singh SK, Ishikawa T, Maurizi MR, Steven AC (2000) Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol Cell 6: 1515–1521 [DOI] [PubMed] [Google Scholar]
- Pan Q, Garsin DA, Losick R (2001) Self-reinforcing activation of a cell-specific transcription factor by proteolysis of an anti-sigma factor in B. subtilis. Mol Cell 8: 873–883 [DOI] [PubMed] [Google Scholar]
- Pan Q, Losick R (2003) Unique degradation signal for ClpCP in Bacillus subtilis. J Bacteriol 185: 5275–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persuh M, Mandic-Mulec I, Dubnau D (2002) A MecA paralog, YpbH, binds ClpC, affecting both competence and sporulation. J Bacteriol 184: 2310–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persuh M, Turgay K, Mandic-Mulec I, Dubnau D (1999) The N- and C-terminal domains of MecA recognize different partners in the competence molecular switch. Mol Microbiol 33: 886–894 [DOI] [PubMed] [Google Scholar]
- Rancour DM, Park S, Knight SD, Bednarek SY (2004) Plant UBX domain-containing protein 1, PUX1, regulates the oligomeric structure and activity of Arabidopsis CDC48. J Biol Chem 279: 54264–54274 [DOI] [PubMed] [Google Scholar]
- Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM (2002) Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol 9: 950–957 [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Glover JR, Singer MA, Lindquist S (1996) HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem Sci 21: 289–296 [PubMed] [Google Scholar]
- Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K (2003) MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci USA 100: 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Maurizi MR (1994) Mutational analysis demonstrates different functional roles for the two ATP-binding sites in ClpAP protease from Escherichia coli. J Biol Chem 269: 29537–29545 [PubMed] [Google Scholar]
- Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, Maurizi MR (2001) Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem 276: 29420–29429 [DOI] [PubMed] [Google Scholar]
- Song HK, Eck MJ (2003) Structural basis of degradation signal recognition by SspB, a specificity-enhancing factor for the ClpXP proteolytic machine. Mol Cell 12: 75–86 [DOI] [PubMed] [Google Scholar]
- Stüdemann A, Noirclerc-Savoye M, Klauck E, Becker G, Schneider D, Hengge R (2003) Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. EMBO J 22: 4111–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D (1998) Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J 17: 6730–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D (1997) Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev 11: 119–128 [DOI] [PubMed] [Google Scholar]
- Turgay K, Persuh M, Hahn J, Dubnau D (2001) Roles of the two ClpC ATP binding sites in the regulation of competence and the stress response. Mol Microbiol 42: 717–727 [DOI] [PubMed] [Google Scholar]
- van den Berg B, Ellis RJ, Dobson CM (1999) Effects of macromolecular crowding on protein folding and aggregation. EMBO J 18: 6927–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wah DA, Levchenko I, Rieckhof GE, Bolon DN, Baker TA, Sauer RT (2003) Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol Cell 12: 355–363 [DOI] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM (1997) The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell 91: 447–456 [DOI] [PubMed] [Google Scholar]
- Watanabe YH, Takano M, Yoshida M (2005) ATP binding to nucleotide binding domain (NBD)1 of the ClpB chaperone induces motion of the long coiled-coil, stabilizes the hexamer, and activates NBD2. J Biol Chem 280: 24562–24567 [DOI] [PubMed] [Google Scholar]
- Weibezahn J, Schlieker C, Bukau B, Mogk A (2003) Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem 278: 32608–32617 [DOI] [PubMed] [Google Scholar]
- Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119: 653–665 [DOI] [PubMed] [Google Scholar]
- Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA (2002) Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat Struct Biol 9: 906–911 [DOI] [PubMed] [Google Scholar]
- Zhang X, Chaney M, Wigneshweraraj SR, Schumacher J, Bordes P, Cannon W, Buck M (2002) Mechanochemical ATPases and transcriptional activation. Mol Microbiol 45: 895–903 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S (2001) The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev 15: 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Results