

Abstract
Parvovirus B19 has been implicated in some cases of acute fulminant non-A, non-B, non-C, non-G liver failure. Our laboratory previously demonstrated that B19 infection of hepatocytes induces apoptosis and that the B19 viral nonstructural protein, NS1, may play a critical role. To study the involvement of NS1 in apoptosis of liver cells, we generated a fusion protein of NS1 with enhanced green fluorescent protein (eGFP) in a system allowing for inducible gene expression. Transfection of the liver-derived cell line HepG2 with the eGFP/NS1 vector allowed expression of the fusion protein, which was visualized by fluorescence microscopy and demonstrated by immunoblotting. The fusion protein localized to discrete domains in the nucleus. Transfection of HepG2 cells with the eGFP/NS1 vector led to apoptosis of 35% of transfected cells, a sevenfold increase over cells transfected with the parent eGFP expression vector. Mutation of the eGFP/NS1 vector to eliminate the nucleoside triphosphate-binding site of NS1 significantly decreased apoptosis, as did treatment of transfected cells with inhibitors of caspase 3 or 9. Neutralization of tumor necrosis factor alpha or Fas ligand had no effect on apoptosis. These results demonstrate that NS1 is sufficient to induce apoptosis in liver-derived cells and that it does so through the initiation of an intrinsic caspase pathway.
Parvovirus B19 is the prototype human parvovirus. B19 is a small, single-stranded DNA virus that is transmitted in blood products or through aerosolized droplets and fomite contamination. B19 infection in children typically manifests as erythema infectiosum, or fifth disease (1). Infection of adults often manifests as arthropathy (26). In patients with chronic hemolytic anemias, such as sickle cell disease or hereditary spherocytosis, B19 infection causes transient aplastic crisis by destroying the erythroid precursor pool (11). Although these are the best-described clinical illnesses caused by B19, the virus has been implicated in a wide spectrum of other illnesses (25).
Acute fulminant liver failure (AFLF) is a potentially fatal disease that may occur as a result of hepatic infection, toxic damage, or liver transplantation. Interestingly, over one third of AFLF cases are accompanied by aplastic crisis (5). Langnas and colleagues found parvovirus B19 DNA by PCR in the native liver from a significant number of patients with AFLF associated with aplastic anemia (18). Karetnyi et al. found B19 virions in AFLF associated with aplastic anemia, as indicated by the presence of viral DNA in a B19 capsid protein capture system. Interestingly, although virions were detected, replicative forms of the viral genome were not, suggesting active infection without replication of the virus in these tissues (17).
We previously demonstrated that infection of HepG2 cells or primary hepatocytes with B19 induces apoptosis in those cells. Although we observed production of NS1 RNA and protein from B19-infected hepatocytes, capsid genes were not expressed, leading us to hypothesize that apoptosis was induced through the action of the NS1 protein. Also, UV-irradiated B19, which is unable to transcribe its genes, did not induce apoptosis, indicating that the input capsid proteins or DNA itself was not responsible for apoptosis after B19 inoculation (30).
NS1 is a 71-kDa, slightly basic protein (7). Although it is named the nonstructural protein, a copy is attached to the viral DNA and is present on the mature virion (8, 9). NS1 is an activating transcription factor for the single promoter of B19 (13, 14). In addition, NS1 nicks the replicative form of the viral genome at the origin of replication, allowing for replication of the viral DNA (10). Purified NS1 protein from Aleutian disease virus, an autonomous parvovirus of mink, binds to and cleaves ATP and acts as a helicase in an ATP-dependent manner (6). The domain responsible for the nucleoside triphosphate (NTP)-binding activity has been identified and shown to be important for cell death induced by NS1 (21).
NS1 is toxic in cell culture systems, and therefore it is very difficult to express. To overcome this problem and investigate the ability of NS1 to induce apoptosis in liver cells, we cloned NS1 under the control of a promoter that is activated upon incubation with the insect steroid ecdysone. The NS1 gene was fused to enhanced green fluorescent protein (eGFP). The combination of an inducible vector tagged with eGFP allows the examination of the effects of NS1 on a single-cell basis at a defined time point. This system overcomes several difficulties usually encountered in work with parvoviral nonstructural proteins. First, it allows regulated expression, which enables examination of the effects of NS1 without concurrent effects from transfection reagents or processes, such as cationic lipids or electroporation. The induction reagent is mild and nontoxic, in contrast to dexamethasone- or heavy metal-inducible systems, both of which can induce apoptosis.
Second, many cell types, including liver cells, are very difficult to transfect. Bulk culture studies may not provide adequate data when transfection efficiencies are low. By fusing NS1 with eGFP, individually transfected cells can be examined. Transfection with the eGFP-only expression vector provides a control for the transfection process and foreign protein expression, at the same time controlling for the effects of eGFP in the cell. Another advantage to this system is that the expression vector can be easily mutated, allowing further investigation of the action of NS1.
The data obtained using the eGFP/NS1 vector demonstrated that NS1 was sufficient to induce apoptosis in transfected cells. NS1-induced apoptosis proceeded through caspase 3- and 9-dependent pathways, as is the case with liver cells infected with B19, and was independent of tumor necrosis factor alpha (TNF-α) or Fas signaling. The unique properties of the inducible eGFP/NS1 fusion protein vector have great potential for further studies of the multiple activities of NS1.
MATERIALS AND METHODS
Cloning of NS1.
NS1 was cloned from B19 viremic serum using PCR. The following primers were used: upper, 5′GTGAGCTAACTAACAGGTATTTATACCGAGCTCGAACATCCTAACA3′, and lower, 5′TAACCTTTTCATAAAATTCCACAAATTGCTGCGAGCTCGCTTTAGC3′. The amplicon incorporates nucleotides 391 to 2535 of the B19 genome (31), with the inclusion of SacI restriction enzyme cut sites at nucleotides 421 and 2498 (GAGCTC). The underlining indicates SacI cut sites. The amplified region encompasses the entire protein-coding region of the NS1 gene.
Two-temperature PCR was performed for 28 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 59.5°C for 2 min 30 seconds. During the last 14 cycles, the annealing/extension step was extended 15 seconds/cycle. The amplicon was analyzed on a 0.5% agarose gel, and the band at 2,144 bp was excised. DNA was purified using a Qiaquick gel extraction kit (QIAGEN, Valencia, CA) and digested with 10 U SacI (Promega, Madison, WI) for 24 h.
Inducible eGFP/NS1 fusion protein DNA was created by inserting the amplified gene for NS1 into the plasmid pIND(GFP)/SP1 (Invitrogen, Carlsbad, CA). The SacI cut site is at the extreme C-terminal end of GFP. The plasmid was digested with 10 U SacI (Promega). The digested plasmid was treated with 1 U shrimp alkaline phosphatase (Promega) for 1 h, and then the shrimp alkaline phosphatase was heat inactivated at 65°C for 1 h. The dephosphorylated plasmid was ligated to the digested amplicon for 12 h at 14°C using 1 U T4 DNA ligase (Invitrogen) and used to transform DH5α chemically competent Escherichia coli cells (Invitrogen). Colonies were screened for correct orientation of the insert by using PCR.
The eGFP/NS1ΔNTP plasmid, a deoxynucleoside triphosphate-binding site deletion mutant, was created by digesting eGFP(SP1)/NS1 with PflMI (New England Biolabs, Beverly, MA). Restriction digests were separated by agarose gel electrophoresis, and the band at 7,393 bp was excised. DNA was extracted with a Qiaquick gel extraction kit. The digested plasmid was incubated with T4 DNA ligase at room temperature for 2 h, and the ligated plasmids were used to transform DH5α chemically competent E. coli cells. Plasmids from transformed colonies were screened for removal of the 426-bp segment containing the deoxynucleoside triphosphate-binding site by digestion with XhoI and BstxI, followed by electrophoresis.
Expression of fusion proteins.
eGFP/NS1, the mutant eGFP/NS1ΔNTP, or parental pIND(GFP)SP1 vector was cotransfected with the ecdysone receptor plasmid pVGRXR into HepG2 cells. Three micrograms of each plasmid was diluted into 125 μl Opti-Mem 1 medium (Invitrogen), along with 3 μl PLUS reagent (Invitrogen), and incubated for 15 min at room temperature. Thirteen microliters of Lipofectamine (Invitrogen) in 125 μl of Opti-Mem 1 was added to the DNA and allowed to form complexes for 15 min at room temperature. The DNA mixture was added to cells in one well of a six-well plate containing 1 ml of Opti-Mem 1 and incubated overnight. The medium was exchanged for growth medium, and protein expression was induced by the addition of ponasterone A, an ecdysone analog, in ethanol stock at 1,000 μg/ml for a final concentration of 10 μg/ml culture medium. Expression was monitored by fluorescence microscopy.
Western blotting.
Cells were lysed in 1% sodium dodecyl sulfate, 4 M urea, and 5% 2-mercaptoethanol. DNA was sheared by passage over a Qiashredder homogenizer column (QIAGEN). Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis through 7.5 to 14% gradient acrylamide gels (Bio-Rad, Hercules, CA). Protein expression was detected by immunoblotting with anti-GFP polyclonal rabbit antiserum (Invitrogen) and ECL+ chemiluminescence (Amersham, Piscataway, NJ).
Detection of apoptosis.
HepG2 cells were grown on glass coverslips and transfected. Twenty-four hours posttransfection, coverslips were removed from culture and covered with 2 μl annexin V-Alexa-fluor 594 (Molecular Probes, Eugene, OR) in 100 μl binding buffer (EMD Biosciences, San Diego, CA). The coverslips were incubated for 15 min at room temperature in the dark and then washed twice with binding buffer. Analysis was performed using fluorescence microscopy. Transfected cells were identified by green fluorescence and examined for apoptosis using a 528- to 553-nm excitation filter and a 600- to 660-nm barrier filter to allow detection of red fluorescence.
Caspase activation and inhibition assays.
To examine transfected cells for caspase 3 and 9 activation, the DEVD-red and LEHD-red systems (EMD Biosciences), respectively, were used. Cells were transfected and induced for expression. Coverslips were overlaid with 1 μl of caspase activity detection substrate in 500 μl of growth medium and incubated for 30 min. Cells were washed three times with wash buffer, and caspase 3 and 9 activities were assayed by immunofluorescence, with positive cells fluorescing red.
For inhibition of caspases, cells were transfected and examined as for annexin V staining. The cell-permeable caspase 3 inhibitor DEVD-FMK, caspase 1, 4, and 5 inhibitor WEHD-FMK, or caspase 9 inhibitor LEHD-FMK (EMD Biosciences) was added to a final concentration of 500 μM 6 h before addition of ponasterone A.
RESULTS
Transfection with eGFP/NS1 construct.
NS1 was cloned into a vector expressing NS1 as a fusion protein with eGFP. To allow for regulated expression, a vector system under the control of an ecdysone-inducible promoter (Invitrogen) was used (Fig. 1A). Ecdysone is a nontoxic insect steroid with no observed effects on mammalian tissues (27). The ecdysone-inducible system is composed of two plasmids: an expression plasmid into which the gene of interest is cloned and expressed under the control of the ecdysone response element and the plasmid pVGRXR, which expresses the modified ecdysone receptor and the mammalian protein RXR. The system utilizes the ecdysone receptor from Drosophila melanogaster, which has been modified to include the VP16 transactivation domain and to recognize the glucocorticoid response element. RXR, a mammalian homologue of the ecdysone receptor binding partner, is also included in the system and binds to the modified ecdysone receptor upon the addition of ponasterone A. This complex binds to the ecdysone response element on the expression plasmid, activating transcription. The expression vector also contains two SP1 enhancer elements. These elements are necessary, since expression is quite low in their absence. This vector system allowed us to precisely regulate the expression of NS1 by the addition of the ecdysone analog ponasterone A to the culture medium.
FIG. 1.

Properties of the eGFP/NS1 plasmid. (A) Plasmid map of eGFP/NS1. The plasmid contains the coding sequence for NS1 fused to the C-terminal end of eGFP, under the control of the ecdysone-inducible response element. Three SP1 enhancers act to increase transcription. This vector is used in tandem with the plasmid pVGRXR, which expresses the heterodimeric ecdysteroid receptor. The inducing agent ponasterone A, a homologue of the insect steroid ecdysone, binds to the ecdysteroid receptor, and the complex activates transcription through binding to the ecdysone-inducible response element. The plasmid is selectable in E. coli with ampicillin and in mammalian cells with Geneticin. BGH, bovine growth hormone. (B) Protein sequence of the NS1 portion of eGFP/NS1 compared to the prototypic sequence (Au) (28) and the sequence of an infectious viral clone (Zhi) (31). Differences between sequences are highlighted in gray.
The DNA sequence of the cloned NS1 was analyzed using an Amersham Megabase Capillary DNA sequencer. Six conservative mutations, two amino acid substitutions, one amino acid deletion, and no insertions or frameshifts were found when the cloned sequence was compared to the published (Au) sequence (31). These mutations were also noticed when the sequence was compared to the NS1 sequence in the infectious clone created by Zhi et al. (34). In addition, four other differences between the cloned NS1 and the Zhi sequence were noted. However, the cloned NS1 and the Au sequence did not differ at these locations (Fig. 1). The cloned DNA sequence included 50 bases of the upstream sequence of viral protein 1 (VP1), as the coding regions in the viral genome overlap slightly and are found in different reading frames.
As an additional control, we prepared a mutated version of eGFP/NS1, eGFP/NS1ΔNTP, which contains a deletion in the NS1 protein from amino acids 259 to 413, encompassing the NTP-binding motif. The mutation was confirmed by sequencing the plasmid. Cells transfected with the parent eGFP vector only, which expresses eGFP upon addition of ponasterone A to the medium, were additionally used as controls for the effects of both transfection and eGFP in our experiments.
The ecdysone-inducible system demonstrated a high level of inducibility, with no detectable protein expression before induction. Maximal expression of eGFP/NS1 was detected 24 h postinduction. The expression level of the NTP motif-deleted protein in transfected cells was similar to that of the parent fusion protein. After removal of the ponasterone A inducing agent, protein levels decreased substantially (Fig. 2).
FIG. 2.
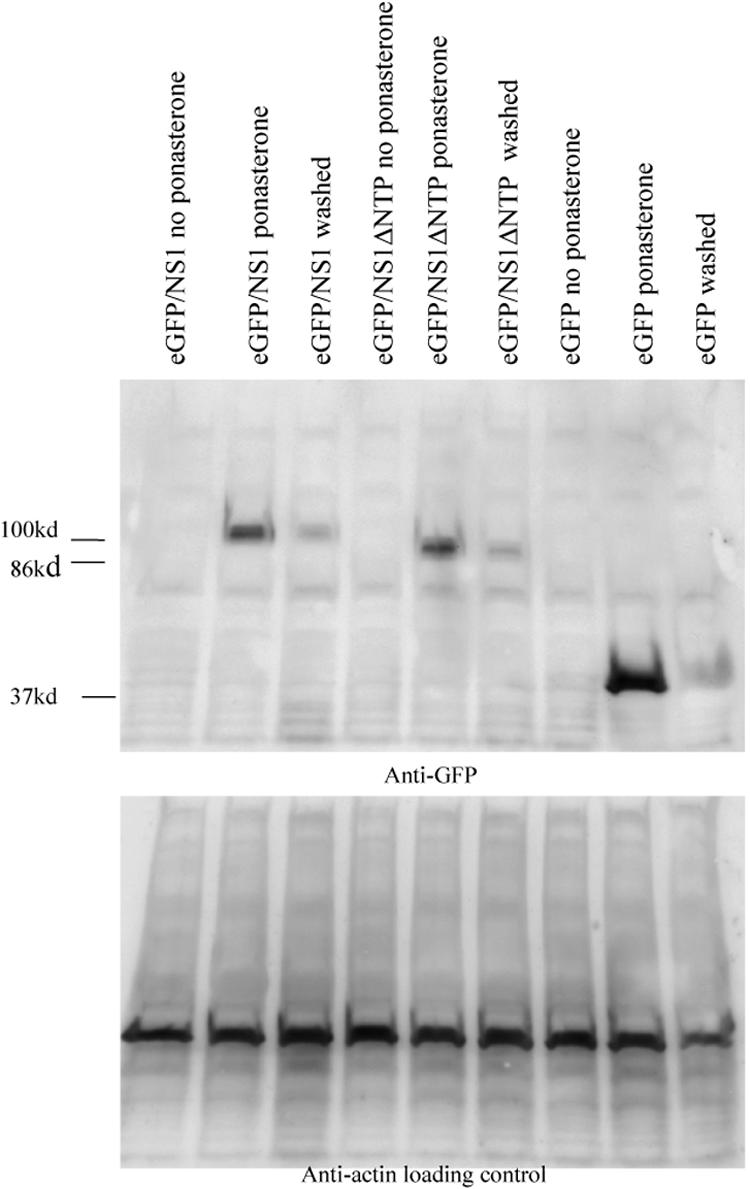
Expression from the ecdysone-inducible system is highly inducible. HepG2 cells were transfected with eGFP/NS1, eGFP/NS1ΔNTP, or eGFP alone. Expression was induced by the addition of ponasterone A to the culture medium. Three conditions are shown here: cells incubated for 24 h without the addition of ponasterone A, cells induced with ponasterone A for 24 h, and cells incubated with ponasterone A for 24 h, washed, and then incubated for 24 h without ponasterone. Protein was detected by Western blotting with anti-GFP antiserum. eGFP/NS1 is detectable at 100 kDa, eGFP/NS1ΔNTP at 95 kDa, and eGFP at 37 kDa.
Localization of eGFP/NS1 fusion protein.
Transfection with any of the three vectors resulted in bright green fluorescence and was achieved in 2% of cells. The eGFP/NS1 and eGFP/NS1ΔNTP proteins localized to punctuate areas of the nucleus, while the eGFP protein from the parental vector was found throughout the cell (Fig. 3). Intracellular localization of the eGFP/NS1 fusion protein (GFP/NS1) and the mutant eGFP/NS1ΔNTP protein was predominantly nuclear, with some cells also demonstrating cytoplasmic and perinuclear staining (Fig. 3). Nuclear localization was observed in 68% of eGFP/NS1-expressing cells, 45% of eGFP/NS1ΔNTP-expressing cells, and none of the eGFP-expressing cells. The nuclear staining was punctuate in nature and similar to that observed upon immunostaining of NS1 in B19-infected cells (28, 30). In parent vector-transfected cells, eGFP was consistently found throughout the cell without preferential nuclear localization.
FIG. 3.

Nuclear localization of wild-type and mutant NS1. (A) eGFP/NS1-, eGFP/NS1ΔNTP-, and eGFP-transfected cells were visualized by fluorescence microscopy. The proteins are visible as green fluorescence. eGFP/NS1- and eGFP/NS1ΔNTP-transfected cells show punctuate nuclear staining, while fluorescence is distributed throughout the cell in the eGFP-transfected cells. (B) True-color image clearly showing punctuate nuclear fluorescence of eGFP/NS1 and widespread fluorescence in eGFP-expressing cells.
Induction of apoptosis.
eGFP/NS1 expression induced apoptosis in a mean of 35% of transfected cells, as measured by annexin V staining (Fig. 4), an approximately fivefold increase over cells expressing eGFP alone. The difference was significant, with a P value of <0.0006 for three experiments. This result demonstrated the ability of NS1 to induce apoptosis in liver-derived HepG2 cells. The percentage of cells undergoing apoptosis was similar to that seen in HepG2 cells and primary hepatocytes infected with B19 virus (30). Transfection of HepG2 cells with eGFP/NS1ΔNTP resulted in a significantly decreased proportion of apoptotic cells compared to eGFP/NS1, with 16% of cells undergoing apoptosis (P < 0.05), confirming an important role for the NTP-binding region in NS1-induced apoptosis. The decreased apoptosis occurred even though nuclear localization and protein expression levels of the mutant protein were comparable to those seen with nonmutated eGFP/NS1.
FIG. 4.

Expression of eGFP/NS1 induces apoptosis in HepG2 cells. Cells expressing either eGFP/NS1, eGFP/NS1ΔNTP, or eGFP were incubated for 24 h and then stained with annexin V conjugated to Alexa-fluor 594 (red). (A) Apoptotic cells stain red around the border of the cell, while eGFP/NS1 or eGFP is detectable by green fluorescence. (B) Quantification of annexin staining experiments. eGFP/NS1 induces significantly more apoptosis than eGFP alone (P < 0.0006) or than the NTP deletion mutant (P < 0.05).
Caspase involvement.
There are multiple apoptotic pathways through which a cell can die, and these pathways are regulated by different caspases. The TNF-α family member-induced cell death pathways activate caspase 8 (3, 23). Caspase 8 is the first of the caspases activated by these pathways and transduces extracellular signals for apoptosis (33). This pathway differs from the endogenous pathways that induce apoptosis, for example, in response to DNA damage or growth factor withdrawal. The latter are transduced through the mitochondria and utilize caspase 9.
B19 infection induced apoptosis in hepatocytes in a caspase 3- and caspase 9-dependent manner (30). To determine whether or not apoptosis in NS1-transfected cells is also caspase 3 and caspase 9 dependent, we transfected HepG2 cells with either eGFP/NS1 or eGFP alone. Caspase 3 and 9 activities were assayed by incubating the cells with the specific detection reagents DEVD-red and LEHD-red, respectively. These are caspase substrates that, when cleaved, cause a red fluorophore to become covalently attached to the active caspase, allowing direct detection of caspase activation. Caspases 3 and 9 were both active in eGFP/NS1-transfected cells, while activity of these caspases was not seen in eGFP-transfected cells. Fifty-two percent of NS1-expressing cells were positive for caspase 9 activity, and 40% were positive for caspase 3 activity (Fig. 5).
FIG. 5.

Caspases 3 and 9 are activated by eGFP/NS1 expression. HepG2 cells were transfected with either eGFP/NS1 or eGFP alone. The caspase activity markers DEVD-red and LEHD-red were used to assay for caspase 3 and caspase 9 activity, respectively. Cells with active caspases fluoresce red in this assay. Cells expressing eGFP/NS1 demonstrated active caspases 3 and 9, while activity of these caspases was not observed in cells expressing eGFP alone.
To determine whether or not caspases 3 and 9 were necessary for apoptosis, HepG2 cells were incubated with specific caspase inhibitors. DEVD-FMK was used to inhibit caspase 3. Caspase 9 was inhibited with LEHD-FMK. Because LEHD-FMK inhibits caspases 1, 4, and 5, although to a lesser extent than caspase 9, cultures were incubated with the caspase inhibitor WEHD-FMK. This caspase inhibitor inhibits caspases 1, 4, and 5 but not 9 (24).
Cultures treated with WEHD-FMK or dimethyl sulfoxide buffer alone demonstrated no decrease in apoptosis, while in those treated with DEVD-FMK or LEHD-FMK there was a significant decrease in apoptosis, almost to background levels (Fig. 6). These findings indicate that NS1 induces apoptosis through a caspase 3- and 9-dependent pathway. The necessity of caspase 9 for the induction of apoptosis suggests NS1 induces apoptosis in HepG2 cells through an intrinsic mechanism, such as DNA damage.
FIG. 6.
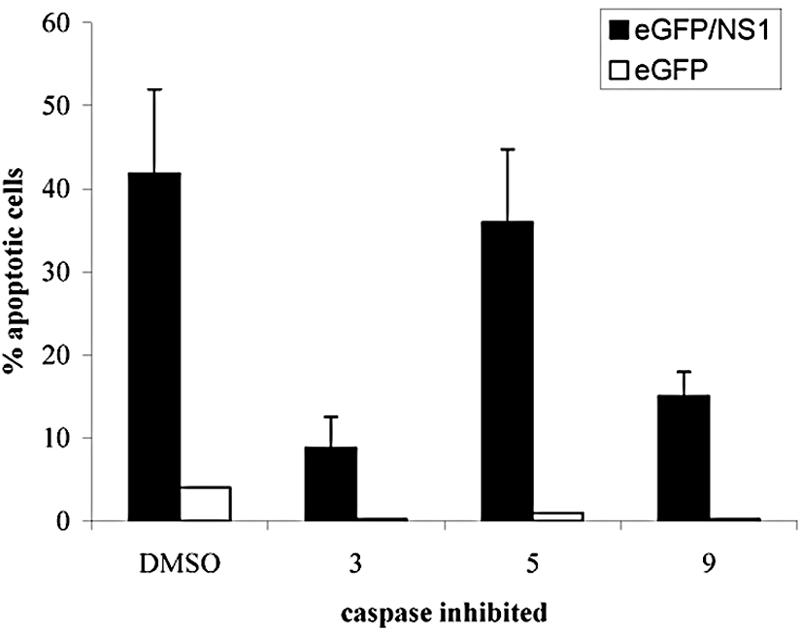
Caspases 3 and 9 are required for NS1-induced apoptosis. Transfected cells were incubated with DEVD-FMK to inhibit caspase 3, LEHD-FMK to inhibit caspase 9, or WEHD-FMK to inhibit caspases 1, 4, and 5. Expression was induced with ponasterone A, and apoptosis was assayed by annexin V staining. DEVD-FMK and LEHD-FMK significantly inhibited apoptosis (P < 0.006 and P < 0.008, respectively), while WEHD-FMK had no effect.
Although caspase 9 was essential for apoptosis, it was possible that TNF-α or the TNF-α receptor family member Fas was also playing a role in NS1-induced apoptosis, as has been observed in erythroid cells (33). Fas is a transmembrane protein that signals through caspase 8. To investigate the role of these molecules in NS1-induced apoptosis of liver cells, transfected cells were treated with antibodies that inhibit signaling by either TNF-α or Fas ligand (Oncogene and BD, respectively). Antibodies were diluted 1:100 in growth medium and added to the cells before NS1 expression was induced. Although the anti-TNF-α antibody was able to inhibit apoptosis induced by addition of recombinant TNF-α, there was no change in the amount of apoptosis in the transfected cells in the presence of either antibody (Fig. 7). It is highly unlikely that TNF-α or Fas plays a significant role in NS1-induced apoptosis of liver cells.
FIG. 7.
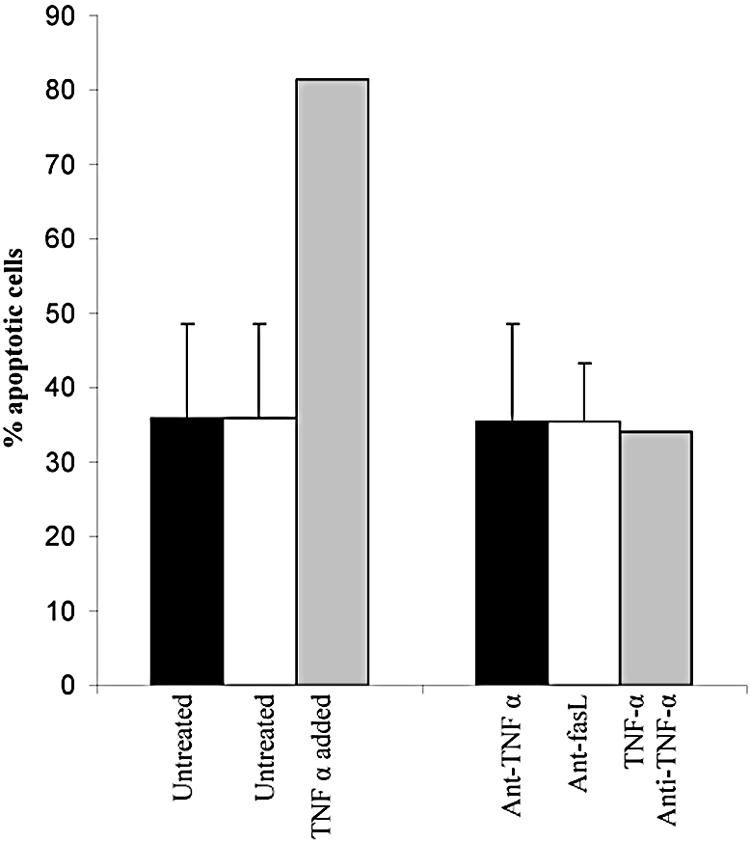
TNF-α and Fas are not involved in NS1-induced apoptosis of hepatocytes. HepG2 cells were transfected with eGFP/NS1, and transfected cells were assayed for apoptosis in the presence or absence of anti-TNF-α or anti-Fas ligand antibodies. Incubation of transfected cells with anti-TNF-α or anti-Fas ligand antibodies had no effect on apoptosis. The lack of inhibition was not due to lack of activity of the antibody, since the anti-TNF-α antibody was able to block apoptosis induced by the addition of recombinant TNF-α in the presence of cycloheximide.
DISCUSSION
Because of the observed toxicity of NS1 (28), observation of the effects of NS1 in transfected cells is difficult. Although stable, inducible cell lines expressing NS1 have been generated, they often express NS1 at low levels and/or require the addition of potentially toxic heavy metals or dexamethasone for induction (19, 22).
The reagents described in this paper are valuable tools for the study of NS1. By providing a mechanism to examine individual transfected cells, the eGFP/NS1 fusion protein expression vector allows the study of the effects of NS1 on cells that are difficult to transfect. The inducible nature of the eGFP/NS1 vector allows for precise control over the expression of NS1 and utilizes as an inducer an insect steroid with no known effects on mammalian cells (27). Furthermore, the ability to easily mutate the expression vector allows for experiments examining the domains and biochemistry of NS1.
The finding that NS1 induces apoptosis in HepG2 cells independently of the presence of full-length viral genomic DNA or other viral proteins confirms that NS1 is sufficient to induce apoptosis. This finding is highly relevant to the demonstrated behavior of B19 in nonpermissive cells, in which NS1 is overexpressed and the structural proteins underexpressed (4, 29, 30). Although productive infection in nonpermissive cells does not occur, the production of NS1 would likely cause the cells to undergo apoptosis.
The mechanisms through which NS1 induces apoptosis are delineated by this study as well. NS1, as a multifunctional protein, could cause cell death through different mechanisms. In erythroid cells, NS1 both induces production of TNF-α and sensitizes cells to killing by TNF-α (15, 32). However, in liver-derived cells, the eGFP/NS1-induced apoptotic pathway does not proceed through TNF-α. NS1 transfection induces activation of caspases 3 and 9, and these caspases are necessary for optimal induction of apoptosis. Involvement of the caspase 9 pathway is typical of apoptosis induced by internal stress, in contrast to apoptosis induced by TNF-α. Further, neutralization of TNF-α with specific antibodies had no effect on apoptosis induced by NS1 in these cells. The absence of a role for TNF-α in NS1-induced apoptosis of hepatocytes is consistent with the observed pathology of AFLF, which is characterized by a massive, noninflammatory dropout of hepatocytes (5). If TNF-α, a powerful proinflammatory cytokine, were involved in AFLF, inflammation would be expected. The disparity between the observed role of TNF-α in B19-induced apoptosis of erythroid cells and its noninvolvement in nonpermissive cells may be a result of many factors, including the transcriptional pattern of the individual cell types, the cells' susceptibility to apoptosis, or other causes.
Some evidence suggests that the use of cell lines to study virus-induced cell death may generate results in which apoptosis occurs in a non-virus-specific manner, depending instead on the default death pathway of the cell line used. In the case of the HepG2 cell line utilized in these experiments, however, other investigators have found that expression of hepatitis B or the hepatitis B X antigen leads to increased apoptosis through TRAIL, a TNF family member (16, 20), rather than through the internal, caspase 8-independent pathway that parvovirus B19 infection (30) or transfection with NS1 initiated. The findings implicating different apoptotic pathways for different viral infections demonstrate that HepG2 cells undergo apoptosis in multiple ways in response to individual viral stimuli. The apoptotic pathway observed upon transfection with NS1 is therefore a specific result of the parvovirus NS1 protein. The mechanism of apoptosis induced by NS1 transfection provides information as to the mechanisms through which NS1 damages the cell.
eGFP/NS1 expression allows assessment of NS1 cellular localization (Fig. 2). Fluorescence studies demonstrated nuclear localization of eGFP/NS1. This pattern of localization further supports the functionality of the expressed NS1 in that nuclear localization signals are intact. Localization of eGFP/NS1 in discrete domains suggests that the fusion protein localizes to autonomous parvovirus replication bodies as previously described for NS1 from minute virus of mice and H-1 parvoviruses (2, 12).
Given the known functions of NS1, the most likely mechanism through which it induces apoptosis is DNA damage. DNA damage-induced apoptosis proceeds through the mitochondrial apoptosis pathway, as does NS1-induced apoptosis. NS1 has nicking and helicase activities that could damage cellular DNA and lead to cell death. Further studies are needed to investigate whether or not NS1 damages cellular DNA and the role of cellular DNA damage in NS1-induced apoptosis.
Acknowledgments
This work was supported in part by the Arthritis Foundation, Central Pennsylvania Chapter.
REFERENCES
- 1.Anderson, M. J., S. E. Jones, S. P. Fisher Hoch, E. Lewis, S. M. Hall, C. L. Bartlett, B. J. Cohen, P. P. Mortimer, and M. S. Pereira. 1983. Human parvovirus, the cause of erythema infectiosum (fifth disease)? Lancet 1:1378. [DOI] [PubMed] [Google Scholar]
- 2.Bashir, T., J. Rommelaere, and C. Cziepluch. 2001. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 75:4394-4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boldin, M. P., T. M. Goncharov, Y. V. Goltsev, and D. Wallach. 1996. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 85:803-815. [DOI] [PubMed] [Google Scholar]
- 4.Brunstein, J., M. Soderlund-Venermo, and K. Hedman. 2000. Identification of a novel RNA splicing pattern as a basis of restricted cell tropism of erythrovirus B19. Virology 274:284-291. [DOI] [PubMed] [Google Scholar]
- 5.Cattral, M. S., A. N. Langnas, R. S. Markin, D. L. Antonson, T. G. Heffron, I. J. S. M. Fox, and B. W. Shaw, Jr. 1994. Aplastic anemia after liver transplantation for fulminant liver failure. Hepatology 20:813-818. [DOI] [PubMed] [Google Scholar]
- 6.Christensen, J., M. Pedersen, B. Aasted, and S. Alexandersen. 1995. Purification and characterization of the major nonstructural protein (NS-1) of Aleutian mink disease parvovirus. J. Virol. 69:1802-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cotmore, S. F., V. C. McKie, L. J. Anderson, C. R. Astell, and P. Tattersall. 1986. Identification of the major structural and nonstructural proteins encoded by human parvovirus B19 and mapping of their genes by procaryotic expression of isolated genomic fragments. J. Virol. 60:548-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotmore, S. F., and P. Tattersall. 1988. The NS-1 polypeptide of minute virus of mice is covalently attached to the 5′ termini of duplex replicative-form DNA and progeny single strands. J. Virol. 62:851-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotmore, S. F., and P. Tattersall. 1989. A genome-linked copy of the NS-1 polypeptide is located on the outside of infectious parvovirus particles. J. Virol. 63:3902-3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotmore, S. F., and P. Tattersall. 2003. Resolution of parvovirus dimer junctions proceeds through a novel heterocruciform intermediate. J. Virol. 77:6245-6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cubel, R. C., M. C. Valadao, W. V. Pereira, M. C. Magalhaes, and J. P. Nascimento. 1992. Aplastic crisis due to human parvovirus B19 infection in hereditary hemolytic anaemia. Rev. Inst. Med. Trop. Sao Paulo 34:479-482. [DOI] [PubMed] [Google Scholar]
- 12.Cziepluch, C., S. Lampel, A. Grewenig, C. Grund, P. Lichter, and J. Rommelaere. 2000. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J. Virol. 74:4807-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doerig, C., P. Beard, and B. Hirt. 1987. A transcriptional promoter of the human parvovirus B19 active in vitro and in vivo. Virology 157:539-542. [DOI] [PubMed] [Google Scholar]
- 14.Doerig, C., B. Hirt, J. P. Antonietti, and P. Beard. 1990. Nonstructural protein of parvoviruses B19 and minute virus of mice controls transcription. J. Virol. 64:387-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu, Y., K. K. Ishii, Y. Munakata, T. Saitoh, M. Kaku, and T. Sasaki. 2002. Regulation of tumor necrosis factor alpha promoter by human parvovirus B19 NS1 through activation of AP-1 and AP-2. J. Virol. 76:5395-5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janssen, H. L., H. Higuchi, A. Abdulkarim, and G. J. Gores. 2003. Hepatitis B virus enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity by increasing TRAIL-R1/death receptor 4 expression. J. Hepatol. 39:414-420. [DOI] [PubMed] [Google Scholar]
- 17.Karetnyi, Y. V., P. R. Beck, R. S. Markin, A. N. Langnas, and S. J. Naides. 1999. Human parvovirus B19 infection in acute fulminant liver failure. Arch. Virol. 144:1713-1724. [DOI] [PubMed] [Google Scholar]
- 18.Langnas, A. N., R. S. Markin, M. S. Cattral, and S. J. Naides. 1995. Parvovirus B19 as a possible causative agent of fulminant liver failure and associated aplastic anemia. Hepatology 22:1661-1665. [PubMed] [Google Scholar]
- 19.Leruez-Ville, M., I. Vassias, C. Pallier, A. Cecille, U. Hazan, and F. Morinet. 1997. Establishment of a cell line expressing human parvovirus B19 non-structural protein from an inducible promoter. J. Gen. Virol. 78:215-219. [DOI] [PubMed] [Google Scholar]
- 20.Liang, X., W. Sun, L. Gao, C. Ma, L. Han, and Y. Chen. 2005. Hepatitis B virus X protein modulates the apoptosis of hepatoma cell line induced by TRAIL. Sci. China C Life Sci. 48:277-286. [DOI] [PubMed] [Google Scholar]
- 21.Momoeda, M., S. Wong, M. Kawase, N. S. Young, and S. Kajigaya. 1994. A putative nucleoside triphosphate-binding domain in the nonstructural protein of B19 parvovirus is required for cytotoxicity. J. Virol. 68:8443-8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita, E., A. Nakashima, H. Asao, H. Sato, and K. Sugamura. 2003. Human parvovirus B19 nonstructural protein (NS1) induces cell cycle arrest at G1 phase. J. Virol. 77:2915-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muzio, M., A. M. Chinnaiyan, F. C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J. D. Bretz, M. Zhang, R. Gentz, M. Mann, P. H. Krammer, M. E. Peter, and V. M. Dixit. 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85:817-827. [DOI] [PubMed] [Google Scholar]
- 24.Mykles, D. L. 2001. Proteinase families and their inhibitors, p. 247-287. In L. M. Schwartz and J. D. Ashwell (ed.), Apoptosis. Academic Press, San Diego, Calif. [DOI] [PubMed]
- 25.Naides, S. J. 2000. Parvoviruses, p. 487-500. In S. Specter, R. L. Hodinka, and S. A. Young (ed.), Clinical virology manual, 3rd ed. ASM Press, Washington, D.C.
- 26.Naides, S. J., L. L. Scharosch, F. Foto, and E. J. Howard. 1990. Rheumatologic manifestations of human parvovirus B19 infection in adults. Initial two-year clinical experience. Arthritis Rheum. 33:1297-1309. [DOI] [PubMed] [Google Scholar]
- 27.No, D., T.-S. Yao, and R. M. Evans. 1996. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 93:3346-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozawa, K., J. Ayub, S. Kajigaya, T. Shimada, and N. Young. 1988. The gene encoding the nonstructural protein of B19 (human) parvovirus may be lethal in transfected cells. J. Virol. 62:2884-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pallier, C., A. Greco, J. Lejunter, A. Saib, I. Vassias, and F. Morinet. 1997. The 3′ untranslated region of the B19 parvovirus capsid protein mRNAs inhibits its own mRNA translation in nonpermissive cells. J. Virol. 71:9482-9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poole, B. D., Y. V. Karetnyi, and S. J. Naides. 2004. Parvovirus B19-induced apoptosis of hepatocytes. J. Virol. 78:7775-7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shade, R. O., M. C. Blundell, S. F. Cotmore, P. Tattersall, and C. R. Astell. 1986. Nucleotide sequence and genome organization of human parvovirus B19 isolated from the serum of a child during aplastic crisis. J. Virol. 58:921-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sol, N., J. Le Junter, I. Vassias, J. M. Freyssinier, A. Thomas, A. F. Prigent, B. B. Rudkin, S. Fichelson, and F. Morinet. 1999. Possible interactions between the NS-1 protein and tumor necrosis factor alpha pathways in erythroid cell apoptosis induced by human parvovirus B19. J. Virol. 73:8762-8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stennicke, H. R., J. M. Jurgensmeier, H. Shin, Q. Deveraux, B. B. Wolf, X. Yang, Q. Zhou, H. M. Ellerby, L. M. Ellerby, D. Bredesen, D. R. Green, J. C. Reed, C. J. Froelich, and G. S. Salvesen. 1998. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 273:27084-27090. [DOI] [PubMed] [Google Scholar]
- 34.Zhi, N., Z. Zadori, K. E. Brown, and P. Tijssen. 2004. Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 318:142-152. [DOI] [PubMed] [Google Scholar]