

Abstract
Hepatitis delta virus (HDV) replication involves processing and accumulation of three RNA species: the genome, its exact complement (the antigenome), and a polyadenylated mRNA that acts as a template for the small delta antigen (δAg), the only protein of HDV and essential for genome replication. In a recently reported experimental system, addition of tetracycline induced synthesis of a DNA-directed source of δAg, producing within 24 h a significant increase in accumulation of newly transcribed and processed HDV RNAs. This induction was used here to study the action of various inhibitors on accumulation. For example, potent and HDV-specific inhibition, in the absence of detected host toxicity, could be obtained with ribavirin, mycophenolic acid, and viramidine. An interpretation is that these inhibitors reduced the available GTP pool, leading to a specific inhibition of the synthesis and accumulation of HDV RNA-directed RNA species. In contrast, no inhibition was observed with l-FMAU (2′-fluoro-5-methyl-β-l-arabinofuranosyl-uridine), alpha interferon, or pegylated alpha interferon. After modifications to the experimental system, it was also possible to examine the effects of three known host RNA polymerase inhibitors on HDV genome replication: amanitin, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), and actinomycin. Of most interest, amanitin at low doses blocked accumulation of HDV RNA-directed mRNA but had less effect on HDV genomic and antigenomic RNAs. Additional experiments indicated that this apparent resistance to amanitin inhibition of genomic and antigenomic RNA relative to mRNA may not reflect a difference in the transcribing polymerase but rather relative differences in the processing and stabilization of nascent RNA transcripts.
The 1,679-nucleotide, single-stranded, circular RNA genome of hepatitis delta virus (HDV) is replicated by RNA-directed RNA transcription mediated by a host polymerase (7). During replication, nascent RNA transcripts are processed to produce three different RNAs. The RNA genome and its exact complement, the antigenome, both arise from greater-than-unit-length RNA linear transcripts that are reduced to unit length by ribozyme processing and then converted to a circular conformation by RNA ligation. An alternative processing of antigenomic RNA transcripts involves 5′ capping and 3′ polyadenylation to produce an mRNA of about 800 nucleotides in length. It is translated to produce a 195-amino-acid species, the small delta antigen (δAg), which is known to be essential for HDV replication (6). The features of HDV RNA transcription and processing have been incorporated into what is referred to as a double rolling-circle model (39).
In order to study HDV replication experimentally, it is possible to infect primary hepatocyte cultures (2, 38). Such culture systems are somewhat inconvenient, but, as yet, infection of established cell lines has not been reported. The replication of the HDV genome can however be studied with cell lines following transfection with HDV RNAs or cDNA (39). Recently, we have derived an experimental system in which HDV genome replication in an established cell line can be induced promptly and significantly in 100% of cells, simply by the addition of tetracycline (TET) (4). As described here, this system has made it easier to examine various agents that might be used as antivirals, that is, inhibitors of HDV genome replication, in the absence of toxic effects on the host cell. Our studies include a demonstration that the accumulation of HDV RNA species can be inhibited by nontoxic concentrations of ribavirin, mycophenolic acid (MPA), and viramidine, a prodrug of ribavirin. Our data are consistent with this action being mediated by depletion of the intracellular GTP pool, and we provide an explanation of how such depletion can specifically interfere with the accumulation of viral rather than host RNA species.
In addition, with a modification of our experimental system, we examined the effects of agents whose actions on HDV RNA accumulation might contribute to a better understanding of the enzymology of HDV replication. The prime example of this is the potent toxin alpha-amanitin, which has been used to show that host RNA polymerase II (Pol II) is required for HDV genome replication. This drug at relatively low doses (1 μg/ml) specifically inhibits host RNA Pol II (12). It has already been applied in several reported studies with cultured cells and cell extracts to obtain data supportive of the role of Pol II in HDV replication (29, 31, 33). However, some data obtained by use of much higher doses of amanitin have been interpreted as evidence that a second polymerase, possibly RNA Pol I, is needed for the transcription of antigenomic RNA (29, 31). As shown here, an alternative interpretation not invoking a second polymerase is that there are significant differences in the abilities of cells to process and accumulate each of the three main HDV RNA species.
In summary, we report here two kinds of applications of an inducible system for HDV genome replication. The first evaluates several antiviral agents, demonstrating in some cases specific inhibition of HDV replication in the absence of detected toxicity to the host cell. The second examines several known inhibitors of host RNA polymerase activity in order to obtain a better understanding of host polymerase involvement in HDV genome replication.
MATERIALS AND METHODS
Inhibitors.
Ribavirin, mycophenolic acid, actinomycin, amanitin, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), and cycloheximide were obtained from Sigma. Viramidine was obtained from Valeant Pharmaceuticals. l-FMAU (2′-fluoro-5-methyl-β-l-arabinofuranosyl-uridine) was obtained from Gilead, formerly Triangle Pharmaceuticals. Alpha interferon and pegylated alpha interferon were obtained from Schering-Plough.
293-HDV and 293-δAg cells.
The human embryonic kidney cell line 293TRex (Invitrogen) was used to create two cell lines as previously described (4). First, we derived and cloned 293-δAg, which expresses a single copy of δAg cDNA under TET control. Second, 293-δAg cells were transfected with an HDV RNA and cloned to produce 293-HDV. These two cell lines were maintained using blasticidin and hygromycin (Invitrogen). δAg expression was induced with TET at 1 μg/ml.
Expression vectors and transfection strategy.
For the studies described in the legend for Fig. 5D to F, 293-δAg cells were transfected with total RNA extracted from HDV-293 cells at 24 h after TET induction. For the studies described in the legend for Fig. 6, three related expression constructs were used. The construct described in the legend for Fig. 6 and used for the studies described in the legend for Fig. 6C and E was made as previously reported (34). Two constructs were derived from it. In one, the 5′ open reading frame (ORF) for δAg was inactivated by a deletion of 2 nucleotides (see Fig. 6B). The other construct replaced the 5′ ORF with fully functional wild-type δAg (see Fig. 6D). All transfections were carried out using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). For all studies described in the legend for Fig. 6, the cells were cotransfected with pSVTVA, a plasmid known to enhance the transcription of RNA species from plasmids containing a simian virus 40 promoter (1, 34).
FIG. 5.

Dose-dependent effects of amanitin, actinomycin, and DRB on the accumulation of processed HDV RNA transcripts. The action of these drugs was assayed under two different experimental situations. In the first strategy (A to C), 293-HDV cells were induced with TET for 24 h in the presence of 30 μM ribavirin, at which time the medium was replaced with medium containing 3H[uridine] and the indicated concentrations of the three inhibitors. After an additional 18 h, total RNA was extracted and assayed as described in the legends for Fig. 1 and 2. In the second experimental strategy (D to F), 293-δAg cells at 24 h after TET induction were transfected with total RNA from TET-induced 293-HDV cells (in order to initiate HDV genome replication). At 4 h after this transfection, the cells were reseeded into smaller identical cultures in the absence of TET and the presence of 3H[uridine] and with the indicated concentrations of the three inhibitors. After an additional 18 h, total RNA was extracted and assayed as described in the legends for Fig. 1 and 2. The color coding is described in the legend for Fig. 2; however, unlike in Fig. 2, here we do not show data for the accumulation of DNA-directed HDV δAg mRNA. (This is because in the present experiments this mRNA was allowed to accumulate not during but before the addition of the inhibitors.) For panels A to C, some of the levels of tRNA actually increased as a correlate with the drug treatments, reaching values as much as 100% more than values with the untreated controls.
FIG.6.

Accumulation of processed DNA-directed antigenomic HDV RNA transcripts. Panel A indicates the region of an expression vector containing greater-than-unit-length sequences of HDV. Following transfection into 293TRex cells, these sequences should be transcribed by Pol II as an antigenomic HDV RNA transcript. The 5′ end corresponds to the known 5′ end of the HDV mRNA. By use of the known poly(A) signal (small open box), this RNA can be processed to release an mRNA comparable to the natural HDV mRNA. Downstream of the poly(A) signal is a unit-length antigenomic RNA sequence flanked by two copies of the antigenomic ribozyme (small open circle). This region of the RNA can be ribozyme processed to release unit-length linear antigenomic RNA, some of which can be further processed by ligation to become unit-length antigenomic RNA circles, like those that are detected during natural HDV replication. It should be noted that the unit-length sequence within the primary transcript does not contain an active poly(A) signal (having been inactivated by conversion of AAUAAA to UUUAAA [34]). Also, the δAg ORF within this sequence is inactivated (by a 2-nucleotide deletion). Three versions of the expression vector were used, and results are shown in panels B to D. The differences were all in terms of the δAg ORF at the 5′ end of the antigenomic RNA transcript. In panel B, the ORF was inactivated by a 2-nucleotide deletion. In panel C, the ORF was subjected to a 36-nucleotide in-frame deletion that produces a protein that cannot support HDV genome replication but is able to support the processing and accumulation of DNA-directed HDV RNA species (25, 32). In panel D, the ORF is the wild-type small δAg, which supported HDV RNA-directed RNA replication. Cells were transfected with each of these specific expression constructs. At 4 h, the cultures were reseeded as identical smaller cultures, and measurements were taken at daily time points up to 6 days, as indicated. For each culture, total RNA was extracted and examined by Northern assay to quantitate both the mRNA (filled circles) and the unit-length antigenomic RNA (open circles). Panel E represents a variation of the experiment described for panel C. At 24 h after the transfection, the cells were reseeded in the indicated concentrations of amanitin. After another 24 h, total RNA was extracted and assayed for the accumulation of the two processed HDV RNA species. The amounts of mRNA and unit-length antigenomic RNAs are indicated by filled and open bars, respectively. These data are normalized relative to those cells not exposed to amanitin.
Total RNA labeling, extraction, and Northern analyses.
In some experiments, during periods of exposure to inhibitors, the cell RNA was labeled by the incorporation of 3H[uridine] (5 μCi/ml; Amersham). All RNA extractions were performed using TriReagent (Molecular Research Center). RNAs were glyoxalated prior to electrophoresis into gels of 1.5% agarose. After electrotransfer, in order to detect 3H[uridine], the charged nylon membrane (Zeta-Probe GT; Bio-Rad) was air-dried prior to direct contact exposure to a special bio-imager screen (BAS-TR-2040; Fuji). Next, the filter was typically subjected to hybridization with 32P-labeled RNA probes to detect genomic and antigenomic HDV RNAs, in each case with quantitation using the bio-imager. However, for studies shown in Fig. 6, the hybridization used a 5′ 32P-labeled oligonucleotide probe. Bio-images were collected and quantitated using Image Gauge v3.3 and prepared for publication using Photoshop v7.0 and Canvas v9.0 software.
RESULTS
Action of antiviral compounds on inducible HDV genome replication.
Figure 1 presents typical data obtained for the inhibitor studies presented subsequently in the manuscript. As previously described, the 293-HDV cells contain a TET-inducible form of the small δAg and a replicating HDV RNA genome that is unable to express any form of δAg (4). Even in the absence of TET, a low level of δAg, which supported a low level of chronic HDV replication, was produced. However, within 24 h of adding TET there was a major increase in δAg synthesis, leading to an increase in the transcription and accumulation of processed HDV RNA species. During this induction period, we added 3H[uridine], which was incorporated into processed host RNAs. Such incorporation was used to monitor drug-associated cell toxicity. In the example shown in Fig. 1, we also added at the time of induction a series of concentrations of ribavirin. At the end of the induction period, total RNAs were extracted and subjected to glyoxalation and agarose gel electrophoresis and then electrotransferred to a charged nylon membrane. By exposure of the membrane to a special bio-imager screen, the 3H was detected (Fig. 1A). We chose the accumulation of 18S rRNA as an indication of transcription by the host RNA polymerase I. Similarly, we used accumulation of 4S tRNA as a measure of transcription by RNA polymerase III. The charged nylon membrane was then used in consecutive hybridizations with 32P RNA probes to detect genomic HDV RNA species (Fig. 1B) and then antigenomic species (Fig. 1C). As can be seen, we detected four processed RNA species: the unit-length HDV genomic RNA (Fig. 1B), the unit-length antigenomic RNA (Fig. 1C), and two species that are less than unit length (Fig. 1C). One of these species was the mRNA for the DNA-directed, TET-inducible δAg transcribed from the integrated HDV cDNA. The other, a faster-migrating species, was the RNA-directed mRNA produced by HDV genome replication. Under extended gel electrophoresis, as indicated here, the HDV RNA-directed mRNA resolved into two bands. As documented elsewhere (4), this probably reflects heterogeneity in the sequence of the poly(A) signal on the replicating HDV RNA (34). [We have shown that the sequence of this poly(A) signal is changed, most likely as a consequence of posttranscriptional RNA editing by a host adenosine deaminase acting on RNA.] It must be noted that this mRNA also contains a 2-nucleotide deletion within the open reading frame, so that no functional small δAg was produced from it. The DNA-directed mRNA was the only source of functional small δAg.
FIG. 1.
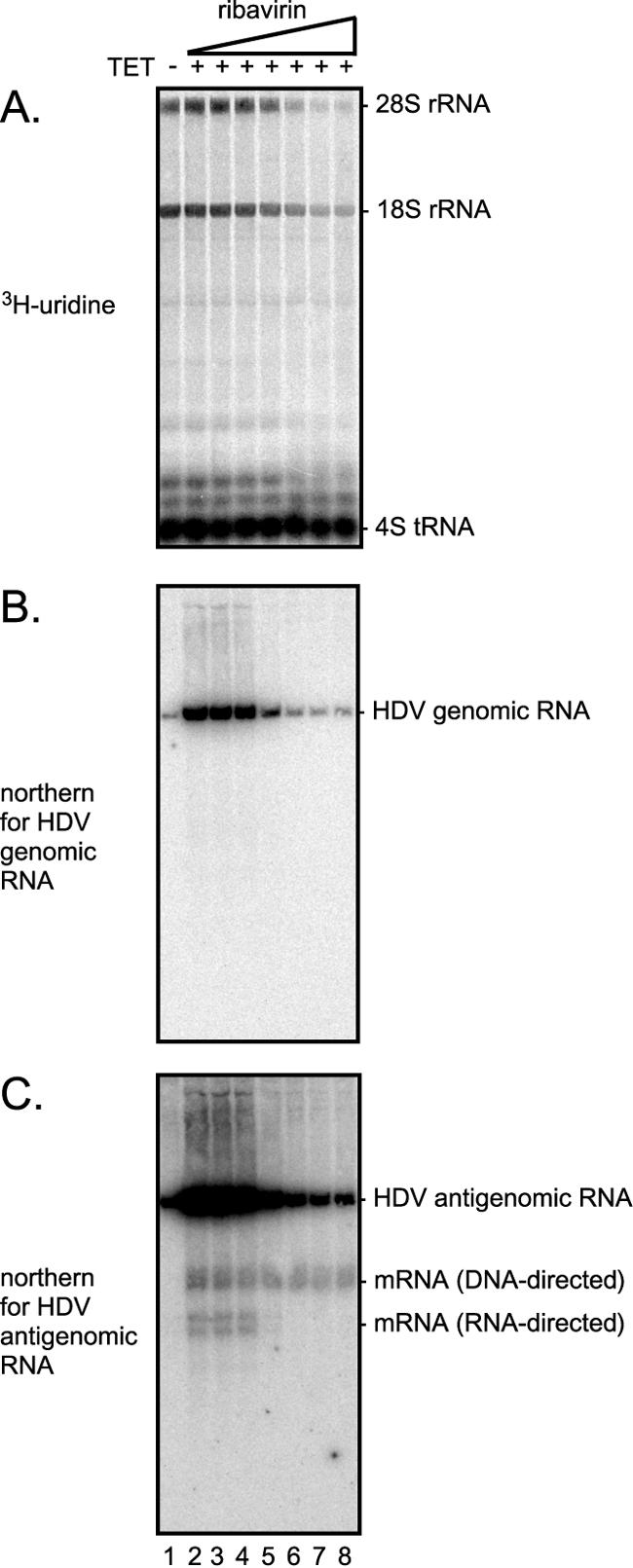
Dose-dependent effects of ribavirin on the accumulation of processed HDV RNA transcripts. Cultures of 293-HDV cells were labeled with 3H[uridine], as indicated, in the presence or absence of TET and different concentrations of ribavirin (3, 10, 30, 100, 300, and 1,000 μM). At 24 h, total RNA was extracted and, after glyoxalation, subjected to agarose gel electrophoresis followed by electrotransfer to a charged nylon membrane. A bio-imager screen was used to quantitate 3H incorporation into RNA species, with results shown in panel A. Then, the membrane was subjected to consecutive hybridizations with 32P-labeled RNA probes, followed by bio-imager quantitation to detect HDV genomic (B) and antigenomic (C) RNA species. Various host and HDV-related species are indicated at the right side.
In the example shown in Fig. 1, it can be seen that with increasing concentrations of ribavirin there was an inhibition of the accumulation of the three HDV RNA-directed processed RNAs. At higher concentrations, there was an inhibition of the 18S rRNA, transcribed by Pol I, but even with the highest concentration tested, there was no significant inhibition of the 4S tRNA, transcribed by Pol III, or the DNA-directed HDV mRNA, transcribed by Pol II.
A quantitation of the data from Fig. 1 is presented in Fig. 2A. It is clear that, the accumulation of the DNA-directed RNA transcripts by Pol I, Pol II, and Pol III was more resistant to inhibition by ribavirin than the HDV RNS-directed RNAs.
FIG. 2.

Dose-dependent effects of ribavirin, MPA, viramidine, l-FMAU, alpha interferon, and pegylated alpha interferon on the accumulation of processed HDV RNA transcripts. The data shown in panel A represent quantitation of the results from Fig. 1. Panels B to F represent quantitations of similar studies using not ribavirin but MPA, viramidine, l-FMAU, alpha interferon, and pegylated alpha interferon, respectively. In all panels, the columns of dark, medium, and light blue refer to accumulation of transcripts by Pol I (18S rRNA), Pol III (tRNA), and Pol II (DNA-directed mRNA for δAg), respectively. Similarly, the red-shaded columns refer to HDV unit-length genomic RNA, unit-length antigenomic RNA, and RNA-directed HDV mRNA (from darkest to lightest red, respectively). For data obtained using 32P-labeled probes, we subtracted the signal detected prior to the drug treatment. In all cases, the data were normalized relative to the accumulations detected in the absence of drug treatment.
Several studies have proposed that the effects of ribavirin on virus replication are mediated primarily by depletion of the GTP pool; it is known that ribavirin inhibits an essential step leading to GTP formation (26, 37). Specifically, ribavirin monophosphate is considered to be a competitive inhibitor of IMP dehydrogenase (IMPDH), the enzyme that converts IMP to xanthine monophosphate.
It is also known that MPA is a direct rather than a competitive inhibitor of IMPDH (36). We tested MPA action on HDV RNA accumulation, with results quantitated and shown in Fig. 2B. We noted that MPA not only gave specific inhibition of the HDV RNA accumulation but also did so at concentrations approximately 30-fold less than were needed for ribavirin.
Although ribavirin is used as an antiviral for several different human infections (26), it is known that the effective doses are close to levels that produce toxicity (15). This side effect is considered a consequence of the accumulation in red blood cells of the phosphorylated forms of ribavirin, which lead to cell toxicity and anemia. Viramidine, a prodrug of ribavirin, has been evaluated (41) as a strategy to avoid this effect, especially when treating patients with virus infections of the liver. Viramidine localizes better to the liver and has less associated toxicity. We tested this drug against HDV replication, with the results presented in Fig. 2C. Again, it was possible to obtain inhibition that was specific for HDV RNAs relative to host RNAs. The concentrations needed were about 10 times higher than for ribavirin. This higher-concentration requirement might be consistent with viramidine being a prodrug for ribavirin and thus requiring activation; however, we cannot exclude other possibilities, such as differences in the abilities of the cells to transport the drug.
Recently, it has been reported that l-FMAU, a nucleoside analog known for its ability to interfere with the replication of hepadnaviruses, can also suppress HDV replication in an infected woodchuck (3). Therefore, we tested l-FMAU in our assay system, with the results quantitated in Fig. 2D. No specific effect on the accumulation of HDV RNAs was detected. Thus, it is most likely that the reported in vivo ability of this drug on HDV particle formation was an indirect effect, mediated by suppression of the necessary helper hepadnavirus.
At this time, the only successful therapy for HDV is one involving long-term treatment with high doses of alpha interferon (13, 14). However, several previous studies have indicated that in cultured cells, interferon treatments do not inhibit the accumulation of HDV RNAs (20, 30). We were able to confirm such findings in our assay system, with results and quantitation shown in Fig. 2E. We also tested pegylated interferon, since in therapies for hepatitis C virus, this form is more biologically active than the unmodified form (40). Again there was no obvious inhibition (Fig. 2F). Thus, as with l-FMAU, we conclude that any in vivo ability of these interferons to suppress HDV accumulation is most likely indirect, probably by an action on the essential helper hepadnavirus.
In the experiments described above, it was found that ribavirin and MPA were both able to inhibit HDV RNA accumulation. A preliminary interpretation of these data is that depletion of the intracellular GTP pool was sufficient to inhibit the accumulation of HDV RNAs. As a further test of this interpretation, we examined whether the actions of these drugs were synergistic. As shown in Fig. 3, we selected suboptimal doses of each drug and applied them separately and together. As can be seen, there was synergy. We also asked whether the actions of these drugs could be reversed by the presence of addition to the medium of an excess of free guanosine (50 μM). As shown, there was almost complete reversal of both inhibitors.
FIG. 3.
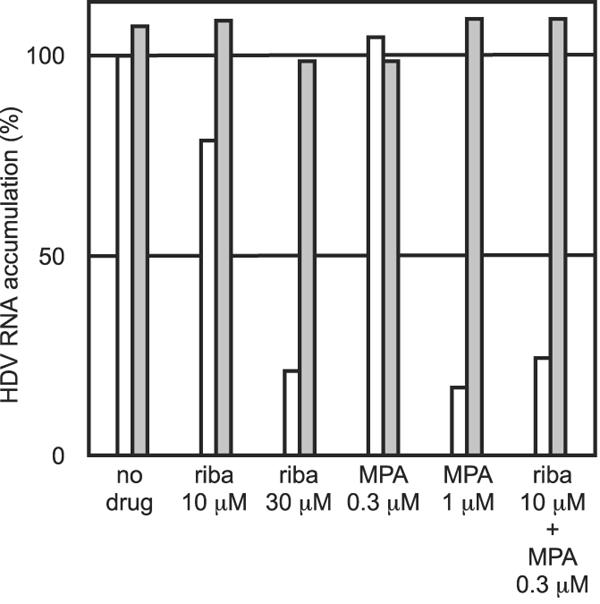
Effects of drug synergy and guanosine rescue on the accumulation of processed genomic HDV RNA transcripts. HDV genome replication in cultures of 293-HDV cells was induced and assayed as described in the legends for Fig. 1 and 2. The inhibitors ribavirin (riba) and MPA were used at the concentrations indicated. The quantitation refers to the accumulation of unit-length genomic HDV RNA. Each drug treatment was performed in the absence (open columns) or presence (filled columns) of 50 μM guanosine.
Several recent studies have offered an alternative model for the action of ribavirin on the replication of RNA viruses. They propose that the inhibition is mediated by what is termed an error catastrophe (9-11, 18, 22). That is, they consider that some ribavirin is actually incorporated into viral RNAs, inhibiting the ability of these RNAs to undergo additional rounds of replication. As a test of this interpretation, we asked whether in an 18-h chase following a 24-h ribavirin treatment there could be the recovery of HDV RNA transcription and the accumulation of processed HDV RNA species. During this chase period, we added the protein synthesis inhibitor cycloheximide (50 μg/ml, a dose sufficient to block all protein synthesis [data not shown]). Thus, all DNA-directed mRNA for the essential δAg was both transcribed and translated in the prior period when the ribavirin was present.
Quantitation of the data from this experiment is presented in Fig. 4. It can be seen that there was extensive recovery during the chase period, even in the presence of cycloheximide. We interpret this as evidence that the necessary amounts of fully functional small δAg were obtained by DNA-directed transcription and translation during the period of ribavirin treatment.
FIG.4.
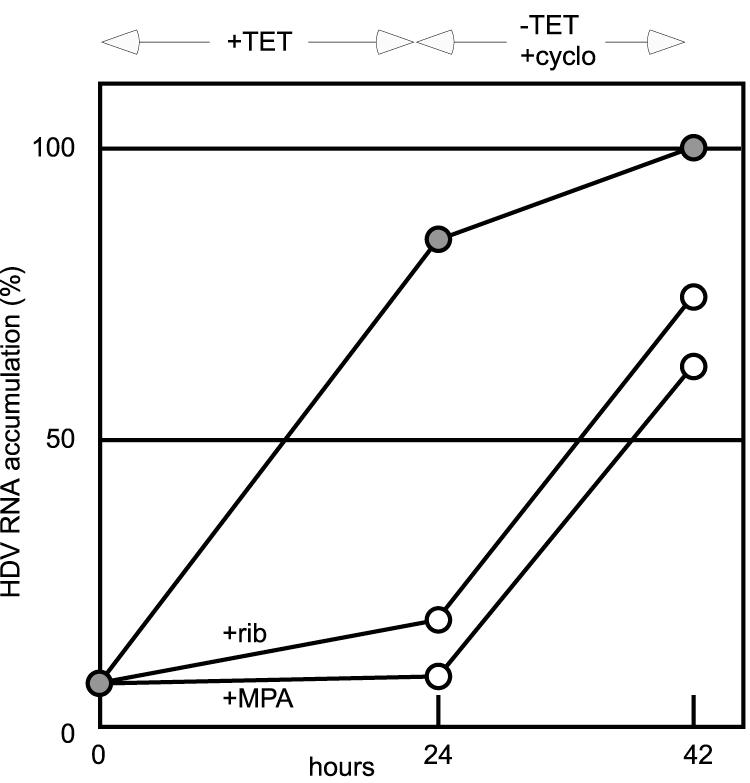
Recovery of the accumulation of processed HDV RNA transcripts following ribavirin treatment. HDV genome replication in cultures of 293-HDV cells was induced and assayed as described in the legends for Fig. 1 and 2. For two of three cultures (open circles), the inhibitors ribavirin (rib) and MPA, as indicated, were used at concentrations of 30 μM and 1 μM, respectively. After 24 h, the medium for each of the three cultures was replaced with medium with no TET and containing 50 μg/ml of cycloheximide (cyclo). After an additional 18 h, total RNA was extracted and Northern analysis was used to quantitate the accumulation of unit-length genomic HDV RNA.
Similar results of genome replication recovery were also obtained with MPA. The latter result has added significance since MPA is not a precursor for incorporation into RNA species. As considered further in the Discussion, the results support a model in which HDV genome replication is reversibly inhibited by GTP pool depletion.
Action of known inhibitors of host RNA polymerases on HDV genome replication.
The above-described results for the reversibility of ribavirin treatment suggested to us that we could extend the scope of our inhibitor studies to include three agents that were already known to inhibit host DNA-directed RNA transcription but whose actions in an experimental system might reveal important aspects of HDV RNA-directed RNA replication. We reasoned that we might obtain results less affected by host cell toxicity if the essential δAg were allowed to accumulate not during but prior to the drug treatment. (This prior expression was needed to separate the effect of the drugs on RNA-directed transcription from their effects on DNA-directed δAg induction and expression.) The reversibility of ribavirin treatment offered us the following strategy. We allowed for 24 h the TET induction and translation of fully functional δAg, in the presence of 30 μM ribavirin, and then removed both the TET and the ribavirin and inhibited the cells for the next 18 h in the presence of 3H[uridine], with or without three drugs. Total RNA was then extracted and assayed, as described in the legends for Fig. 1 and 2, for the accumulation of host and viral RNAs.
Specifically, the three drugs we tested were amanitin, DRB, and actinomycin D, with results summarized in Fig. 5A to C, respectively.
In addition, to test whether or not some artifact might be introduced by the pretreatment with ribavirin, we examined each of the three inhibitors with a second strategy, with results summarized in Fig. 5D to F. In this second strategy, we used 293-δAg cells that contain the TET-inducible δAg but do not contain replicating HDV RNA. These cells were induced with TET for 24 h to allow expression of δAg, and then TET was removed and HDV replication was initiated by transfecting these cells with total RNA that had been extracted from 293-HDV cells undergoing HDV replication (19). (This provided a good source of HDV RNA for efficient initiation of HDV genome replication.) Four hours later, these cells were reseeded into smaller identical cultures, which were incubated for the next 18 h with 3H[uridine] and various concentrations of the three inhibitors. Both prior and new results obtained with each of the three inhibitors are presented below.
(i) Amanitin.
This extremely toxic substance obtained from mushrooms has been used for some time to distinguish the three different host DNA-directed RNA polymerases (12). It has also been used by us and by others to help determine the host polymerase(s) involved in HDV RNA-directed RNA synthesis (21, 29, 31, 33). It is known that, of the three nuclear DNA-directed RNA polymerases, Pol II is the most sensitive (showing inhibition even at a concentration of 1 μg/ml), Pol III 10 times less sensitive, and Pol I resistant to even 100 μg/ml. While there is agreement that the synthesis of HDV mRNA and unit-length genomic RNAs is sensitive to low doses of amanitin, two studies from the same lab report that that even doses of 100 μg/ml do not block the accumulation of unit-length antigenomic RNA (29, 31). This has been interpreted as evidence that Pol I rather than Pol II is used for this transcription (21). With this controversy in mind, we examined the action of amanitin on the ability of HDV RNA accumulation to recover following ribavirin pretreatment.
The results obtained with amanitin are presented in Fig. 5A and D. In both situations, more than 90% of the accumulation of the HDV RNA-directed mRNA species was sensitive to treatment with amanitin at 1 μg/ml. In contrast, the accumulation of the unit-length genomic and antigenomic RNAs was apparently more resistant, and higher doses were needed to obtain comparable inhibition. For reasons we do not understand, this apparent resistance was stronger for the experiment shown in Fig. 5A than for that shown in Fig. 5D. However, as is explained subsequently, further experiments presented in Fig. 6 provided an alternative to the interpretation that such resistance was because the polymerase involved was other than Pol II.
(ii) DRB.
Host DNA-directed Pol II transcription is inhibited by DRB; the mechanism involves p-TEFb, a positive elongation factor that phosphorylates the carboxyl terminal domain of the large subunit of Pol II. DRB inhibits this phosphorylation (44). In contrast, certain in vitro studies suggest that δAg can block the inhibitory effects of DRB on Pol II transcription of both DNA and RNA templates (42, 43). Furthermore, a prior report indicated that, in vivo, DRB action on HDV could not be separated from toxic effects on the host cell (33). The results shown here are consistent with those prior studies. That is, in the two in vivo situations, we found that DRB had no specific effect on the accumulation of HDV RNAs (Fig. 5B and E). At the highest doses tested, an inhibition was observed, but prior to this there was inhibition of the accumulation of host 18S rRNA, implying that the effect on HDV was due to cell toxicity.
(iii) Actinomycin.
At low doses (e.g., 0.04 μg/ml), this drug has been reported to block the accumulation of processed rRNA transcripts (35). Also, with much higher doses and for short periods of time, many RNA viruses undergoing RNA-directed RNA transcription can continue to replicate largely in the absence of host DNA-directed RNA transcription (16). This is thought to be because actinomycin binds to double-stranded DNA but not to double-stranded RNA (17). (Actually, for HDV, we do not know whether the RNA-directed transcription is from a 100% double-stranded RNA template or makes use of the ability of the unit-length circular genomic and antigenomic RNAs to fold, via intramolecular base pairing involving 74% of all nucleotides, to form an unbranched rod-like structure that resembles double-stranded RNA.) It has been reported that HDV transcription and the accumulation of processed HDV RNAs can continue even in the presence of 50 μg/ml of actinomycin (28). The results of our studies are summarized in Fig. 5C and F. Note first that 0.01 μg/ml of actinomycin was more than sufficient to inhibit the accumulation of 18S rRNA. The accumulation of the three HDV RNA species was more resistant, but certainly as little as 1 μg/ml of actinomycin was sufficient to cause major inhibition of both viral and host RNA species. Perhaps others were able to detect continued HDV RNA synthesis in the presence of 50 μg/ml actinomycin because the treatment time was much shorter (28).
We next returned to the issue of how the accumulation of the unit-length genomic and antigenomic RNAs of HDV could be more resistant to amanitin than the HDV mRNA (Fig. 5A and D). We realized that the accumulation of processed HDV RNA species is the consequence not only of RNA-directed RNA transcription but also of the posttranscriptional RNA processing and of the stability of such RNAs. It was also understood that the unit-length RNAs and the mRNA are processed by different methods, and by their different natures, it was likely that these RNAs might have different stabilities. It was also apparent from our earlier studies that the presence or absence of δAg could impact the accumulation of processed unit-length HDV RNAs (24).
To address these differences in the context of amanitin sensitivity, we began by designing the following strategy to compare the processing and stability of two different forms of antigenomic HDV RNA transcripts. As represented in Fig. 6A, we transfected 293TRex cells with a DNA expression vector that would use Pol II to express an antigenomic RNA. The initiation site of this RNA was the same as that for the natural 5′ end of the HDV mRNA. Following this sequence, as expected for a multimeric transcript of antigenomic HDV RNA, was a poly(A) signal, the antigenomic ribozyme, and then a complete unit-length antigenomic RNA sequence. This strategy was based on one used in a previous study of the processing of antigenomic HDV RNA transcripts (34). It can be seen that such an RNA transcript could lead to the accumulation of two different processed HDV RNAs: a polyadenylated mRNA and a unit-length antigenomic RNA. Our initial strategy was to use Northern analyses to measure the molar amounts of accumulation of these two species as a function of the time after transfection.
Three additional modifications need to be noted. (i) The poly(A) signal near the 3′ end of the nascent transcript was inactivated by a 2-nucleotide substitution (34). We did this to increase the proportion of RNA transcripts that would be processed to unit-length RNA species. (ii) The open reading frame near the 3′ end was inactivated by a 2-nucleotide deletion. This modification was made to eliminate this region as a source of δAg. (iii) As described below, the open reading frame near the 5′ end was considered in three formats.
(a) First, if the 5′ ORF was inactivated, we obtained the results shown in Fig. 6B. That is, there was accumulation of mRNA but there were relatively smaller amounts of unit-length antigenomic RNA. The latter result was consistent with our previous findings that accumulation of unit-length RNAs depends upon the presence of δAg (23, 32).
(b) Second, if the 5′ ORF was one that produced a form of delta protein with a small internal deletion that was stably expressed but did not support HDV replication (32), we obtained the results shown in Fig. 6C. In this case, we detected a significant increase in the accumulation of unit-length species, although it should be noted that there were still relatively larger amounts of mRNA.
(c) Third, if this ORF made a fully functional small δAg, which would support HDV replication, we obtained the results shown in Fig. 6D. At day 6, for example, the unit-length antigenomic RNA was 50-fold more abundant than the mRNA. Of course, HDV replication was now providing additional RNA-directed sources of both unit-length antigenomic RNA and mRNA.
It can be seen that for all three formats the relative amounts of mRNA to unit-length antigenomic RNA were different and that, in each case, the ratio changed with time. For example, in the second scenario (Fig. 6C), the ratio at day 1 versus that at day 6 changed 22-fold. Yet, in all of these experiments, the initial template was the same and the same Pol II transcribed it.
We conclude that all of the differences in the accumulation of the HDV mRNA relative to the unit-length antigenomic RNA were due to differences in processing and stability rather than in the polymerase used for transcription.
With this interpretation in mind, we next tested the effect of different concentrations of amanitin on the accumulation of the two processed HDV RNA species. Cells were transfected with the construct described above for Fig. 6C, that is, the one that does not lead to HDV RNA-directed replication. After 24 h, the cells were reseeded and exposed to different concentrations of amanitin for the next 24 h, and then Northern analyses were used to quantitate the mRNA and unit-length antigenomic RNA species. As shown in Fig. 6E, the accumulation of unit-length RNA was up to four times more resistant to amanitin than was the accumulation of the mRNA. However, this difference was not because of different polymerases. That is, we conclude it was due to the drug treatment that exposed differences in the posttranscriptional RNA processing and/or stability of the two RNAs.
DISCUSSION
The experiments presented here have used an experimental system of TET-induced HDV RNA-directed RNA replication (4) to characterize the susceptibility of replication to a range of inhibitors. From the use of internal controls for the ability of treated cells to process and accumulate host RNA species during such treatments, it has in some cases been possible to determine treatment conditions that seem to act specifically on the HDV RNAs. However, this discussion is limited to two important questions. First, how it is that such specificity can be achieved, especially in a situation where the virus is using a host-encoded polymerase? Second, how is it that data from studies using the inhibitor amanitin have led to an interpretation that two different host polymerases are needed for HDV replication (21)?
(i) Ribavirin, a guanosine analog, is well known to have broad-spectrum antiviral activity against a wide range of animal viruses. In cultured cells, it is known to block the replication of positive- and negative-stranded RNA viruses and some DNA viruses (26, 37). It has previously been reported to inhibit HDV replication in cultured primary woodchuck hepatocytes (8). In our studies, the inhibition of HDV replication could be obtained in the absence of detected effects on host RNA metabolism (Fig. 1 and 2A). Similar specific inhibition was also obtained with MPA (Fig. 2B) and the ribavirin prodrug viramidine (Fig. 2C). For the following reasons, we conclude that in our experiments these drugs are acting primarily via depletion of the intracellular GTP pool. (a) Others have shown by using the concentration ranges applied here that ribavirin and MPA have an exact correlation between GTP pool depletion and inhibition of replication of RNA viruses other than HDV (26). (b) While antiviral mechanisms other than the GTP pool depletion have been proposed for ribavirin (15), no such ambiguity exists for MPA. It is a direct inhibitor of IMPDH and it cannot be incorporated into RNA. (c) As we have shown here, inhibition by ribavirin and MPA was not only synergistic but could be obviated by exogenous addition of an excess of guanosine (Fig. 3). That is, the guanosine was acting downstream of the IMPDH block and allowing the formation of GTP.
It could be argued that GTP pool depletion causes a different form of misincorporation, specifically, the misincorporation of CTP, ATP, or UTP at locations requiring GTP. We do not consider this a significant problem in our experimental system, because the DNA-directed mRNA transcribed by Pol II in the presence of either ribavirin or MPA was still able to be translated into δAg, which was apparently fully functional in its ability to subsequently support the accumulation of HDV RNA species (Fig. 4). Nevertheless, there is still the formal possibility that some level of misincorporation, either produced by GTP depletion or even provoked by ribavirin incorporation, could have led to HDV RNAs that were either prematurely terminated or were completed but became replication defective. With other experimental systems, it has been noted that such error-prone replication can reduce infectivity (10, 11, 22). However, in our experimental system, after reversal of the inhibition, we should be able to achieve “recovery” by using new error-free transcription from the replication-competent templates.
The following is our explanation of how such GTP depletion could have a selective effect on the accumulation of viral RNAs relative to the host RNAs. It is based on the fact that the accumulation of viral RNAs depends upon an exponential process. That is, to achieve maximal accumulation, some of the first RNAs produced must in turn become templates for the transcription and processing of more RNAs, etc. This exponential process, which does not apply to the host DNA-directed RNAs, is thus more susceptible to perturbations that are compounded to create a significant decrease in the total RNA that can be accumulated. We would further propose that such interference with an exponential process probably applies to many of the situations where ribavirin and the related compounds can lower the accumulation of viral RNAs achieved for replication as initiated in cultured cells.
Furthermore, it is possible that the explanation given above is a major contributor to the antiviral actions observed in vivo. It is relevant that the in vivo concentrations achieved during ribavirin therapy are about 15 μM (27), which is close to our experimental value of inhibition of HDV by 30 μM (Fig. 2A). It should also be noted that for treatments of chronic infections, such as hepatitis C virus, ribavirin alone has little effect unless accompanied by a second antiviral agent, typically alpha interferon (15).
(ii) The studies of the action of amanitin on the accumulation of processed HDV RNAs presented here may have solved what has been a major source of confusion for those studying HDV replication. As reviewed elsewhere, there is much evidence that Pol II is involved in aspects of HDV RNA-directed RNA synthesis (5). However, it has been reported that in some experimental situations the accumulation of processed unit-length antigenomic RNAs continues after treatments with amanitin at concentrations of 100 μg/ml, a dose much higher than the 1-μg/ml dose typically sufficient to block transcription by Pol II. This has led to the interpretation that a polymerase other than Pol II is needed for the transcription of antigenomic RNA (21, 29, 31). In the present study, we have also found that in some situations the accumulation of processed antigenomic RNA can be somewhat more resistant to amanitin than the accumulation of HDV mRNA. (We also showed that the accumulation of unit-length genomic RNA could also be more resistant [Fig. 5A and D].) However, in further experiments, we were able to separate the transcription of antigenomic RNAs from the subsequent events of posttranscriptional processing and stabilization (Fig. 6). We found that processed unit-length antigenomic RNA continued to accumulate longer than the mRNA. This was true even when the two RNAs were derived from identical precursor pools and transcribed in the same cells and by the same Pol II. These differences in RNA accumulation were not only time dependent but were further altered by the presence and absence of forms of δAg (Fig. 6B to D). Furthermore, this relative difference in accumulation could be accentuated by treatment with increasing concentrations of amanitin (Fig. 6E). In summary, these new studies support the simpler interpretation that the transcription of HDV RNAs is dependent upon just one host polymerase, Pol II. Further studies are needed to determine how this redirection from DNA to RNA templates is achieved.
Acknowledgments
J.T. was supported by grants AI-26522 and CA-06927 from the NIH and by an appropriation from the Commonwealth of Pennsylvania.
J.C. designed and performed all of the experiments. All authors contributed to the analysis and writing of the manuscript. Glenn Rall and William Mason gave valuable comments on the manuscript. Thanks go to Jim Wu at Valeant for providing viramidine and to Michael Roggendorf for discussions regarding his earlier studies of ribavirin treatments.
REFERENCES
- 1.Asselbergs, F. A. M., and P. Grand. 1993. A two-plasmid system for transient expression of cDNAs in primate cells. Anal. Biochem. 209:327-331. [DOI] [PubMed] [Google Scholar]
- 2.Barrera, A., B. Guerra, H. Lee, and R. E. Lanford. 2004. Analysis of host range phenotypes of primate hepadnaviruses by in vitro infections of hepatitis D virus pseudotypes. J. Virol. 78:5233-5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casey, J., P. J. Cote, I. A. Toshkov, C. K. Chu, J. L. Gerin, W. E. Hornbuckle, B. C. Tennant, and B. E. Korba. 2005. Clevudine inhibits hepatitis delta virus viremia: a pilot study of chronically infected woodchucks. Antimicrob. Agents Chemother. 49:4396-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, J., S. O. Gudima, C. Tarn, X. Nie, and J. M. Taylor. 2005. Development of a novel system to study hepatitis delta virus genome replication. J. Virol. 79:8182-8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, J., X. Nie, S. Gudima, and J. Taylor. Replication of the hepatitis delta virus genome. In K. Hefferon (ed.), Recent advances in RNA virus replication, in press. Transworld Research Network, Kerala, India.
- 6.Chao, M., S.-Y. Hsieh, and J. Taylor. 1990. Role of two forms of the hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J. Virol. 64:5066-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, P.-J., G. Kalpana, J. Goldberg, W. Mason, B. Werner, J. Gerin, and J. Taylor. 1986. Structure and replication of the genome of hepatitis δ virus. Proc. Natl. Acad. Sci. USA 83:8774-8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi, S. S., R. Rasshofer, and M. Roggendorf. 1989. Inhibition of hepatitis delta virus RNA replication in primary woodchuck hepatocytes. Antivir. Res. 12:213-222. [DOI] [PubMed] [Google Scholar]
- 9.Crotty, S., and R. Andino. 2002. Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect. 4:1301-1307. [DOI] [PubMed] [Google Scholar]
- 10.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 98:6895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crotty, S., D. Maag, J. J. Arnold, W. Zhong, J. Y. Lau, Z. Hong, R. Andino, and C. E. Cameron. 2000. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6:1375-1379. [DOI] [PubMed] [Google Scholar]
- 12.de Mercoyrol, L., C. Job, and D. Job. 1989. Studies on the inhibition by α-amanitin of single-step addition reactions and productive RNA synthesis catalysed by wheat-germ RNA polymerase II. Biochem. J. 258:165-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farci, P. 2003. Delta hepatitis: an update. J. Hepatol. 39(Suppl. 1):S212-S219. [DOI] [PubMed] [Google Scholar]
- 14.Farci, P., T. Roskams, L. Chessa, G. Peddis, A. P. Mazzoleni, R. Scioscia, G. Serra, M. E. Lai, M. Loy, L. Caruso, V. Desmet, R. H. Purcell, and A. Balestrieri. 2004. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 126:1740-1749. [DOI] [PubMed] [Google Scholar]
- 15.Feld, J. J., and J. H. Hoofnagle. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967-972. [DOI] [PubMed] [Google Scholar]
- 16.Flint, S. J., L. W. Enquist, V. R. Racaniello, and A. M. Skalka. 2004. Principles of virology: molecular biology, pathogenesis, and control of animal viruses, 2nd ed. ASM Press, Washington, D.C.
- 17.Goldberg, I. H., and E. Reich. 1964. Actinomycin inhibition of RNA synthesis directed by DNA. Fed. Proc. 23:958-964. [PubMed] [Google Scholar]
- 18.Graci, J. D., and C. E. Cameron. 2002. Quasispecies, error catastrophe, and the antiviral activity of ribavirin. Virology 298:175-180. [DOI] [PubMed] [Google Scholar]
- 19.Gudima, S. O., J. Chang, and J. M. Taylor. 2005. Reconstitution in cultured cells of replicating HDV RNA from pairs of less than full-length RNAs. RNA 11:90-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilan, Y. M., A. Klein, J. Taylor, and R. Tur-Kaspa. 1992. Resistance of hepatitis delta virus replication to alpha interferon treatment in transfected human cells. J. Infect. Dis. 166:1164-1166. [DOI] [PubMed] [Google Scholar]
- 21.Lai, M. M. 2005. RNA replication without RNA-dependent RNA polymerase: surprises from hepatitis delta virus. J. Virol. 79:7951-7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanford, R. E., D. Chavez, B. Guerra, J. Y. N. Lau, Z. Hong, K. M. Brasky, and B. Beames. 2001. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 75:8074-8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazinski, D. W., and J. M. Taylor. 1994. Expression of hepatitis delta virus RNA deletions: cis and trans requirements for self-cleavage, ligation, and RNA packaging. J. Virol. 68:2879-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazinski, D. W., and J. M. Taylor. 1995. Intracellular cleavage and ligation of hepatitis delta virus genomic RNA: regulation of ribozyme activity by cis-acting sequences and host factors. J. Virol. 69:1190-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazinski, D. W., and J. M. Taylor. 1993. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 67:2672-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leyssen, P., J. Balzarini, E. De Clercq, and J. Neyts. 2005. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 79:1943-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl, K., L. Stahle, A. Bruchfeld, and R. Schvarcz. 2005. High-dose ribavirin in combination with standard dose peginterferon for treatment of patients with chronic hepatitis C. Hepatology 41:275-279. [DOI] [PubMed] [Google Scholar]
- 28.Macnaughton, T. B., and M. M. Lai. 2002. Genomic but not antigenomic hepatitis delta virus RNA is preferentially exported from the nucleus immediately after synthesis and processing. J. Virol. 76:3928-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macnaughton, T. B., S. T. Shi, L. E. Modahl, and M. M. Lai. 2002. Rolling circle replication of hepatitis delta virus RNA is carried out by two different cellular RNA polymerases. J. Virol. 76:3920-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNair, A. N. B., J. Monjardino, D. Cheng, H. C. Thomas, and I. M. Kerr. 1993. Hepatitis delta virus and the interferon system. Prog. Clin. Biol. Res. 382:161-164. [PubMed] [Google Scholar]
- 31.Modahl, L. E., T. B. Macnaughton, N. Zhu, D. L. Johnson, and M. M. C. Lai. 2000. RNA-dependent replication and transcription of hepatitis delta virus RNA involve distinct cellular RNA polymerases. Mol. Cell. Biol. 20:6030-6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moraleda, G., S. Seeholzer, V. Bichko, R. Dunbrack, J. Otto, and J. Taylor. 1999. Unique properties of the large antigen of hepatitis delta virus. J. Virol. 73:7147-7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moraleda, G., and J. Taylor. 2001. Host RNA polymerase requirements for transcription of the human hepatitis delta virus genome. J. Virol. 75:10161-10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nie, X., J. Chang, and J. M. Taylor. 2004. Alternative processing of hepatitis delta virus antigenomic RNA transcripts. J. Virol. 78:4517-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perry, R. P., and D. E. Kelley. 1970. Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J. Cell. Physiol. 76:127-139. [DOI] [PubMed] [Google Scholar]
- 36.Sintchak, M. D., M. A. Fleming, O. Futer, S. A. Raybuck, S. P. Chambers, P. R. Caron, M. A. Murcko, and K. P. Wilson. 1996. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 85:921-930. [DOI] [PubMed] [Google Scholar]
- 37.Smee, D. F., M. Bray, and J. W. Huggins. 2001. Antiviral activity and mode of action studies of ribavirin and mycophenolic acid against orthopoxviruses in vitro. Antivir. Chem. Chemother. 12:327-335. [DOI] [PubMed] [Google Scholar]
- 38.Taylor, J., W. Mason, J. Summers, J. Goldberg, C. Aldrich, L. Coates, J. Gerin, and E. Gowans. 1987. Replication of human hepatitis delta virus in primary cultures of woodchuck hepatocytes. J. Virol. 61:2891-2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor, J. M. 2006. Hepatitis delta virus. Virology 344:71-76. [DOI] [PubMed] [Google Scholar]
- 40.Tine, F., M. Attanasio, F. Russo, and L. Pagliaro. 2005. A decade of trials of interferon-alpha for chronic hepatitis C. A meta-regression analysis. Contemp. Clin. Trials 26:179-210. [DOI] [PubMed] [Google Scholar]
- 41.Wu, J. Z., C. C. Lin, and Z. Hong. 2003. Ribavirin, viramidine and adenosine-deaminase-catalysed drug activation: implication for nucleoside prodrug design. J. Antimicrob. Chemother. 52:543-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi, K., and H. Handa. Hepatitis delta antigen and RNA polymerase II. In H. Handa and S. Yamada (ed.), Hepatitis delta virus, in press. Landes Bioscience, Georgetown, Tex.
- 43.Yamaguchi, Y., J. Filipovska, K. Yano, A. Furuya, N. Inukai, T. Narita, T. Wada, S. Sugimoto, M. M. Konarska, and H. Handa. 2001. Stimulation of RNA polymerase II elongation by hepatitis delta antigen. Science 293:124-127. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi, Y., T. Wada, and H. Handa. 1998. Interplay between positive and negative elongation factors: drawing a new view of DRB. Genes Cells 3:9-15. [DOI] [PubMed] [Google Scholar]