

Abstract
The recent solution of the crystal structure of a fragment of the vesicular stomatitis virus matrix (M) protein suggested that amino acids 121 to 124, located on a solvent-exposed loop of the protein, are important for M protein self-association and association with membranes. These residues were mutated from the hydrophobic AVLA sequence to the polar sequence DKQQ. Expression and purification of this mutant from bacteria showed that it was structurally stable and that the mutant M protein had self-association kinetics similar to those of the wild-type M protein. Analysis of the membrane association of M protein in the context of infection with isogenic recombinant viruses showed that both wild-type and mutant M proteins associated with membranes to the same extent. Virus expressing the mutant M protein did show an approximately threefold-lower binding affinity of M protein for nucleocapsid-M complexes. In contrast to the relatively minor effects of the M protein mutation on virus assembly, the mutant virus exhibited growth restriction in MDBK but not BHK cells, a slower induction of apoptosis, and lower viral-protein synthesis. Despite translating less viral protein, the mutant virus produced more viral mRNA, showing that the mutant virus could not effectively promote viral translation. These results demonstrate that the 121-to-124 region of the VSV M protein plays a minor role in virus assembly but is involved in virus-host interactions and VSV replication by augmenting viral-mRNA translation.
Virus assembly is a complex process involving a number of important steps. For enveloped viruses, the viral genome must be packaged into a nucleoprotein (nucleocapsid), which must be brought to the host membrane. This nucleocapsid must acquire a lipid bilayer by budding from the host membrane (for a review, see reference 14). For negative-strand RNA viruses of the order Mononegavirales and for the families Orthomyxoviridae and Retroviridae, these assembly functions appear to be carried out largely by viral matrix, or M, proteins (28, 32). This multifunctional nature of the M protein makes it a critical player in virus assembly, which has led to a great deal of interest in determining how M proteins carry out such different functions. M proteins from different viruses have very different sequences and structures (32), a fact that has led to few initial clues about how they carry out their multiple functions and has highlighted the importance of studying individual M proteins to shed light on the wider class of functional analogs.
The M protein of vesicular stomatitis virus (VSV) is a particularly well-studied example of this class of proteins. VSV M protein has been shown to play several roles in the virus assembly pathway. VSV M is responsible for condensing the viral nucleocapsid, causing the collapse of the loosely coiled, flexible nucleocapsid into the characteristic tightly coiled, bullet-like shape found in assembled virions (23, 25, 26). Mutagenesis and proteolysis studies have shown that the interaction of M protein with nucleocapsid requires an N-terminal sequence of the protein (2, 17). The N-terminal sequence (amino acids 1 to 50) is protease accessible in assembled nucleocapsid-M (NCM) complexes (17) and, by using a photoactivatable membrane probe, has been shown to associate with cellular membranes (20). The sequence PPPY contained within the N-terminal sequence is involved in a late stage of budding, allowing the release of budded virions from the membrane (15).
In addition to the N-terminal sequence, several different regions in the middle of the protein sequence have been implicated in virus assembly, but these studies have yet to be extended to an understanding of whether these sequences cooperate and how they contribute to viral assembly and budding in an intact virus. In particular, proteolysis studies have suggested the contribution of a 4-amino-acid (aa) region at aa 121 to 124 in membrane association (9).
A detailed understanding of why these regions are important has been hampered to some extent by the lack of structural information to indicate which residues of the M protein are part of the interior of the folded protein and which residues are located in solvent-exposed regions that can participate in membrane association and/or protein-protein interactions. This problem has been significantly reduced with the recent solution of the crystal structure of an M protein fragment (10). The ability to obtain crystals of the M protein required proteolytic removal of both the N-terminal sequence (aa 1 to 47) and the hydrophobic sequence aa 121 to 124 to prevent M protein self-association, but the resulting crystal structure represents a large portion of the M protein sequence. The structure showed that the M protein fragments formed a compactly folded structure with two alpha helices cradled in a beta sheet (Fig. 1A). The structure indicated that residues 121 to 124 lie on a solvent-exposed loop. Based on this crystallographic data and the properties of the cleaved M protein fragment, it was proposed that the aa 121-to-124 sequence played a role in both M protein self-association and M protein association with membranes (9, 10). To determine the importance of this region in the context of a nonproteolyzed M protein and during VSV infection, we mutated the region from a hydrophobic sequence to a polar sequence and analyzed its association with NCM complexes and with membranes in both the presence and the absence of other viral components. Our findings do not support a strong role for this region in membrane association. Instead, we have found that mutating amino acids 121 to 124 of VSV M alters the virus-host interaction so that viral mRNAs are translated less efficiently and the induction of apoptosis is delayed. While a role for the M protein in regulating apoptosis has been reported previously (8, 19), this is the first report of a role for the M protein in controlling the translation of viral mRNAs.
FIG. 1.

Mutation of aa 121 to 124 of VSV M protein does not affect self-association. (A) Image of theoretical model (based on M crystal structure; see Materials and Methods) illustrating the location of the presumptive loop region. (B) Diagram of wt M protein (top) and a mutant used in this study illustrating placement and sequence of mutations. (C) Coomassie staining of purified recombinant M proteins. Lane 1 is purified virions as a marker for the migration of wild-type M protein. The star denotes a proteolytic fragment. mut, mutant. (D) Stopped-flow analysis of self-association of M. The graph shows changes in light-scattering intensity following a shift of the salt concentration from 250 mM to either 65 or 130 mM NaCl. The traces are representative of four experiments each from two separate protein preparations. Wild-type traces are shown in black, and hydrophobic-loop mutant traces are in gray. The inset shows the first 12 seconds of each run to illustrate the lag seen in 130 mM NaCl samples.
MATERIALS AND METHODS
Chemicals, unless otherwise stated, were purchased from Fisher Scientific. Thapsigargin was purchased from Calbiochem. Antibodies against eIF2α and phospho-eIF2α, were purchased from Cell Signaling Technologies. Okadaic acid and microcystin were purchased from Alexis Pharmaceuticals. Ni-nitrilotriacetic acid beads were obtained from QIAGEN, and phosphocellulose (P11) was obtained from Whatman. Swiss-Model (30) was used to generate the coordinates of the M protein fragment from aa 51 to 227. This model includes amino acids 122 to 127, which are missing in the crystal structure of the matrix protein.
Mutagenesis and bacterial expression of M protein.
The gene encoding the M protein from the Orsay strain of VSV (2) was subcloned into the pET21d bacterial expression vector (Novagen). The sequence encoding aa 121 to 124 was mutagenized using a previously described protocol (29). BL21(DE3) pLYSs cells were transformed with the expression vector, and 500 nM IPTG (isopropyl-β-d-thiogalactopyranoside) was added when the cells reached an optical density at 600 nm of 0.6. Following expression at 37°C for 3 h, the cells were spun down, lysed in 1/100 volume of lysis buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 250 mM NaCL, 20 mM imidazol), and purified using Ni-nitrilotriacetic acid beads. Following adsorption onto the beads, the M protein was eluted in Ni elution buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 400 mM imidazole). The resulting eluted protein was diluted and passed over a P11 column and then eluted off of the P11 column using a high-salt buffer (24). The resulting purified protein was dialyzed against 10 mM Tris, pH 7.5, 250 mM NaCl.
Stopped-flow analysis of M self-association.
Purified wt or mutant M protein was diluted to 170 μg/ml in 10 mM Tris, pH 7.5, 650 mM NaCl and placed in the sample syringe of an Applied Photophysics stopped-flow spectrophotometer (22). The NaCl concentration was shifted to 65 mM or 135 nM NaCl by 10-fold dilution in the appropriate buffer, and light scattering was analyzed as a function of time after dilution as described previously (22).
Recovery of recombinant viruses.
M genes encoding wild-type (wt) M protein (Orsay strain) and 121-to-124 mutant M protein were subcloned into an infectious cDNA clone described previously (19). Recombinant viruses were recovered as described previously (19). For all experiments, BHK cells were cultured in Dulbecco's modified Eagle medium (DMEM) (Gibco-BRL) containing 7% fetal bovine serum (FBS) and 2 mM glutamine. The cells were grown to 80 to 90% confluence and were infected with virus in DMEM with 2% FBS at a multiplicity of infection (MOI) of 10 PFU/cell in a small volume (500 μl per well for a six-well dish). At 1 h postinfection, the culture volume was doubled by the addition of DMEM plus 2% FBS.
Assays for M protein exchange with NCM complexes.
The ability of radiolabeled wt or mutant M protein to exchange with unlabeled M protein in virion NCM complexes was assayed as described previously (7). Briefly, in vitro transcription and translation (Promega TNT) was used to produce [35S]methionine-labeled M protein. Radiolabeled M protein was mixed with increasing concentrations of nonradiolabeled purified virions, which served as a source of nucleocapsid-M complexes. Triton X-100 was added to permeabilize the virion envelope and, following a 5-min incubation time to allow equilibrium exchange of M protein, the NCM complexes were pelleted by centrifugation. The supernatants and pellets were then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and phosphorimager analysis.
Membrane association assays.
For analysis of M protein in the absence of other viral components, cells were infected with vaccinia virus expressing T7 RNA polymerase (VTF7-3) at an MOI of 20, and then 60 min postinfection, the cells were transfected with appropriate pET21d-M plasmids and allowed to express M protein for 6 h. For analysis of M protein in the presence of other viral components, cells were infected with recombinant viruses expressing recombinant wt (rwt) or mutant M protein at an MOI of 10 for 4 h. For both conditions, the cells were then starved for methionine for 10 min and then incubated in media containing [35S]methionine (200 μCi/ml) for 60 min. The cells were then washed and lifted from the dish by scraping it. The cells were resuspended in homogenization buffer and lysed using a Dounce homogenizer (100 to 125 strokes). The postnuclear lysates were then adjusted to 60% sucrose and were fractionated on a 60-40-10% sucrose gradient by centrifugation at 35,000 × g for 16 h at 4°C. Fractions were then collected from the top, M protein was immunoprecipitated using 23H12 antibody (19), and immunoprecipitates were separated on a 10% SDS-PAGE gel and exposed using a phosphorimager.
Pulse-chase analysis of virus assembly.
Cells were infected with rwt or a hydrophobic-loop mutant at an MOI of 10; 4 h postinfection, the cells were starved for methionine for 10 min and then incubated with media containing [35S]methionine for 60 min. The radioactive media were replaced with nonradioactive media. At 30, 60, and 90 min postlabeling, the media were removed and lysates of cells were made. Virions were purified through a 15% sucrose cushion, and then virion and cell lysate fractions were run on an SDS-PAGE gel and analyzed by phosphorimager analysis. The total amounts of radioactivity in each fraction were back-calculated based on the phosphorimager analysis.
Northern blotting.
Blotting was performed as described previously (4). Briefly, infected or mock-infected cells were lysed in 60-mm dishes with Trizol (Invitrogen); 5 μg of total RNA from each sample was glyoxylated and separated on a 1% agarose gel. Following ethidium bromide staining to check for loading, RNA was transferred onto nitrocellulose membranes (Genescreen Plus; NEN) and probed using a 32P-labeled M gene cDNA probe, and the results were determined by phosphorescence imaging and analyzed using Imagequant software.
Immmunoblotting.
Infected or mock-infected cells were lysed in six-well dishes using 400 μl EBC buffer containing 1 mM PMSF (phenylmethylsulfonyl fluoride), 1 mM benzamidine, 100 nM okadaic acid, and 100 nM microcystin (4). The lysates were spun at 10,000 × g for 8 min in a refrigerated centrifuge, and 360 μl of the supernatant was added to 40 μl of 10× sample buffer for SDS-PAGE. Equal volumes of lysate were electrophoresed on 12% SDS-PAGE gels. Following electrophoresis, the gels were electroblotted onto nitrocellulose membranes and blocked in Tris-buffered saline (pH 7.5) plus 5% dry milk. Antibodies were diluted as recommended by the manufacturers.
Metabolic labeling and determination of the rate of protein synthesis.
BHK cells were mock infected or infected with VSV and then labeled with [35S]methionine for 10 min at various times postinfection, as described above. Following labeling, the cells were washed three times with phosphate-buffered saline and lysed in 400 μl RIPA buffer for 10 min at 4°C. The lysates were spun at 10,000 × g for 8 min. Supernatant (360 μl) was added to 40 μl 10× SDS-PAGE sample buffer, and 10 μl was electrophoresed on a 12% SDS-PAGE gel. The gels were stained with Coomassie blue to determine their protein contents, and then they were analyzed by phosphorescence imaging (Molecular Dynamics, Inc.). The phosphorescence intensities were analyzed using Imagequant software. The rate of viral-protein synthesis was determined from the experiments by quantitation of the radioactivity in the viral N/P and M bands in each lane. The rate of host protein synthesis was determined by quantitation of the radioactivity between the L and G bands and the N/P and M bands and below the M band. For all types of experiment, three separate experiments were analyzed in this manner to determine an average.
Cell rounding/blebbing and caspase 3 assays.
Cells were grown to approximately 50% confluence in six-well dishes, infected with wt control virus or hydrophobic-mutant virus, and placed on a Zeiss inverted microscope with a six-well stage. Images were taken every 15 min using OpenLab (Improvision). Movies were made from streamed images, and occurrences of cell rounding and blebbing were tabulated using the time stamp record. Caspase 3 assays were done using a caspase 3 activity kit from R&D. Analysis was done according to the manufacturer's instructions.
RESULTS
Earlier studies suggested that amino acids 121 to 124 are important for the self-association of M protein and the association of M protein with membranes (9, 10). In the wild-type M protein, the aa 121-to-124 region sequence, AVLA, is hydrophobic and is likely to be a loop region, based on the crystal structure and a theoretical model (Fig. 1A). Because these previous experiments utilized proteolytic fragments of the M protein, we wanted to determine the importance of this region for self-aggregation, membrane association, and the association of M with nucleocapsid in the context of the full-length protein. We mutated the AVLA sequence to DKQQ (Fig. 1B), reasoning that this would replace hydrophobic residues with hydrophilic residues and that the charged Asp and Lys residues would mimic the positive N-terminal and negative C-terminal charges that would be generated following proteolysis in the region.
Hydrophobic-loop mutations do not effect M aggregation.
We were able to bacterially express and purify milligram amounts of the M protein as a C-terminal His-tagged protein using a two-step column procedure that removed contaminating bacterial proteins (Fig. 1C). These preparations contained minor amounts of a 21-kDa proteolytic fragment of M that is common in purified M protein preparations. The presence of this fragment does not affect the self-association of intact M protein (11). This purified M protein was stable for up to a week in a high-salt (250 mM NaCl) buffer. We assessed the ability of the purified M protein to self-associate under conditions of reduced salt concentration. A solution of wt or hydrophobic-loop mutant M (φ mutant) protein in high-salt buffer was mixed with a low-salt buffer so that the final NaCl concentration was either 65 mM or 130 mM. Under these lower-salt conditions, M protein will spontaneously self-associate to form aggregates. We determined the time course of self-association by stopped-flow analysis of light-scattering intensity. Stopped-flow analysis allowed us to observe the changes in light scattering caused by the formation of large M protein aggregates on a time scale of milliseconds. At 130 mM salt (Fig. 1D, lower traces), there was a brief lag before significant aggregation occurred (Fig. 1D, inset), and most of the aggregation had occurred within 40 seconds. At the lower salt concentration of 65 mM, the lag phase was not evident, and aggregation occurred to a greater extent (Fig. 1D, upper traces). The wt and mutant M proteins showed virtually identical aggregation characteristics in this assay, demonstrating that in the context of the full-length protein, this region does not dramatically affect the aggregation of the M protein.
Effect of the hydrophobic-loop mutations on M protein association with NCM complexes or cellular membranes.
Using an M protein exchange assay that measures the reversible binding of M protein to the NCM complexes at physiological ionic strength, we also determined whether mutations in the hydrophobic loop, aa 121 to 124, altered the ability of the M protein to interact with NCM complexes. There are both reversible and irreversible steps in M protein association with the nucleocapsid (21); the exchange assay tests the reversible step by determining the ability of radiolabeled M protein to be recruited onto nucleocapsids that have already been assembled with unlabeled M protein. In this assay, NCM complexes derived from purified virus are incubated with [35S]methionine-labeled M protein, and following a short incubation time to allow the exchange of labeled M protein with nonradiolabeled M protein, complexes are pelleted and analyzed by SDS-PAGE and phosphorimaging. If the labeled M protein is capable of associating with NCM complexes, a portion of the radiolabeled protein will be found in the pellet following centrifugation, while the labeled M protein that is not exchanged will remain in the supernatant. With increasing amounts of added NCM complex, more M protein should be found in the pellet.
Because previous reports showed that the N terminus of the VSV M protein was important for nucleocapsid-M association (2), we wanted to determine how modification of either the N or C terminus would affect the M-NCM interaction. Figure 2A shows controls for the association of His-tagged M protein with NCM complexes. Wild-type M protein lacking a His tag showed significant association with NCM complexes in this assay (Fig. 2A, top), while the negative control MN1, which lacks aa 4 to 21, which are important for NCM binding (2, 16), did not associate in this assay (Fig. 2A, upper middle). When we tested M protein that was tagged at the N terminus (His6-M), we found that the modified M protein did not associate with NCM complexes (Fig. 2A, lower middle). This was also true of N-terminal glutathione S-transferase and thioredoxin fusions (data not shown), suggesting that the free N terminus of M protein is important for NCM association.
FIG. 2.

Nucleocapsid and membrane association of wild-type and hydrophobic-loop mutant M proteins. (A) Phosphorimages of M protein exchange assays determining the association of 35S-labeled, in vitro-translated M protein with nucleocapsid-M complexes (see Materials and Methods). Shown are supernatant (S) and pellet (P) fractions for 10, 25, and 50 μg/ml of added NCM. MN1 is a negative control M protein lacking aa 4 to 21, His6-M is an N-terminally His-tagged M, and M-His6 is a C-terminally tagged M protein. (B) M protein exchange assays for wt M (c-terminally His tagged) and a hydrophobic-loop mutant using 6, 15, and 36 μg/ml added NCM complex. (C) The membrane association of radiolabeled M protein in transfected cells was determined by fractionation of cell lysates on sucrose flotation gradients. The autoradiograph shows radiolabeled M protein levels in individual fractions (1 [top] to 9 [bottom]) of the sucrose gradient. (D) Quantitation of total M protein in each fraction. The filled bars are wt M protein, and the white bars are hydrophobic-loop mutant M. The data shown are the means ± standard deviations of three separate experiments.
In contrast to His6-M, an M protein that had a C-terminal His tag (M-His6) was able to interact with NCM complexes at a level that was similar to that of untagged M protein (Fig. 2A, bottom), showing that the His tag at the C terminus does not interfere with M protein association with nucleocapsids. This allowed us to use C-terminally His-tagged wild-type and mutant M proteins to analyze M protein interactions with NCM complexes. Figure 2B shows the results of experiments using 35S-labeled, C-terminally His-tagged wt and φ mutant M proteins in this assay. Both wild-type (top) and the φ mutant (bottom) M proteins showed almost identical associations with NCM complexes in the assay, with similar amounts of 35S-labeled M protein in the pellet and the supernatant.
We also determined the abilities of both the wild-type and the φ mutant M proteins to associate with cellular membranes. Cells were infected with vaccinia virus expressing the T7 polymerase to allow expression of protein from our T7-based M expression vectors. One hour after infection, the cells were transfected with wt and mutant M plasmids. Four hours after transfection, the cells were labeled with [35S]methionine, lysed in a buffer lacking detergent, and separated on a sucrose gradient by centrifugation. Fractions from the sucrose gradient were then solubilized with detergent, and M protein was immunoprecipitated from each fraction. In this assay, the M protein associated with membranes floats to the top of the gradients (fractions 2 and 3), while M protein that is not membrane associated will remain near the bottom of the gradient. Both wild-type and mutant M proteins showed an association with membranes, indicated by M protein being found in fractions 2 and 3 (Fig. 2C). Quantitation of the amount of M present in each fraction as a percentage of the whole in three separate experiments is shown in Fig. 2D. A noticeable difference between the wt and the hydrophobic mutant was evident from this analysis. Mutant M protein was associated to a lesser extent than wt M protein. The total mutant M protein associated with membrane fractions was 57% ± 14% (mean ± standard deviation) that of wt M protein, and this difference was statistically significant (P < 0.05). These data suggest that, in the absence of other viral components, the aa 121-to-124 region of M protein plays a role in the association of M with membranes.
Association of M protein with membranes during infection with recombinant mutant virus does not depend on the hydrophobic-loop region.
The finding that mutation of the hydrophobic loop had an effect on the membrane association of M protein in transfection assays led us to determine the effect of this mutation in the context of infection with a recombinant mutant virus. We constructed infectious cDNAs encoding a mutant M protein and an isogenic control that expressed the wild-type M protein. From these cDNAs, we rescued infectious virus expressing the wild-type M protein or the mutant M protein. Sequencing of reverse transcription-PCR products proved that no additional mutations in the M gene had occurred during the recovery (data not shown). The association with cellular membranes of wt or mutant M protein expressed by the recombinant viruses was determined in experiments similar to those in Fig. 2D, except that virus-infected cells were analyzed rather than transfected cells. When the results of five independent experiments were analyzed (Fig. 3A), they showed differences from the behavior of M protein expressed in the absence of other viral components. First, it appeared that a smaller fraction of wt M associated with membranes during virus infection than with transfected cells. Second, the reduced association with the membrane that was previously seen in the φ mutant was not evident in the context of a virus infection, leading to the conclusion that defects in membrane association are not apparent in the virus expressing the mutant M protein.
FIG. 3.

Characterization of recombinant M protein mutant virus. (A) Membrane association of M protein from wt VSV (black bars) or protein from hydrophobic mutant VSV (white bars) was determined by fractionation of infected-cell lysates on sucrose flotation gradients. The error bars indicate standard deviations. (B) Association of wt and hydrophobic-mutant M proteins with NCM complexes from wild-type or mutant virions. The graph represents the quantitation of three separate experiments ± standard deviations. The solid lines represent experiments in which nucleocapsid-M complex was derived from wild-type control virus, and the dotted lines represent experiments in which NCM was derived from mutant virus. Wild-type control (▪); hydrophobic mutant (○).
We also used the recombinant mutant virus to test the effect of the hydrophobic-loop mutation on M protein binding to mutant NCM complexes. These experiments were similar to those shown in Fig. 2B but included NCM complexes from both wt VSV and the φ mutant virus. When NCM complexes derived from recombinant wt VSV were used, the wt and mutant M proteins showed equal abilities to associate with these NCM complexes at all concentrations (Fig. 4B), similar to the results seen in Fig. 2B. When NCM complexes from the recombinant mutant virus were used in this assay, we found a different response. Both the wild-type and mutant M proteins showed significantly lower apparent affinities for mutant than for wild-type NCM complexes (Fig. 3B). These results show that the association of M protein with NCM complexes is reduced when the hydrophobic loop is mutated; furthermore, it shows that this defect is not at the level of association of individual M protein molecules with the nucleocapsid, as the mutant M protein associated well with NCM complexes from wild-type virus. Rather, the defect was apparent only when NCM complexes from the mutant virus were used, suggesting that mutating the aa 121-to-124 region of the M protein affects either the cooperativity of M protein association with the nucleocapsids or a higher order of binding, such as nucleocapsid condensation.
FIG. 4.
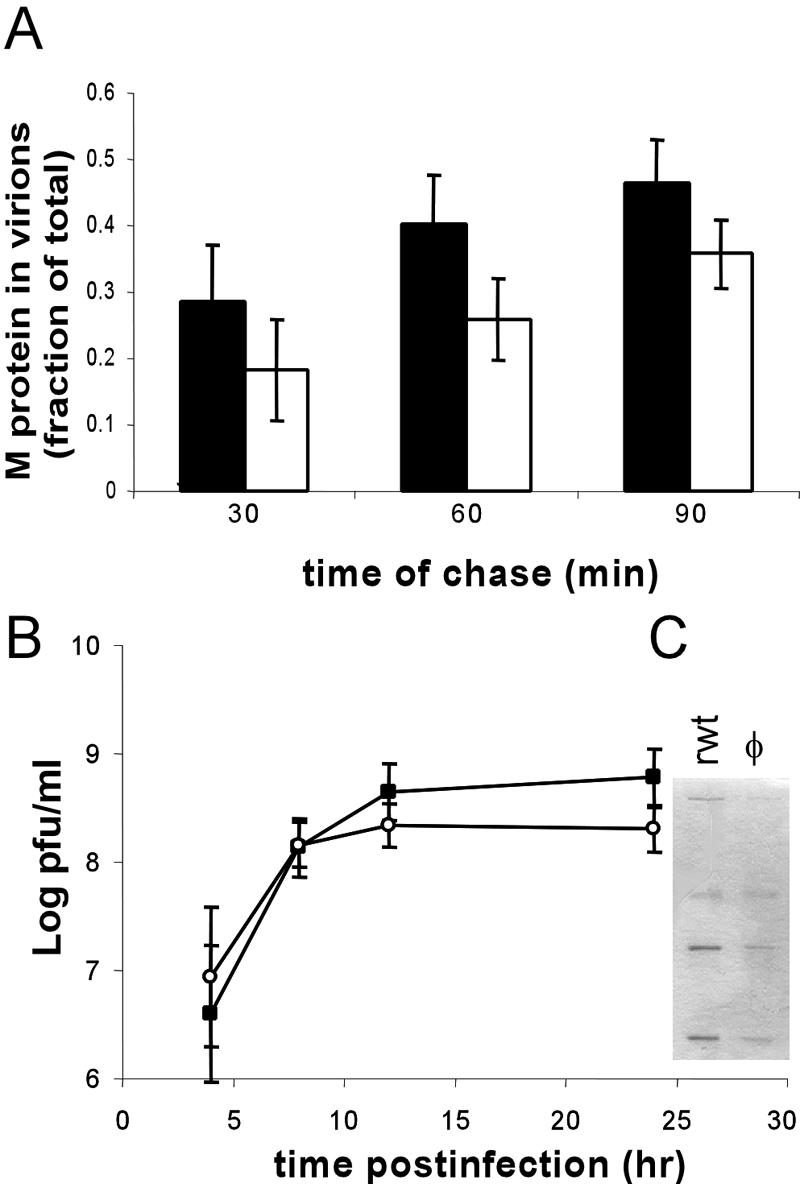
(A) Pulse-chase analysis of M protein incorporated into virions. The black bars are wild-type VSV, and the white bars are the hydrophobic-loop mutant. The bars represent the amount of radiolabeled M incorporated into virions over the amount of radiolabeled M present in virions plus cells. (B) Virus titers of recombinant wild-type VSV and recombinant VSV expressing the hydrophobic-loop mutant. Wild-type control (▪); hydrophobic mutant (○). The inset shows an SDS-PAGE gel of purified virus from media of infected cells (24 h p.i.). All experiments are the averages of three separate experiments ± standard deviations, except for B, which is five separate experiments.
To test whether this defect in NCM binding seen in the φ mutant led to a defect in budding, cells were infected with wt or mutant virus and then labeled with [35S]methionine for 60 min at 6 h postinfection (a time when only viral proteins are synthesized) and then chased for 30, 60, or 90 min. Following the chase period, media and cell lysates were obtained, and the amounts of [35S]methionine-labeled M protein in budded virions (released into the media) and in cell lysates (inside the cell) were determined. The amount of M protein in budded virions was then expressed as a fraction of the labeled M in virions plus the amount in cell lysates. The data from three of these experiments are depicted in Fig. 4A. For the wild-type virus, the amount of M protein incorporated into virions increased steadily from just under 30% in the first half hour to almost 50% at 90 min. The virus expressing the hydrophobic-loop mutant showed a similar steady increase in the amount of M protein incorporated into virions over this time scale, but a slightly smaller fraction of the total M was incorporated (20 to 38%). While this trend was seen at all times, it was not significant within the margin of error of the assay.
Single-step virus growth experiments (Fig. 4B) showed that the virus containing the hydrophobic-loop mutations grew to a slightly lower titer (2 × 108 for the mutant virus versus 6 × 108 for the wt virus), but this was not statistically significant within the margin of error of the plaque assay. To further investigate the small change in the virus titer, we purified virions from the media of rwt- and φ mutant virus-infected cells, and the virion proteins were analyzed by SDS-PAGE and Coomassie blue staining to determine whether there were differences in the amounts or the ratios of virus protein (Fig. 4C). Consistent with the small drop in virus titer observed in the single-step growth curve, we did see a difference in the total amount of virus proteins, with supernatants from the rwt virus-infected cells showing approximately two to three times more viral protein per ml of medium than the φ mutant virus-infected cells. There was no difference in the composition of the virus proteins between the mutant viruses, and the ratios of M protein to G and N proteins were the same in both viruses (not shown).
Viruses with the φ mutant M protein show defects in viral translation.
Contrary to expectations based on previous data (9, 10), the recombinant M protein mutant virus showed only minor defects in M protein function related to virus assembly (Fig. 3C and 4). None of these had a large effect on virus growth (Fig. 3B). However, following the recovery of the recombinant mutant virus, two dramatic phenotypes were noted: (i) the mutant virus showed defects in translation of viral mRNA and (ii) the virus showed a slower induction of apoptosis in BHK cells. The defect in viral translation is illustrated in Fig. 5. Cells were infected at an MOI of 10 with recombinant wild-type VSV or with the mutant virus expressing the M protein hydrophobic-loop M mutant. At increasing times postinfection, cells were pulse-labeled with [35S]methionine and analyzed by SDS-PAGE and phosphorimaging (Fig. 5A). The synthesis of host proteins is apparent as a ladder of dark bands in mock-infected cells and in lysates from early times postinfection, while viral proteins become apparent beginning at 4 h postinfection (p.i.) and are largely the only proteins synthesized by 8 h p.i. The recombinant wild-type virus and the virus expressing the φ mutant M protein showed similar abilities to suppress host protein synthesis. Quantitation of the effect of virus infection on host protein synthesis (Fig. 5B) by analyzing three separate experiments showed that both viruses strongly inhibited host gene expression, with equal kinetics. Strikingly, however, the hydrophobic-loop mutant appeared to synthesize viral protein at a lower rate (Fig. 5A). Quantitation of viral-protein synthesis (Fig. 5C) showed that the rate of viral-protein synthesis was lower than that seen in the wild-type control at all times and was only 10 to 20% of the rate of the wild-type VSV at 8 h p.i., when viral-protein synthesis is near maximal in cells infected with wild-type virus (3). This was true both in BHK and in HeLa cells (data for BHK cells only are shown for simplicity). To determine whether the change in promoting viral-protein synthesis could be caused by a subset of the mutations in the hydrophobic loop, we generated and recovered a second mutant virus that contained only two amino acid changes rather than four (AVLA→AVQQ). We then determined the rate of protein synthesis of the rwt, φ mutant, and AVQQ mutant at 8 h p.i. and found that the AVQQ mutant showed strong viral-protein synthesis at that time, similar to the wild-type virus (Fig. 4D).
FIG. 5.

Protein synthesis changes in cells infected with recombinant wt and hydrophobic-mutant viruses. (A) Cells were mock infected or infected with recombinant wt control or hydrophobic (φM)-mutant VSV for the indicated times and labeled with 35S methionine for 10 min. The cell lysates were electrophoresed on a 10% SDS-PAGE gel. A phosphorescence image of a representative gel is shown. (B) Quantification of host protein synthesis following virus infection. The error bars indicate standard deviations. (C) Quantification of viral-protein synthesis following virus infection. (D) Cells were mock infected or infected with wt control, φM mutant, or a 2-amino-acid mutation (AVLA→AVQQ) for 8 h and then labeled and treated as described for panel A.
One possible reason for lower viral-protein synthesis in the φ mutant virus was a lower level of viral mRNA. The levels of mRNA were determined by Northern blot analysis of M mRNA from mock-infected cells or cells infected with either wild-type or mutant virus for 6 or 8 h. These experiments (Fig. 6A) showed that there was a greater accumulation of viral mRNA in cells infected with the recombinant mutant virus than in those infected with the wild-type control. Cells infected with the mutant virus accumulated at least twice as much mRNA as those infected with the wild-type virus, despite the fact that the rate of viral-protein synthesis was only half that of the wild-type virus, demonstrating that mRNAs produced by this mutant virus had a much lower translation efficiency than mRNAs produced by wt virus.
FIG. 6.

Analysis of RNA accumulation, eIF2α phosphorylation, and mutation dominance in hydrophobic-mutant viruses. (A) Total RNA was prepared from cells mock infected or infected with wild-type control or hydrophobic-mutant VSV. The RNA was separated on a 1% agarose gel and transferred to a nitrocellulose membrane, and mRNA was detected using a 32P-labeled probe against M protein mRNA. (B) Phosphorylation of eIF2α following virus infection. Extracts from mock- or virus-infected cells were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and probed for phospho-eIF2α (Phos eIF2α), total eIF2α, and actin. (C) Cells were infected with wild-type control, hydrophobic-mutant virus, or both at the MOIs indicated above each lane; 8 h p.i., the cells were labeled with [35S]methionine and treated as described for panel A.
A possible explanation for this reduced level of viral-protein synthesis in cells infected with the recombinant mutant virus is the early stimulation of eIF2α phosphorylation as a host response to block viral-protein synthesis. Analysis of eIF2α phosphorylation (Fig. 6B) in BHK cells following infection with either wt or mutant virus showed that there was little if any difference in the phosphorylation of this translation initiation factor between the wild-type and mutant viruses. Both viruses induced very little eIF2α phosphorylation at early times postinfection, with increased phosphorylation becoming detectable above the mock-infected control only at 8 h p.i., after the defect in viral-protein synthesis was apparent.
An additional question about the decreased protein synthesis phenotype of the φ mutant was whether the weak viral-protein translation represented a loss of function or whether the φ mutation caused a dominant block of virus translation through an unknown mechanism. To test this, we performed coinfections with the wild-type virus and the φ mutant virus. Our expectations were that if the φ mutant virus was causing a dominant block of virus translation, the coinfection would display the φ mutant translation phenotype. If the φ mutant mutation resulted in a loss of function, the coinfection would show strong protein synthesis, like the wild-type virus. Figure 6C shows an autoradiograph of [35S]methionine-labeled protein extracts from wt VSV and φ mutant VSV coinfections labeled at 8 h p.i. Coinfections were done using the φ mutant at a constant MOI of 10, while the rwt virus used to coinfect was varied from an MOI of 10 to 1. In cells that were infected with rwt virus alone, robust viral-protein synthesis was seen, and in viruses infected with the φ mutant virus, reduced viral-protein synthesis was observed, as expected. In the cells that were coinfected with the wt and φ mutant viruses, the strong protein synthesis phenotype was always observed, demonstrating that the protein synthesis defect appears to be a loss of function in this M mutant virus that does not interfere with the strong protein synthesis encouraged by the wild-type virus.
Virus with φ mutant M protein shows slower induction of CPE and cell death.
A second unanticipated phenotype of the φ mutant virus was that cells infected with the virus showed delayed cytopathic effect (CPE) (cell rounding) following infection. This is illustrated in Fig. 7A, which shows images of BHK cells infected with either the hydrophobic mutant virus or the wild-type control virus at 10 h p.i. Almost all cells infected with the wild-type virus had rounded at this time, while among the cells infected by the φ mutant virus there were still a large number of cells that had not yet become rounded. Immunofluorescent labeling of these cells for expression of VSV G protein showed that all cells were infected and expressing viral proteins (data not shown). We used time-lapse microscopy to image and count the cells that were undergoing cell rounding and other signs of cell death. Quantitation of three experiments is shown in Fig. 7B for cell rounding and 7C for membrane blebbing and cell rupture. There was a small lag in cell rounding seen in cells infected with the hydrophobic-mutant virus. While 50% of cells infected with wild-type virus showed cell rounding by 4 h p.i., a similar proportion of rounding in cells infected with the hydrophobic-loop mutant did not occur until 15 to 20 h p.i. (Fig. 7B), a time at which 90% of wild-type control-infected cells were rounded. The onset of membrane blebbing showed an even greater lag. In cells infected by wild-type virus, 50% of the cells showed signs of membrane blebbing and cell rupture by 35 h p.i. and all had undergone membrane blebbing by 48 h p.i. (Fig. 7C). In contrast, cells infected with virus expressing the φ mutant M protein showed little membrane blebbing at 35 h p.i., and only 40% of these cells blebbed by 48 h p.i. Taken together, these data suggested that while the φ mutant M protein-expressing virus was still capable of inhibiting host cell gene expression, there was a significant delay in the induction of other host responses, such as cell rounding and death. This was confirmed by analysis of caspase 3 activity using a fluorogenic caspase 3 substrate. These experiments (shown in Fig. 7D) showed that there was less caspase 3 activity in the φ mutant virus-infected cells at 8, 12, and 24 h p.i.
FIG. 7.

Apoptosis induction in cells infected with wt and hydrophobic-mutant recombinant viruses. (A) Phase-contrast images of BHK cells infected with hydrophobic-mutant virus (left) or wt control virus (right) at an MOI of 10 for 10 h. (B) Cells were infected with wild-type control or hydrophobic-mutant virus and imaged by time-lapse microscopy (see Materials and Methods) for 48 h. Cell rounding is expressed as a fraction of the total number of cells in the field and as a function of time. (C) The cumulative fraction of cells entering apoptosis as determined by membrane blebbing and cell lysis consistent with apoptosis plotted as a function of time. The data in panels B and C are averages of three separate experiments with >150 cells per field. (D) Cells were mock infected or infected with wild-type control virus or hydrophobic-mutant virus for 8, 12, and 24 h. The cells were then lysed an analyzed for caspase 3-like activity using a fluorogenic substrate. The data are the averages of six separate experiments ± standard deviations.
Virus with φ mutant M protein shows decreased replication in MDBK cells.
The finding that the mutant VSV grew to titers similar to those of the wt in BHK cells but displayed decreased protein synthesis led us to characterize the growth of VSV in MDBK cells. Replication of VSV in BHK cells has been shown to be very robust, and mutants of VSV that synthesize less protein than wild-type VSV often show little or no attenuation in these cells (12). Other cells, however, more effectively restrict the replication of attenuated viruses (31). To determine whether the φ mutant virus displayed attenuation, we performed single-cycle and multiple-cycle growth experiments on the wild-type and φ mutant viruses in MDBK cell lines. Cells were infected at an MOI of 10 for single-cycle experiments and at an MOI of 0.1 for multiple-cycle experiments. Plaque assays of supernatants from either high or low MOI infection taken at 24 h p.i. (Fig. 8A) showed that wt virus grew to titers of ∼3 × 107, while the mutant virus grew to titers of 1 × 106 or 5 × 105, respectively, demonstrating that there was significant attenuation of virus growth in this mutant. Analysis of virus growth by Western blotting of cell extracts at 24 h p.i. (data not shown) showed significant accumulation of VSV G protein in cells infected with wt VSV but almost no detectable G protein in cells infected with the mutant virus. Consistent with this, MDBK cells infected with the wild-type virus at high and low MOIs showed significant CPE at 24 h p.i. MDBK cells infected with the φ mutant virus showed some CPE, but it was significantly lower than that seen with wild-type virus at both high and low MOIs (Fig. 8B).
FIG. 8.

Replication of wt and φ mutant virus in MDBK cells. Cells were infected with VSV or the φ mutant recombinant virus at an MOI of 10 or 0.1. At 24 h p.i., the cells were analyzed as follows. (A) Virus titers were determined from cells infected at high or low MOI. Wild-type control (▪); hydrophobic mutant (□). The titers shown are the means (plus standard deviations) of the results from three separate experiments using media from cells at 24 h p.i. (B) Phase-contrast imaging showing rwt-infected cells in the top images and cells infected by mutant virus in the bottom images.
DISCUSSION
The results presented here show that aa 121 to 124 of the VSV M protein play an important functional role in regulating viral translation and the induction of apoptosis in the context of a viral infection. The original aim of these studies—to test the role of this region in controlling self-association and association with cellular membranes—has led to the conclusion that this domain plays a minimal role in both of these processes. By analyzing M protein association using stopped-flow assays, we were able to document the initial self-association events of purified M protein and to show that there was very little difference in the rate of association (Fig. 1D). This argues that the hydrophobic nature of the aa 121-to-124 loop does not contribute significantly to the self-association of M protein. It is still possible that the region plays a role in conformational changes or a similar structurally important event leading to self-association, but the hydrophobic nature of the loop is not critical for this process.
Analyses of M protein association with membranes led to two different conclusions, depending on the experimental design. Wt or mutant M proteins expressed by recombinant viruses did not behave differently in their association with membranes. This suggests that the hydrophobic loop plays a minor role in interacting with the membrane during virus infection, contrary to previous hypotheses (10). However, expression of M protein in the absence of other viral components did show that the mutant M protein was reduced in membrane association, results which were similar to earlier reports (10). We found that analyzing M protein association with membranes in the absence of other viral components can lead to higher levels of association of wt M with the membrane, which may make minor differences in the mutant M protein more apparent. Alternatively, factors beyond the M protein's inherent affinity for membranes control the level of M protein found associated with membranes during virus infection. This hypothesis is particularly attractive, since previous experiments suggested that there is a regulation of M protein association with the nucleocapsid at the membrane (7). This regulation could be at the level of interactions with viral or host components that control membrane association.
An interesting discovery with this virus was that the mutation of the aa 121-to-124 hydrophobic loop of M protein changed the behavior of NCM complexes derived from the mutant virus. Both wild-type and mutant M proteins showed a decreased ability to associate with NCM complexes from mutant viruses. There was a fivefold decrease in the half-maximal binding in this assay when mutant NCM complexes were used. This decrease was not enough to affect the assembly of the virus, since there was little if any change in viral titers. This is consistent with results with another mutant M protein with a twofold decrease in affinity for NCM complexes that had no effect on assembly (21). These results suggest that regions beyond the N terminus of M are important for the assembly and stability of the NCM complex. The mechanism of this change in M protein exchange is not clear, as it could either reflect the fact that NCM complexes containing mutant M protein are in a different conformation or that the mutant M protein now lacks an important region for the cooperative association of M protein with nucleocapsids. Further biochemical and biophysical studies will be required to determine the exact nature of this alteration.
Incorporating the aa 121-to-124 mutation into a recombinant virus uncovered a previously unrecognized function of M protein in controlling virus translation. As illustrated in Fig. 5, mutation of the hydrophobic loop caused the virus to increase mRNA production, but there was less viral-protein synthesis. Of particular interest is the fact that the mutation appears to result in a loss of M function (illustrated by the coinfection experiments) (Fig. 6C) and of the kinetics of viral-protein synthesis in the mutant virus. In cells infected with the wild-type VSV, viral-protein synthesis increased even as host protein synthesis was decreasing. In cells infected with the mutant virus, translation of viral messages started out at lower levels than that of the wild-type virus and decreased as host protein synthesis decreased (Fig. 5B and C), suggesting that in the wild-type virus M protein acts to maintain translation of viral messages, while host messages are inhibited.
When these data are considered along with previous studies that have shown the M protein to be important for blocking host protein synthesis, they suggest that there are two ways in which the M protein interacts with the host translation apparatus. The M protein is important for blocking host protein synthesis (1, 4), but it is also a translational activator for viral mRNA, allowing or facilitating high levels of viral-protein synthesis. The high levels of mRNA evident in the M mutant virus (Fig. 6A) are likely an adaptive response of the virus—an effort to produce more protein in response to low levels of translation. The behavior of the φ mutant virus is similar to that of the previously identified S2 VSV mutant. This virus also showed an increased expression of viral mRNA and a decreased synthesis of viral protein (6), the first demonstration that a VSV factor was important for allowing viral-mRNA translation. The mutations that caused the S2 phenotype have not been published, but our results suggest that they may reside near or within the hydrophobic loop of the M protein.
In its failure to activate translation of viral mRNA, the φ mutant virus is analogous to protein synthesis-deficient mutants in another virus system, namely, adenovirus. Temperature-sensitive mutants of the 100,000-molecular-weight (100K) protein in adenovirus show a loss of viral late-gene expression, even though high levels of mRNA are synthesized (13). Recent studies have shown that the 100K protein becomes an integral part of an adenovirus-specific eIF4F complex during late viral infection (5) and link late viral mRNA to this translation initiation complex and to driving its translation (33). Given the similarities in mutation effects (mutation leading to an increase in viral-mRNA accumulation but a decrease in the rate of protein synthesis) on the adenovirus 100K proteins and the φM mutation, it is attractive to speculate that M protein may play a role in facilitating VSV mRNA translation.
The φM mutant virus also showed a delayed induction of apoptosis. This delay could be due to two phenomena. Earlier reports showed that the ability of M protein to induce apoptosis was linked to its ability to inhibit host gene expression (19). The aa 121-to-124 M protein mutant retains this ability, so it is unlikely that changes in apoptosis are due to this aspect of M function. In the context of a virus infection of BHK cells, the inhibition of host gene expression by M protein leads to slower apoptosis (8, 18). It is possible that the slightly increased inhibition of host gene expression seen at 8 h p.i. (Fig. 5A and B) further delays apoptosis in these cells. Additionally, it is likely that the lower levels of viral protein slow the induction of apoptosis, since other viral components in addition to the M protein are involved in the induction of apoptosis in VSV-infected cells (19).
The deficiencies noted in the mutant virus likely contribute to the attenuated growth that we see in MDBK cells (Fig. 7). Cells infected with the φ mutant virus showed less CPE, lower levels of protein accumulation, and a 1- to 2-log-unit loss in viral titer compared to wild-type recombinant VSV (Fig. 7B). The fact that this attenuation was noticeable in MDBK cells but less significant in BHK cells is similar to other VSV mutants. The S2 mutant discussed above and the host range-restricted mutants that are defective in mRNA methylation (12, 31) show similar phenotypes, growing well in BHK cells but poorly in other cell types. Both of these mutants have translational defects equal to or more severe than those seen in the φ mutant virus, suggesting that the translation defect is responsible for this attenuation. Mutants in Thieler's virus that have translation defects also show a similar pattern of strong growth in BHK cells and restricted growth in other cell types (27), so cell-type differences may play a role in the abilities of different RNA virus mRNAs to gain access to the translation apparatus.
The findings presented here, combined with earlier work on the importance of the M protein in apoptosis and inhibition of host gene expression, highlight the complex role of M in viral replication and pathogenesis. Mutations like the one described here may have a primary effect (such as changing viral or host translation) that leads to secondary effects that influence virus-host interactions, like apoptosis, or virus assembly events, like M-nucleocapsid association. Understanding these roles will involve further efforts to delineate the specific functions and interactions that M controls but will provide important insight into how virus proteins directly and indirectly influence virus-host interactions and virus replication.
Acknowledgments
We thank Griffith Parks for his critical reading of the manuscript.
This work was supported by grants RO1 AI015892 and RO1 AI052304 to D.S.L.
REFERENCES
- 1.Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S. Lyles. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 77:4646-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black, B. L., R. B. Rhodes, M. McKenzie, and D. S. Lyles. 1993. The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. J. Virol. 67:4814-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connor, J. H., and D. S. Lyles. 2005. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 280:13512-13519. [DOI] [PubMed] [Google Scholar]
- 4.Connor, J. H., and D. S. Lyles. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177-10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuesta, R., Q. Xi, and R. J. Schneider. 2000. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBO J. 19:3465-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis, N. L., and G. W. Wertz. 1980. A VSV mutant synthesizes a large excess of functional mRNA but produces less viral protein than its wild-type parent. Virology 103:21-36. [DOI] [PubMed] [Google Scholar]
- 7.Flood, E. A., M. O. McKenzie, and D. S. Lyles. 2000. Role of M protein aggregation in defective assembly of temperature-sensitive M protein mutants of vesicular stomatitis virus. Virology 278:520-533. [DOI] [PubMed] [Google Scholar]
- 8.Gaddy, D. F., and D. S. Lyles. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudier, M., Y. Gaudin, and M. Knossow. 2001. Cleavage of vesicular stomatitis virus matrix protein prevents self-association and leads to crystallization. Virology 288:308-314. [DOI] [PubMed] [Google Scholar]
- 10.Gaudier, M., Y. Gaudin, and M. Knossow. 2002. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 21:2886-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudin, Y., A. Barge, C. Ebel, and R. W. Ruigrok. 1995. Aggregation of VSV M protein is reversible and mediated by nucleation sites: implications for viral assembly. Virology 206:28-37. [DOI] [PubMed] [Google Scholar]
- 12.Grdzelishvili, V. Z., S. Smallwood, D. Tower, R. L. Hall, D. M. Hunt, and S. A. Moyer. 2005. A single amino acid change in the l-polymerase protein of vesicular stomatitis virus completely abolishes viral mRNA cap methylation. J. Virol. 79:7327-7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayes, B. W., G. C. Telling, M. M. Myat, J. F. Williams, and S. J. Flint. 1990. The adenovirus L4 100-kilodalton protein is necessary for efficient translation of viral late mRNA species. J. Virol. 64:2732-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayakar, H. R., E. Jeetendra, and M. A. Whitt. 2004. Rhabdovirus assembly and budding. Virus Res. 106:117-132. [DOI] [PubMed] [Google Scholar]
- 15.Jayakar, H. R., K. G. Murti, and M. A. Whitt. 2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol. 74:9818-9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaptur, P. E., M. O. McKenzie, G. W. Wertz, and D. S. Lyles. 1995. Assembly functions of vesicular stomatitis virus matrix protein are not disrupted by mutations at major sites of phosphorylation. Virology 206:894-903. [DOI] [PubMed] [Google Scholar]
- 17.Kaptur, P. E., R. B. Rhodes, and D. S. Lyles. 1991. Sequences of the vesicular stomatitis virus matrix protein involved in binding to nucleocapsids. J. Virol. 65:1057-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopecky, S. A., and D. S. Lyles. 2003. Contrasting effects of matrix protein on apoptosis in HeLa and BHK cells infected with vesicular stomatitis virus are due to inhibition of host gene expression. J. Virol. 77:4658-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenard, J., and R. Vanderoef. 1990. Localization of the membrane-associated region of vesicular stomatitis virus M protein at the N terminus, using the hydrophobic, photoreactive probe 125I-TID. J. Virol. 64:3486-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyles, D. S., and M. O. McKenzie. 1998. Reversible and irreversible steps in assembly and disassembly of vesicular stomatitis virus: equilibria and kinetics of dissociation of nucleocapsid-M protein complexes assembled in vivo. Biochemistry 37:439-450. [DOI] [PubMed] [Google Scholar]
- 22.Lyles, D. S., M. O. McKenzie, and R. R. Hantgan. 1996. Stopped-flow, classical, and dynamic light scattering analysis of matrix protein binding to nucleocapsids of vesicular stomatitis virus. Biochemistry 35:6508-6518. [DOI] [PubMed] [Google Scholar]
- 23.Lyles, D. S., M. O. McKenzie, P. E. Kaptur, K. W. Grant, and W. G. Jerome. 1996. Complementation of M gene mutants of vesicular stomatitis virus by plasmid-derived M protein converts spherical extracellular particles into native bullet shapes. Virology 217:76-87. [DOI] [PubMed] [Google Scholar]
- 24.McCreedy, B. J., Jr., and D. S. Lyles. 1989. Distribution of M protein and nucleocapsid protein of vesicular stomatitis virus in infected cell plasma membranes. Virus Res. 14:189-205. [DOI] [PubMed] [Google Scholar]
- 25.Newcomb, W. W., and J. C. Brown. 1981. Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J. Virol. 39:295-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newcomb, W. W., G. J. Tobin, J. J. McGowan, and J. C. Brown. 1982. In vitro reassembly of vesicular stomatitis virus skeletons. J. Virol. 41:1055-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilipenko, E. V., A. P. Gmyl, S. V. Maslova, E. V. Khitrina, and V. I. Agol. 1995. Attenuation of Theiler's murine encephalomyelitis virus by modifications of the oligopyrimidine/AUG tandem, a host-dependent translational cis element. J. Virol. 69:864-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pornillos, O., J. E. Garrus, and W. I. Sundquist. 2002. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 12:569-579. [DOI] [PubMed] [Google Scholar]
- 29.Sarkar, G., and S. S. Sommer. 1990. The “megaprimer” method of site-directed mutagenesis. BioTechniques 8:404-407. [PubMed] [Google Scholar]
- 30.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simpson, R. W., J. F. Obijeski, and M. P. Morrongiello. 1979. Conditional lethal mutants of vesicular stomatitis virus. III. Host range properties, interfering capacity, and complementation patterns of specific hr mutants. Virology 93:493-505. [DOI] [PubMed] [Google Scholar]
- 32.Timmins, J., R. W. Ruigrok, and W. Weissenhorn. 2004. Structural studies on the Ebola virus matrix protein VP40 indicate that matrix proteins of enveloped RNA viruses are analogues but not homologues. FEMS Microbiol. Lett. 233:179-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xi, Q., R. Cuesta, and R. J. Schneider. 2004. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 18:1997-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]