

Abstract
Herpes simplex virus (HSV) glycoprotein heterodimer gE/gI is necessary for virus spread in epithelial and neuronal tissues. Deletion of the relatively large gE cytoplasmic (CT) domain abrogates the ability of gE/gI to mediate HSV spread. The gE CT domain is required for the sorting of gE/gI to the trans-Golgi network (TGN) in early stages of virus infection, and there are several recognizable TGN sorting motifs grouped near the center of this domain. Late in HSV infection, gE/gI, other viral glycoproteins, and enveloped virions redistribute from the TGN to epithelial cell junctions, and the gE CT domain is also required for this process. Without the gE CT domain, newly enveloped virions are directed to apical surfaces instead of to cell junctions. We hypothesized that the gE CT domain promotes virus envelopment into TGN subdomains from which nascent enveloped virions are sorted to cell junctions, a process that enhances cell-to-cell spread. To characterize elements of the gE CT domain involved in intracellular trafficking and cell-to-cell spread, we constructed a panel of truncation mutants. Specifically, these mutants were used to address whether sorting to the TGN and redistribution to cell junctions are necessary, and sufficient, for gE/gI to promote cell-to-cell spread. gE-519, lacking 32 C-terminal residues, localized normally to the TGN early in infection and then trafficked to cell junctions at late times and mediated virus spread. By contrast, mutants gE-495 (lacking 56 C-terminal residues) and gE-470 (lacking 81 residues) accumulated in the TGN but did not traffic to cell junctions and did not mediate cell-to-cell spread. A fourth mutant, gE-448 (lacking most of the CT domain), did not localize to cell junctions and did not mediate virus spread. Therefore, the capacity of gE/gI to promote cell-cell spread requires early localization to the TGN, but this is not sufficient for virus spread. Additionally, gE CT sequences between residues 495 and 519, which contain no obvious cell sorting motifs, are required to promote gE/gI traffic to cell junctions and cell-to-cell spread.
Herpes simplex virus (HSV) commonly infects mucosal and ocular epithelial tissues, causing oral and genital lesions. During primary infection in epithelial tissues, HSV enters sensory and autonomic neurons where the virus replicates and can establish latency. Periodic reactivation in neurons leads to transient replication and spread along neuronal axons, leading to the reinfection of epithelial tissues. This cycle of HSV spread in epithelial tissues, entry into neurons, spread to sensory ganglia, and return to epithelial tissues involves directed intracellular transport to specific cell surfaces and extremely rapid spread between cells (reviewed in reference 23). As evidence of the speed of this process, HSV can spread from a single infected cell to over 250 cells in the cornea within 48 h (34). HSV is largely cell associated and spreads across cell junctions and resists the effects of high concentrations of virus-neutralizing antibodies. Titers of antibodies do not predict the severity of disease (8). We demonstrated that HSV cell-to-cell spread in cultured epithelial cells involves a process by which progeny virions are targeted specifically to epithelial cell junctions (24). By virtue of being sorted to epithelial cell junctions there is preferential movement of virus between cells rather than into extracellular fluids. There is also evidence for the directed spread of HSV in the nervous system. HSV particles are preferentially sorted into sensory axons and move in the direction of epithelial tissues rather than into dendrites and toward the central nervous system (40). Once HSV reaches epithelial and neuronal cell junctions, virus particles can spread to adjacent cells through interactions with cellular receptors that preferentially accumulate at these junctions (25, 36).
HSV cell-to-cell spread requires three viral glycoproteins, gD, gB, and gH/gL, that are also required for the related process by which extracellular virions enter cells (3, 16, 27, 37). However, in contrast to these envelope glycoproteins, HSV glycoprotein gE/gI promotes cell-to-cell spread without any obvious role in the entry of extracellular particles (11, 12). HSV gE/gI complexes form quickly, and most gE is bound to gI and vice versa, consistent with the view that gE/gI functions entirely or primarily as a heterodimer in HSV-infected cells (2, 18, 21, 22), despite suggestions that gE can function apart from gI (38, 45). HSV gE- or gI-null mutants display markedly reduced spread between cultured epithelial and neuronal cells and in epithelial and neuronal tissues (11-13, 34). For example, HSV gE-null mutants spread to between 4 and 6% of the epithelial cells compared to wild-type HSV infection of the cornea (34), and the spread of gE−/gI− double mutants is reduced to 2% (D. C. Johnson, unpublished data). Additionally, gE mutants display markedly reduced spread between neurons in the retina as well as from the retina to retinorecipient regions of the brain (12).
HSV gE/gI appears to participate in at least two processes that promote cell-to-cell spread. The extracellular (ET) domains of gE/gI are necessary for spread, probably to promote virus movement across epithelial cell junctions (7, 24). This was based on observations that gE/gI accumulates at cell junctions late in infection, apparently tethered there in a manner similar to that of various cell adhesion molecules (30, 48), and that when expressed in trans, gE/gI can interfere with cell-to-cell spread (7). Moreover, small insertion mutations in the gE ET domain that do not reduce cell surface expression, incorporation into virions, or complex formation with gI can abolish the capacity of gE/gI to promote cell-to-cell spread (35). We hypothesized that gE/gI can promote cell-to-cell spread by binding receptors that are selectively expressed at cell junctions. However, the relatively large cytoplasmic (CT) domain of gE is involved in a second process that promotes cell-to-cell spread. Mutants lacking the gE CT domain behave much like gE-null mutants, with dramatically reduced spread between cultured epithelial cells and within the corneal epithelium (35, 48).
The gE CT domain promotes extensive accumulation of gE/gI in the trans-Golgi network (TGN) in HSV-infected cells at early times of infection and when gE/gI is expressed by transfection or with virus vectors (1, 13, 30, 48). Swapping the gE CT domain in place of the gD CT domain caused gD to accumulate in the TGN, but this was not the case with the gI CT domain (30). The TGN and endosomes serve as intracellular compartments where secondary envelopment of HSV occurs (9, 14, 39, 43, 47, 49). HSV gE/gI and gD serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm; nucleocapsids accumulate in massive quantities in the cytoplasm when both gE and gD are deleted (14). Thus, gE/gI both accumulates extensively in the TGN at times when virus particles are being assembled and, in conjunction with gD, promotes envelopment there. Related to these observations, the gE CT domain is required for the specific sorting of enveloped virions formed in the TGN to epithelial cell junctions (24). When gE, or just the gE CT domain, was deleted, virions trafficked to apical surfaces rather than to lateral cell junctions. These observations supported a working model in which gE/gI affects intracellular sorting decisions promoting HSV envelopment into subdomains of the TGN from which cargo, in this case, virus particles, is transported specifically to lateral cell surfaces (24).
Coincident with the relocalization of HSV virions from sites of envelopment in the TGN to lateral cell surfaces late in infection, gE/gI is redistributed from the TGN to epithelial cell junctions (30, 48, 49). This process appears to involve global rearrangement or redistribution of the TGN because HSV gB and host TGN proteins (TGN46 and carboxypeptidase D) also moved to the plasma membrane (49). This redistribution was specifically to lateral cell surfaces, not to apical surfaces, and did not require the assembly of enveloped virions in the TGN. Apparently, there are viral proteins that function to redistribute enveloped virus particles from the TGN to cell junctions, and other TGN components follow.
In polarized epithelial cells, the TGN and endosomes are the major compartments in which membrane and secreted proteins are sorted to either basolateral or apical cell surfaces (reviewed in references 5, 28, and 31). Decisions are made as to whether cellular proteins are sequestered into subdomains of the TGN that then give rise to vesicles, frequently coated with clathrin as well as other proteins, which are directed to basolateral or apical cell surfaces. The cytoplasmic domains of cargo proteins interact with cytosolic adaptor molecules, e.g., the clathrin adaptors AP-1 and AP-3 and PACS-1, that promote the coating of vesicles and direct transport (15, 44). Clathrin adaptors recognize tyrosine (YXXØ, where Ø is a larger hydrophobic amino acid) and dileucine motifs in the cytoplasmic domains of membrane proteins, among other signals. We previously demonstrated that a clathrin adaptor, AP-1B, selectively expressed in polarized epithelial cells functions to sort alphaherpesvirus to cell junctions (24).
The gE CT domain is 106 amino acids in length and contains several obvious TGN sorting motifs, and these are also found in pseudorabies virus (PRV) and varicella-zoster virus homologues (20, 26, 28, 29, 33, 41). The HSV gE CT domain contains two tyrosine motifs (YXXØ) as well as an acidic cluster of amino acids, which is phosphorylated (48). Tyrosine motifs bind the μ1 and μ2 components of AP-1 and AP-2 clathrin adaptors to promote incorporation into clathrin-coated transport vesicles (17). The HSV gI CT domain also contains a dileucine motif at the C terminus that likely binds the β1 component of AP-1 clathrin adaptors. We demonstrated that an epithelium-specific component of the AP-1 complex, μ1B, was important for directing PRV particles to lateral cell surfaces (24). The gE CT domain also interacts with the acidic cluster-binding protein PACS-1 (C. Crump and G. Thomas, personal communication) that directs molecules to the TGN (17, 44). TGN sorting sequences, e.g., tyrosine motifs, can also function in the endocytosis of HSV gE/gI from the cell surface (1, 30). However, the majority of HSV gE/gI may accumulate in the TGN through internal recycling loops rather than by endocytosis, as with other TGN proteins (17, 44).
In this study, we constructed truncation mutants of the gE CT domain. These mutants allowed us to address whether the sorting of gE/gI to the TGN and redistribution to cell junctions was necessary, and sufficient, for gE/gI to promote cell-to-cell spread. A mutant that did not accumulate in the TGN was unable to mediate cell-to-cell spread. In addition, mutants that localized to the TGN but that could not redistribute to cell junctions were also defective for virus spread. Therefore, both processes are necessary for cell-to-cell spread of different sequences in the gE ET domain responsible for determining TGN localization and redistribution to cell junctions.
MATERIALS AND METHODS
Cells and viruses.
HaCaT cells and Vero cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). ARPE-19 cells were grown in DMEM/F-12 medium containing 10% FBS. F-BAC, an HSV type 1 (HSV-1) strain F derivative with a bacterial artificial chromosome (BAC) inserted into the tk gene (19), and F-BAC mutants were propagated and titered on Vero cells.
Mutagenesis of the gE gene.
A plasmid, pUC US7/8PA gE-448, in which all but three residues the gE CT domain were removed has been described previously (35). Plasmids containing additional gE CT domain truncations were constructed by using PCR with a template involving plasmid pUC-US7/8 (48). Specifically, two stop codons and an StuI restriction site were inserted after residue 470, 495, or 519. In all cases, the sense oligonucleotide GCAGGCGGCCTCCGTCAATCTG, corresponding to codons 340 to 346, was employed. To construct gE-470, the antisense oligonucleotide TTTAGGCCTCTATTAGCTGTCGGCCACGCGAATGTA, corresponding to codons 463 to 469 preceding two stop codons and a StuI site, was used. For gE-495, the antisense oligonucleotide CAAAGGCCTTTATTATCTCTCCGGGGGGGCCAG, corresponding to codons 489 to 494 preceding two stop codons and an StuI site, was used. The antisense oligonucleotide used to construct gE-519 was CAAAGGCCTTTATTAACGGGGGTATACAGACGG, corresponding to codons 513 to 518 preceding two stop codons and an StuI site. Following PCR, the DNA product was digested with restriction endonucleases MluI and StuI, and bands of the correct size were purified and ligated into pUC-US7/8PA that had been digested with MluI and StuI. The plasmids that produced pUC-US7/8PA gE-470, pUC-US7/8PA gE-495, and pUC-US7/8 gE-519 were sequenced and then digested with restriction endonucleases PacsI and AscI, and the mutated US8 genes were purified and ligated into plasmid pSTPA, a plasmid used to shuttle sequences into the HSV BAC (35), creating the plasmids pSTPA gE-470, pSTPA gE-495, and pSTPA gE-519. To construct a gE-null mutant, the following oligonucleotides were employed: (i) ATGGATCGCGGGGCGGTGGTGGGGTTTCTTATTGTGTAGGCTGGGAGCTGCTTTC, corresponding to codons 1 to 9 of the US8 gene followed by a stop codon and the priming site corresponding to plasmid pKD4 (10), and (ii) TTACCAGAAGACGGACGAATCGGAGGACATATGAATATCCTCCTTAG, corresponding to codons 543 to 551 of the gE gene followed by the reverse priming site for pKD4. PCR using these two oligonucleotides generated a kanamycin gene cassette flanked by 36 bp of the gE gene (denoted gE/kanamycin cassette) that was used to recombine the mutation directly into F-BAC.
Construction of F-BAC mutants.
Plasmids pSTPA gE-470, pSTPA gE-495, and pSTPA gE-519 contain a temperature-sensitive origin of replication, the SacB gene, encoding sucrose sensitivity, and a kanamycin resistance gene (35) and were used to shuttle recombinant forms of gE into the HSV BAC using a modified protocol based on that described previously by Horsburgh et al. (19). In brief, shuttle plasmids were electroporated into RR1 bacteria containing the HSV-1 BAC (19), and transformants were incubated and restreaked onto plates containing chloramphenicol (20 μg/ml) and kanamycin (30 μg/ml) at 43°C. Following this procedure, individual colonies were streaked onto chloramphenicol- and sucrose (5%, wt/vol)-containing plates and incubated at 30°C. Colonies that were replica plated onto both kanamycin and chloramphenicol plates to confirm the loss of the shuttle plasmid and the presence of mutations were confirmed by PCR amplification of appropriate regions and restriction analyses. Mutant F-BACΔgE, lacking gE coding sequences, was created using a protocol modified from that described previously by Datsenko and Wanner (10). Briefly, pKD46, a plasmid encoding ampicillin resistance and red recombinase, was transferred into RR1 bacteria containing F-BAC. F-BAC- and pKD46-containing bacteria were electroporated with the gE/kanamycin gene cassette, 1 ml of SOC medium (0.5% yeast extract, 2.0% tryptone, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 20 mM MgSO4, 20 mM glucose) was added for 1 h, and chloramphenicol (20 μg/ml) was then added and incubated at 25°C overnight. The following day, the transformation mixture was plated onto chloramphenicol/kanamycin plates and incubated overnight at 37°C. Recombinant colonies were confirmed by screening for kanamycin resistance and sensitivity to ampicillin and by PCR and restriction analysis. Plasmid pCP20, which encodes ampicillin resistance, and FLP recombinase were introduced into bacteria containing F-BAC. Transformants were selected at 30°C on chloramphenicol (20 μg/ml) and ampicillin (100 μg/ml) plates, and resistant colonies were streaked onto chloramphenicol plates and grown at 43°C to induce FLP recombinase genes. Colonies were replica plated onto chloramphenicol, kanamycin, and ampicillin plates, and colonies that were Chlr Amps Kans were analyzed by PCR and restriction analyses to confirm the loss of both the US8 gene and the kanamycin cassette. F-BAC that contained mutant gE was sequenced across, upstream, and downstream of the mutation site.
Derivation of HSV from BAC DNA.
Bacteria containing F-BAC were grown in LB medium containing chloramphenicol, and BAC DNA was purified using alkaline lysis, isopropanol precipitation, and phenol-chloroform extraction. Vero cells in Opti-MEM medium lacking serum were transfected with HSV-BAC DNA using Lipofectamine (Invitrogen, Carlsbad, CA), and cytopathic effects of viruses were observed after 4 to 5 days.
Radiolabeling of cells, virus purification, and immunoprecipitation of gE.
Vero cells were infected with HSV using 10 PFU/cell and labeled from 6 to 9 h postinfection with [35S]methionine/cysteine (75 μCi/ml) in medium lacking methionine and cysteine. Radiolabeled cells were lysed in 1% NP-40-0.5% deoxycholate buffer containing 2 mg/ml bovine serum albumin and 1.0 mM phenylmethylsulfonyl fluoride, and gE/gI, gD, or gB was immunoprecipitated with monoclonal antibody (MAb) 3114 (anti-gE), DL6 (anti-gD), or 15βB2 (anti-gB) as described previously (48). Radiolabeled HSV-1 particles were prepared from cell culture supernatants harvested after 18 h by centrifugation through 30% sucrose at 75,000 × g for 1 h in a Beckman SW41 rotor. Virus pellets were disrupted in buffer containing 2% sodium dodecyl sulfate and subjected to electrophoresis and Western blotting using anti-gE MAb 3114.
Cell-to-cell spread in cultured epithelial cells.
Spread of F-BAC mutants in cultured HaCaT and ARPE-19 cells was determined as described previously (48). Briefly, cells were infected with a low multiplicity of virus, and after 2 h, the virus was removed and cells were overlaid with medium containing 0.4% human gamma globulin for 72 h. The cells were fixed in phosphate-buffered saline (PBS) containing 4% paraformaldehyde for 20 min, washed, permeabilized with 0.2% Triton X-100 in PBS for 10 min, and then stained with rabbit anti-HSV-1 serum (Dako, Copenhagen, Denmark) and donkey anti-rabbit immunoglobulin G (IgG) antibodies conjugated with horseradish peroxidase (Amersham, Arlington, IL). Plaques were visualized after the addition of peroxidase substrate 3,3′-diaminobenzidine hydrochloride (Sigma). The area of 10 plaques was determined by using NIH Image software.
Single-step HSV replication.
HaCaT cells were infected with HSV using 10 PFU/cell. After 2 h, virus was removed, and cells were washed for 1 min with 0.1 M Na citrate, pH 3.0, and then washed and overlaid with medium containing 1% FBS. At various times, the cells were scraped from dishes and pelleted and either the cells and medium or medium alone was frozen at −70°C and subsequently thawed and sonicated, and virus was then titered using Vero cells.
Immunofluorescence microscopy.
HaCaT cells or ARPE-19 cells growing on glass coverslips were infected with HSV using 10 PFU/cell, and after 6 h or 11.5 h, the cells were washed and fixed in PBS containing 4% paraformaldehyde for 20 min. Cells were then washed in PBS, permeabilized with 0.2% Triton X-100 in PBS for 20 min, and washed twice with PBS containing 0.02% Tween 20 (PBS-T) before incubation with PBS-T containing 2% normal goat serum for 1 h. Cells were incubated with sheep anti-TGN46 (Serotec) or rabbit anti-β-catenin (Sigma) antibodies simultaneously with anti-gE MAb 3114 for 2 h, washed three times with PBS-T, and incubated with Cy5-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) or with Alexa-594 donkey anti-sheep IgG simultaneously with Alexa-488 goat anti-mouse IgG (Molecular Probes, Eugene OR). The cells were washed, mounted using ProLong (Molecular Probes, Eugene, OR), and photographed using a Nikon TE200 inverted fluorescent confocal microscope with an Applied Precision Deltavision wide-field image restoration system, and images were processed by deconvolution software. In the case of Cy5-conjugated secondary antibodies, the blue signal was converted to a red signal.
Binding of IgG-coated RBC to HSV-infected cells.
Sheep red blood cells (RBC) were labeled with 51Cr (100 to 150 μCi/ml) and then incubated with rabbit anti-sheep RBC IgG, as described previously (18). HaCaT cells infected with HSV for 14 h were incubated with saturating amounts of IgG-coated RBC for 2 h at 37°C in DMEM with 1% FBS. The cells were incubated for 10 to 15 min on ice, washed three to four times with PBS containing 1% FBS, and then lysed in NP-40-deoxycholate buffer, and the radioactivity was measured.
RESULTS
Construction of HSV-1 gE CT domain mutants.
The HSV gE CT domain affects intracellular trafficking of gE/gI, enhances the movement of virions to epithelial cell junctions, and promotes virus spread (11-13, 24, 48). Figure 1 depicts the gE CT domain showing identifiable TGN sorting motifs including two tyrosine motifs at residues 463 and 472, two serine-rich residues at positions 476 and 477 that are phosphorylated (48), and a cluster of acidic residues between residues 478 and 484 that likely interacts with PACS-1 (C. Crump and G. Thomas, personal communication).
FIG. 1.

Map of the gE CT domain and truncation mutants. The HSV-1 gE CT domain is 106 residues and C terminal to the 25-amino-acid transmembrane (TM) domain, which ends at amino acid 445. There are three arginine (R) residues next the transmembrane domain; two tyrosine motifs (YXXØ) at positions 463 to 466 and 472 to 475; three serines that are phosphorylated at residues 476, 477, and 503; and a cluster of acidic residues located between residues 476 and 484. Mutations in the CT domain were constructed by inserting two tandem stop codons in place of residue 519, 495, 470, or 448. The numbering of gE includes the signal sequence and is based on the previously published sequence of HSV-1 strain 17 gE (29), whereas HSV-1 strain F gE (used here) contains two additional residues in the ET domain (48). Wt, wild type.
To characterize regions of the gE CT domain that are important for intracellular sorting and cell-to-cell spread, we truncated the CT domain by inserting tandem stop codons. Mutations were transferred into the HSV genome using a BAC copy of HSV-1 strain F (F-BAC) (19). In mutant gE-448, two stop codons were inserted into the gE CT domain C terminal to three arginine residues that are adjacent to the membrane so that the vast majority of the CT domain was removed (Fig. 1). Mutant gE-470 has two stop codons replacing residue 470 so that only the membrane-proximal YXXØ motif is present and all potential phosphorylation sites are removed. In gE-495, both tyrosine motifs, the acidic cluster, two of three phosphorylation sites, and an additional 11 amino acids are present. Mutant gE-519 contains all but the final 32 amino acids of the gE CT. F-BACΔgE is an additional mutant lacking all gE coding sequences constructed using the BAC system.
Expression of mutant gE proteins and incorporation into virions.
To characterize the expression of gE mutants, Vero cells were infected and then radiolabeled with [35S]methionine/cysteine. Cell extracts were denatured to disrupt the gE/gI complex, and gE was immunoprecipitated using MAb 3114 and subjected to electrophoresis and autoradiography. F-BAC expressed immature (66-kDa) and mature (80-kDa) forms of gE as observed previously (35). Mutants gE-519, gE-495, gE-470, and gE-448 expressed immature and mature gE molecules of the sizes predicted (Fig. 2A). As expected, cells infected with F-BACΔgE expressed no gE protein. Normal amounts of gB and gD proteins were expressed by all mutants (Fig. 2B and C). As with other mutants lacking all gE CT domain sequences (35, 48), the present gE mutants complexed normally with gI as measured by immunoprecipitation with MAb 3063 that requires an epitope produced only in the gE/gI complex (data not shown).
FIG. 2.

Analysis of mutant gE expression and incorporation into virions. Vero cells were infected with F-BAC (wild type), F-BACΔgE (gE null), gE-448, gE 470, gE-495, or gE-519 for 9 h and then radiolabeled with [35S]methionine/cysteine for 3 h. Glycoproteins gE (A), gB (B), and gD (C) were immunoprecipitated from cell extracts using MAb 3114, MAb 15βB2, and MAb DL6, respectively, and subjected to electrophoresis. (D) HaCaT cells were infected with various viruses, and cell culture supernatants were collected at 16 h and centrifuged through a sucrose cushion for 1 h. Virus pellets were solubilized and subjected to electrophoresis, and gE was detected by Western blotting with MAb 3114. A nonspecific protein was detected even in the absence of gE (F-BACΔgE).
To ensure that mutant gE proteins were incorporated into the virion envelope, HaCaT cells were infected with each virus, cell culture supernatants were harvested, and virions were partially purified by centrifugation through a sucrose cushion. Pelleted virus particles were examined by Western blotting using anti-gE MAb 3114. In Fig. 2D, bands corresponding to the size of mature gE, as appropriate for each construct, were observed. In addition to gE, there was a cross-reacting protein of approximately 72 kDa present in all lanes, including F-BACΔgE. These results demonstrated that cells infected by these mutant viruses produced the appropriate gE molecules that were processed to mature forms and incorporated into virion envelopes.
Cell-to-cell spread of HSV-1 gE CT domain mutants.
HSV-1 mutants lacking either gE or just the gE CT domain form plaques that contain 15 to 20% of the number of epithelial cells compared with plaques formed by wild-type HSV-1 (35, 48). This was observed with HaCaT cells, a human keratinocyte cell line that mimics the cells infected in human mucosa, as well as with ARPE-19 cells, a human retinal epithelial cell line that also forms extensive cell junctions. Mutants F-BACΔgE, gE-448, gE-470, and gE-495 all formed plaques that encompassed 16 to 18% of the number of HaCaT cells compared with plaques formed by F-BAC (Fig. 3A). By contrast, F-BAC gE-519 formed large plaques that were similar to plaques produced by wild-type F-BAC (Fig. 3A). Similar results were obtained with ARPE-19 cells (Fig. 3B). With both epithelial cell lines, there was a distinct division between large plaques formed by gE-519 and wild-type HSV-1 and small plaques formed by gE-448, gE-470, and gE-495 and the gE-null mutant. Therefore, truncation of the C-terminal 56 residues of gE, a region that does not contain obvious TGN sorting motifs in mutant gE-495, abolished the capacity of gE/gI to mediate cell-to-cell spread, yet the removal of 32 residues in mutant gE-519 did not.
FIG. 3.

Cell-to-cell spread of HSV gE CT domain mutants. Human HaCaT keratinocytes (A) or ARPE-19 retinal epithelial cells (B) were infected with F-BAC, F-BACΔgE, gE-448, gE-470, gE-495, or gE-519 using a low multiplicity of infection (30 to 100 PFU/35-mm dish). After 72 h, cells were stained with anti-HSV polyclonal antibodies and peroxidase-conjugated secondary antibodies. Ten plaques were photographed, and NIH Image software was used to calculate plaque areas. The numbers shown in each panel are the average areas ± standard deviations for each set of plaques compared to F-BAC plaques arbitrarily set at 100.
Replication of gE CT domain mutants.
To ensure that the defects observed in plaque formation by gE CT domain mutants were not due to defects in virus replication, we characterized the production of infectious virus and movement into extracellular compartments. At various times after infection, total virus (cells and medium combined) or virus in cell culture supernatants was harvested from HaCaT cells and titered on Vero cells. All five mutants produced similar total amounts of progeny virus compared to that of F-BAC (Fig. 4A), consistent with the conclusion that replication was not altered. However, F-BACΔgE and F-BAC gE-448 produced significantly more virus in cell culture supernatants at early times of infection (Fig. 4B), as has previously been reported for gE-null and CT domain mutants (24). Specifically, there was ≈45-fold more F-BAC gE-448 and F-BACΔgE in the media compared with wild-type HSV 10 h after infection. Mutants gE-470 and gE-519 did not display increased virions in growth medium. Note that standard deviations were uniformly very small, so that in most cases, error bars were covered by the symbols (Fig. 4). At late times of infection, e.g., after 14 h, the differences in the quantities of virus in cell culture supernatants disappeared, likely because cell junctions were blown apart and apical and basolateral compartments were mixed. Coupled with previous observations (24), we concluded that that virions produced by a gE mutant lacking the entire CT domain (gE-448) were missorted toward apical cell surfaces, whereas gE-470, gE-495, and gE-519 were not.
FIG. 4.

Replication of F-BAC mutants. HaCaT cells were infected with F-BAC, F-BACΔgE, gE-448, gE-470, gE-495, or gE-519 using 5 PFU/cell for 2 h, and extracellular virus was then inactivated by washing the cells briefly with 0.1 M Na citrate, pH 3.0. At various times, the combined cells and medium (A) or medium alone (B) was harvested, frozen, and sonicated, and HSV-1 was titered on Vero cells by plaque titration. The standard deviations are shown as bars and can be seen in a few cases, but for most points, these bars are small and are covered by the symbols. WT, wild type.
Accumulation of gE/gI in the TGN.
gE/gI accumulates predominately in the TGN at early times after HSV-1 infection (6 h) (13, 30, 48, 49). gE/gI TGN localization appears to be important for virus assembly and as a first step towards the selective sorting of enveloped particles to cell junctions, which promotes cell-to-cell spread. To examine whether gE CT domain mutants accumulated in the TGN at early times, confocal microscopy was used to compare gE/gI localization to TGN46, a cellular component of the TGN (20). In HaCaT cells, wild-type gE/gI (F-BAC) (Fig. 5A, green) was found extensively in a perinuclear location colocalizing with TGN46 (red) 6 h after infection. Similarly, gE/gI was predominately perinuclear and colocalized with TGN46 in cells infected with mutants gE-519, gE-495, and gE-470 (Fig. 5A). With wild-type HSV and mutants gE-519, gE-495, and gE-470, there were also smaller, more punctuate gE/gI vesicles that were nearer the plasma membrane, and a fraction of these did not contain TGN46. Still, the majority of gE/gI was present in the TGN. By contrast, gE/gI expressed in gE-448-infected cells was significantly different. There was little perinuclear accumulation, most cytoplasmic vesicles containing gE/gI were more peripheral, and few vesicles contained TGN46 and gE/gI localized to plasma membranes, both lateral and apical (Fig. 5A).
FIG.5.


Accumulation of gE/gI in the TGN. HaCaT cells (A) or ARPE-19 cells (B) were infected with F-BAC, F-BACΔgE, gE-448, gE-470, gE-495, or gE-519. After 6 h, the cells were fixed, permeabilized, and then stained with MAb 3114 (anti-gE [green]) and stained simultaneously with sheep anti-TGN46 (red). They were then washed and stained with secondary antibodies, Alexa 488-conjugated goat anti-mouse IgG and Alexa 594-conjugated donkey anti-sheep IgG, and characterized by confocal microscopy.
These results were extended to include ARPE-19 cells. Again, wild-type gE/gI and that of mutants gE-519, gE-495, and gE-470 primarily accumulated in a perinuclear location colocalizing with TGN46 at 6 h (Fig. 5B). There was also a smaller fraction of gE/gI that was found in more peripheral vesicles, some that did not stain with TGN46 antibodies. As in HaCaT cells, gE/gI produced in gE-448-infected cells was found in vesicles throughout the cytoplasm, which largely did not contain TGN46, as well as on lateral and apical cell surfaces. We concluded that mutants gE-519, gE-495, and gE-470 largely retain the ability to localize to the TGN. By contrast, gE-448, lacking the majority of the CT domain, did not accumulate in the TGN.
Redistribution of gE/gI to lateral cell junctions.
gE/gI accumulates at lateral cell surfaces colocalizing with the cell junction marker β-catenin late in HSV infection (30, 48, 49). We sought to determine if gE/gI redistributed to lateral cell junctions when the gE CT domain was truncated and whether this correlated with cell-to-cell spread. In HaCaT cells infected with F-BAC and mutant gE-519, gE/gI largely accumulated at cell junctions substantially colocalizing with β-catenin (Fig. 6A). However, in cells infected with gE-495 and gE-470, gE/gI was much less extensively redistributed to cell junctions and instead was found largely in cytoplasmic vesicles. A fraction of these cytoplasmic vesicles were just under lateral plasma membranes adjacent to, but not overlapping with, β-catenin (Fig. 6A). By contrast, gE/gI accumulated at cell junctions and was also present in vesicles distributed throughout the cytoplasm in cells infected with gE-448 (Fig. 6A). We previously reported that a mutant lacking the gE CT domain was present on apical surfaces by constructing z-axis confocal images (48), and a similar phenotype was observed with this mutant (data not shown).
FIG.6.


Redistribution of gE/gI to lateral junctions. HaCaT cells (A) or ARPE-19 cells (B) were infected with F-BAC, F-BACΔgE, gE-448, gE-470, gE-495, or gE-519 for 11.5 h. The cells were fixed, permeabilized, and then stained with MAb 3114 (anti-gE [green]) and stained simultaneously with rabbit anti-β-catenin (red) antibodies. Cells were then washed and stained with secondary antibodies, Alexa 488-conjugated goat anti-mouse IgG and Cy5-conjugated goat anti-rabbit IgG.
In ARPE-19 cells, wild-type gE and gE-519 were again predominately at cell junctions, although there were also cytoplasmic vesicles containing gE/gI (Fig. 6B). We have found that gE/gI generally does not traffic as extensively to cell junctions in ARPE-19 cells compared with HaCaT cells. In ARPE-19 cells infected with F-BAC gE-495 and gE-470, gE/gI accumulated in tight perinuclear bundles and was rarely found at cell junctions. Interestingly, there was substantial β-catenin present in the cytoplasmic vesicles that contained gE/gI in these cells (Fig. 6B). Again, the gE/gI produced in gE-448-infected cells was found on all cell surfaces and in membrane vesicles throughout the cytoplasm, while β-catenin remained at cell junctions (Fig. 6B). We concluded that gE/gI produced in gE-519-infected cells redistributes to cell junctions, while gE/gI produced in gE-495 and gE-470-infected cells does not accumulate at junctions and instead remains in cytoplasmic vesicles.
Distribution of gE CT domain mutants onto apical surfaces.
HSV gE/gI binds the Fc domains of IgG on the surfaces of HSV-infected cells (2, 21, 22). We recently reported that epithelial cells infected with HSV mutants lacking the CT domain bound approximately fivefold more IgG-coated sheep RBC than cells infected with wild-type HSV-1 (35). We concluded that the removal of the gE CT domain promotes increased traffic to apical cell surfaces instead of to lateral cell surfaces and produces increased IgG binding at apical surfaces. Thus, this increased Fc receptor activity is a good quantitative measure of mislocalization of gE/gI to apical surfaces. We examined the binding of 51Cr-labeled IgG-coated RBC to cells infected with gE CT domain mutants. HaCaT cells infected with gE-448 exhibited 430% increased binding of IgG-coated RBC compared with cells infected with wild-type HSV F-BAC (Fig. 7). Cells infected with mutants gE-470, gE-495, and gE-519 all bound IgG-coated RBC in a manner similar to that of cells infected with F-BAC. Therefore, gE-519, gE-495, and gE-470 are not extensively present on apical cell surfaces, confirming the results of confocal experiments.
FIG. 7.
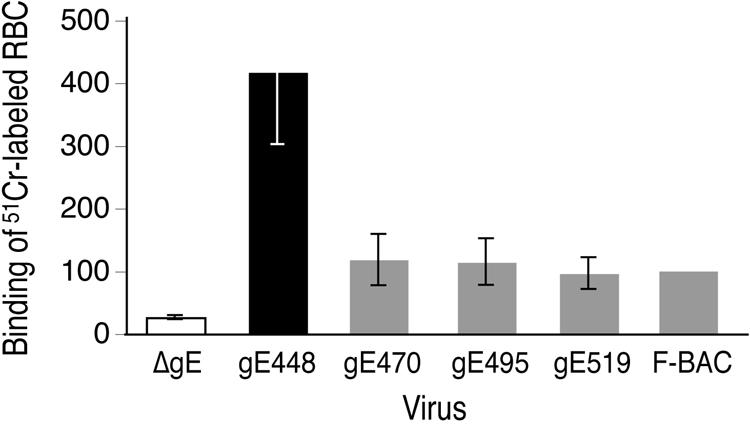
Binding of IgG-coated RBC to cells infected with gE CT domain mutants. HaCaT cells were infected with F-BAC, F-BACΔgE, gE-448, gE-470, gE-495, and gE-519 using 5 PFU/cell for 10 h. The cells were incubated with 51Cr-labeled, IgG-coated RBC for 2 h at 37°C and then washed, and total radioactivity bound to cells was determined. The amount of radioactivity bound by F-BAC-infected cells was arbitrarily set at 100, and triplicate wells infected with F-BACΔgE, gE-448, gE-470, gE-495, and gE-519 were compared to this value.
DISCUSSION
Fundamental to the correct subcellular localization of HSV proteins and subsequent assembly, directed egress of virions, and cell-to-cell spread is the virus' capacity to usurp elements of the host cell trafficking systems. HSV proteins are sorted to the TGN for assembly, and virions are then directed to cell junctions, processes that increase the spread of virus to neighboring cells (24, 48). Most studies of epithelial cell-to-cell spread have involved cultured cells. However, characterization of HSV spread from sensory neurons into the corneal epithelium produced evidence for the directed transport of virus. HSV particles moved specifically onto apical surfaces of less-differentiated, basal epithelial cells spreading toward the surface of the cornea, and after reaching squamous epithelial cells, the virus then spread laterally (32). In neurons, similar or related sorting decisions that determine whether virions transit down axons, remain in neuron cell bodies, or move into dendrites are made. HSV and PRV gE/gI mutants spread poorly between cultured epithelial and neuronal cells and within corneal and neuronal tissues in vivo, and it appears that gE/gI functions to direct HSV particles toward other cells or down neuronal axons (6, 12, 24, 35, 42, 46).
HSV gE/gI accumulates in the TGN in early phases of infection and is then specifically sorted to cell junctions in late phases of infection and appears to be tethered there (30, 48, 49). This sorting of gE/gI correlates with the directed sorting of HSV virions to cell junctions (24). HSV-1 mutants lacking the gE CT domain do not localize gE/gI to the TGN well, and virions are subsequently mislocalized to apical cell surfaces and into cell culture supernatants. These mutants spread poorly, exhibiting similar defects in spread as do gE-null mutants (35, 48). The model that was tested here suggests that the gE CT domain promotes localization of gE/gI to TGN subdomains that sort gE/gI and, by extension, virions to cell junctions (23, 24). Specifically, we tested whether TGN accumulation and sorting to cell junctions required the same gE CT domain sequences and whether one or both of these processes were required for cell-to-cell spread.
Three of the gE CT domain truncation mutants, gE-470, gE-495, and gE-519, produced gE/gI molecules that largely accumulated in the TGN, colocalizing with TGN46, at early times of infection (Fig. 8). By contrast, gE-448, which lacks the majority of the CT domain and forms small plaques, produced gE/gI that was found throughout the cytoplasm as well as on both apical and lateral cell surfaces. This supports the conclusion that TGN accumulation is necessary in order for gE/gI to promote cell-to-cell spread.
FIG. 8.

Cartoon summarizing traffic of gE mutants. In cells infected with mutant gE-519 and wild-type HSV-1, gE/gI is initially found in the TGN but the moves to cell junctions, and these viruses spread well. gE/gI expressed in gE-470- and gE-495-infected cells localizes initially to the TGN and then remains primarily in cytosolic vesicles, some underlying the plasma membrane, and these viruses spread poorly. In gE-448-infected cells, gE/gI does not accumulate in the TGN and remains distributed throughout the cytoplasm (cyto) and on apical and cell surfaces, and this mutant spreads poorly. nuc, nucleus.
While wild-type HSV and gE-519 produced gE/gI that redistributed from the TGN to cell junctions at late times, gE/gI produced in cells infected with mutants gE-495 and gE-470 remained in cytoplasmic vesicles, frequently underlying the plasma membrane (Fig. 8). Since mutants gE-470 and gE-495 exhibited a similar deficiency in cell-to-cell spread, we concluded that TGN localization is necessary, but not sufficient, for gE/gI-mediated spread and that gE/gI also must redistribute to cell junctions for the virus to efficiently spread. Related to this, we found that virus particles produced in gE-448-infected cells were missorted apically so that 45-fold-more infectious virus was observed in cell culture supernatants at 10 h compared with wild-type HSV. By contrast, gE-470 and gE-495 produced particles that were not released into cell culture supernatants at these early times, yet these viruses did not spread well. This is consistent with the notion that gE-470 and ge-495 produce gE/gI dimers that do not traffic to cell junctions and that virions produced are not shed into apical compartments but may instead accumulate in cytoplasmic vesicles.
gE TGN sorting motifs, those that can be recognized based on the primary sequence, all cluster to a 20-residue region in the N-terminal half of the gE CT domain (Fig. 1). Surprisingly, truncation of all of these sequences except the tyrosine motif at residue 463 produced a gE/gI heterodimer (gE-470) that retained much of its ability to accumulate in the TGN, yet a mutant lacking the gE CT domain (gE-448) did not. Apparently, HSV gE/gI can accumulate in the TGN with just one gE tyrosine motif (gE-470), supporting the view that there is ample redundancy built into the gE CT domain in terms of TGN localization motifs. Previous studies of the PRV gE CT domain indicated that the replacement of both tyrosine motifs reduced spread in cultured epithelial cells and, to some extent, into the brain (41). Again, these studies were consistent with the notion that sorting and spread depend on a number of different motifs in the gE CT domain that are redundant.
Redistribution of gE/gI to cell junctions at late times of infection required sequences in the C-terminal half of the gE CT domain between residues 495 and 519 (Fig. 1). No recognizable sorting motifs are present in this region, although this region is proline rich. Eight of the 10 gE CT domain prolines are present between residues 487 and 519. The accumulation of gE/gI just under the plasma membrane in gE-495- and gE-470-infected cells may suggest that the movement of gE/gI from the TGN to lateral cell surfaces involves endosomes as intermediates. There are distinct recycling loops between the TGN and endosomes and between endosomes and the plasma membrane (17). Thus, with these mutants, it is possible that gE/gI becomes trapped in endosomal compartments and is unable to reach the plasma membrane or is rapidly internalized. However, analysis of this is complicated, as HSV disrupts the Golgi apparatus, the TGN, and possibly endosomes, dispersing cellular components to other compartments and the plasma membrane (4, 49). As such, the normal architecture of post-Golgi membrane compartments is lost, making determinations of the character of these gE/gI-containing vesicles difficult.
These results add important details to our model for how gE/gI facilitates cell-to-cell spread. In this hypothesis, gE/gI promotes envelopment into specific subdomains of the TGN that then sort nascent virions to cell junctions. Here, we showed that TGN accumulation is not sufficient and that gE/gI apparently functions at some post-TGN site and must move to cell junctions in order to mediate cell-to-cell spread. These data are consistent with the notion that gE-495 and gE-470 have defects in the delivery of virions to cell junctions. However, this requires substantiation by electron microscopy, studies that are in progress. It is difficult to understand how the gE CT domain can alter the traffic of viral glycoproteins and particles after gE/gI is incorporated into the virion envelope so that the gE CT domain is buried inside the virus particle. One possibility is that gE/gI, which is not part of the virion envelope but is present in membrane vesicles that enclose newly enveloped virions, might couple to cell sorting machinery and thereby move both gE/gI and virions to cell junctions. By this model, the gE CT domain affects sorting in both TGN and post-TGN compartments. A related hypothesis suggests that gE/gI accumulates in the TGN, promotes envelopment there, and also interacts with other HSV proteins in a process that unmasks cryptic sorting sequences in the gE CT domain that promote delivery to cell junctions. This model is attractive because it may explain the requirement for both the gE ET and CT domains in cell-to-cell spread and may explain why gE/gI remains at the TGN compartment when other HSV proteins are not present. However, these models await further validation through identification and characterization of gE/gI-interacting proteins.
Acknowledgments
We are most grateful to Todd Wisner for technical assistance, advice, and critically reading the manuscript. We thank Aurelie Snyder of the Microbiology Core Facility for valuable expertise with confocal microscopy and deconvolution microscopy, Tiffani Howard for graphics, and Jeff Vandehey for help submitting this paper online. A.F. is especially grateful to J.C.
The work was supported by grants from the National Institute for Allergy and Infections Diseases (AI 73996) and the National Eye Institute (EY11245).
REFERENCES
- 1.Alconada, A., U. Bauer, B. Sodeik, and B. Hoflack. 1999. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 73:377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell, S., M. Cranage, L. Borysiewicz, and T. Minson. 1990. Induction of immunoglobulin G Fc receptors by recombinant vaccinia viruses expressing glycoproteins E and I of herpes simplex virus type 1. J. Virol. 64:2181-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai, W. Z., S. Person, S. C. Warner, J. H. Zhou, and N. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 61:714-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campadelli, G., R. Brandimarti, C. Di Lazzaro, P. L. Ward, B. Roizman, and M. R. Torrisi. 1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 90:2798-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campo, C., A. Mason, D. Maouyo, O. Olsen, D. Yoo, and P. A. Welling. 2005. Molecular mechanisms of membrane polarity in renal epithelial cells. Rev. Physiol. Biochem. Pharmacol. 153:47-99. [DOI] [PubMed] [Google Scholar]
- 6.Ch'ng, T. H., and L. W. Enquist. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 79:10875-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins, W. J., and D. C. Johnson. 2003. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell-to-cell spread. J. Virol. 77:2686-2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corey, L., and P. G. Spear. 1986. Infections with herpes simplex viruses (1). N. Engl. J. Med. 314:686-691. [DOI] [PubMed] [Google Scholar]
- 9.Dasgupta, A., and D. W. Wilson. 2001. Evaluation of the primary effect of brefeldin A treatment upon herpes simplex virus assembly. J. Gen. Virol. 82:1561-1567. [DOI] [PubMed] [Google Scholar]
- 10.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J. Rainbow, and D. C. Johnson. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dingwell, K. S., L. C. Doering, and D. C. Johnson. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69:7087-7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dingwell, K. S., and D. C. Johnson. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933-8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farnsworth, A., K. Goldsmith, and D. C. Johnson. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 77:8481-8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folsch, H., H. Ohno, J. S. Bonifacino, and I. Mellman. 1999. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell 99:189-198. [DOI] [PubMed] [Google Scholar]
- 16.Forrester, A., H. Farrell, G. Wilkinson, J. Kaye, N. Davis-Poynter, and T. Minson. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu, F., C. M. Crump, and G. Thomas. 2001. Trans-Golgi network sorting. Cell. Mol. Life Sci. 58:1067-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanke, T., F. L. Graham, V. Lulitanond, and D. C. Johnson. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vectors expressing glycoproteins E and I. Virology 177:437-444. [DOI] [PubMed] [Google Scholar]
- 19.Horsburgh, B. C., M. M. Hubinette, D. Qiang, M. L. MacDonald, and F. Tufaro. 1999. Allele replacement: an application that permits rapid manipulation of herpes simplex virus type 1 genomes. Gene Ther. 6:922-930. [DOI] [PubMed] [Google Scholar]
- 20.Humphrey, J. S., P. J. Peters, L. C. Yuan, and J. S. Bonifacino. 1993. Localization of TGN38 to the trans-Golgi network: involvement of a cytoplasmic tyrosine-containing sequence. J. Cell Biol. 120:1123-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson, D. C., and V. Feenstra. 1987. Identification of a novel herpes simplex virus type 1-induced glycoprotein which complexes with gE and binds immunoglobulin. J. Virol. 61:2208-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson, D. C., M. C. Frame, M. W. Ligas, A. M. Cross, and N. D. Stow. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson, D. C., and M. T. Huber. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, D. C., M. Webb, T. W. Wisner, and C. Brunetti. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krummenacher, C., I. Baribaud, R. J. Eisenberg, and G. H. Cohen. 2003. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J. Virol. 77:8985-8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurten, R. C. 2003. Sorting motifs in receptor trafficking. Adv. Drug Deliv. Rev. 55:1405-1419. [DOI] [PubMed] [Google Scholar]
- 27.Ligas, M. W., and D. C. Johnson. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matter, K., and I. Mellman. 1994. Mechanisms of cell polarity: sorting and transport in epithelial cells. Curr. Opin. Cell Biol. 6:545-554. [DOI] [PubMed] [Google Scholar]
- 29.McGeoch, D. J., A. Dolan, S. Donald, and F. J. Rixon. 1985. Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J. Mol. Biol. 181:1-13. [DOI] [PubMed] [Google Scholar]
- 30.McMillan, T. N., and D. C. Johnson. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellman, I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12:575-625. [DOI] [PubMed] [Google Scholar]
- 32.Ohara, P. T., A. N. Tauscher, and J. H. LaVail. 2001. Two paths for dissemination of herpes simplex virus from infected trigeminal ganglion to the murine cornea. Brain Res. 899:260-263. [DOI] [PubMed] [Google Scholar]
- 33.Olson, J. K., and C. Grose. 1997. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 71:4042-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polcicova, K., P. S. Biswas, K. Banerjee, T. W. Wisner, B. T. Rouse, and D. C. Johnson. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 102:11462-11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polcicova, K., K. Goldsmith, B. L. Rainish, T. W. Wisner, and D. C. Johnson. 2005. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: evidence for gE/gI receptors. J. Virol. 79:11990-12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richart, S. M., S. A. Simpson, C. Krummenacher, J. C. Whitbeck, L. I. Pizer, G. H. Cohen, R. J. Eisenberg, and C. L. Wilcox. 2003. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 77:3307-3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roop, C., L. Hutchinson, and D. C. Johnson. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saldanha, C. E., J. Lubinski, C. Martin, T. Nagashunmugam, L. Wang, H. van Der Keyl, R. Tal-Singer, and H. M. Friedman. 2000. Herpes simplex virus type 1 glycoprotein E domains involved in virus spread and disease. J. Virol. 74:6712-6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens, J. G., and M. L. Cook. 1973. Latent infections induced by herpes simplex viruses. Cancer Res. 33:1399-1401. [PubMed] [Google Scholar]
- 41.Tirabassi, R. S., and L. W. Enquist. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 73:2717-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomishima, M. J., G. A. Smith, and L. W. Enquist. 2001. Sorting and transport of alpha herpesviruses in axons. Traffic 2:429-436. [DOI] [PubMed] [Google Scholar]
- 43.van Genderen, I. L., R. Brandimarti, M. R. Torrisi, G. Campadelli, and G. van Meer. 1994. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology 200:831-836. [DOI] [PubMed] [Google Scholar]
- 44.Wan, L., S. S. Molloy, L. Thomas, G. Liu, Y. Xiang, S. L. Rybak, and G. Thomas. 1998. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94:205-216. [DOI] [PubMed] [Google Scholar]
- 45.Wang, F., W. Tang, H. M. McGraw, J. Bennett, L. W. Enquist, and H. M. Friedman. 2005. Herpes simplex virus type 1 glycoprotein E is required for axonal localization of capsid, tegument, and membrane glycoproteins. J. Virol. 79:13362-13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whealy, M. E., J. P. Card, A. K. Robbins, J. R. Dubin, H. J. Rziha, and L. W. Enquist. 1993. Specific pseudorabies virus infection of the rat visual system requires both gI and gp63 glycoproteins. J. Virol. 67:3786-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whiteley, A., B. Bruun, T. Minson, and H. Browne. 1999. Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J. Virol. 73:9515-9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wisner, T., C. Brunetti, K. Dingwell, and D. C. Johnson. 2000. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J. Virol. 74:2278-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wisner, T. W., and D. C. Johnson. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-Golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519-11535. [DOI] [PMC free article] [PubMed] [Google Scholar]