

Abstract
Hepatitis C virus (HCV) is a human pathogen affecting nearly 3% of the world’s population1. Chronic infections can lead to cirrhosis and liver cancer. The RNA replication machine of HCV is a multi-subunit membrane-associated complex. The non-structural protein NS5A is an active HCV replicase component2,3, a pivotal regulator of replication2,4, and a modulator of cellular processes spanning from innate immunity to dysregulated cell growth5,6. NS5A is a large phosphoprotein (56–58 kDa) with an amphipathic α-helix at its N-terminus that promotes membrane association7–9. Following this helix, NS5A is organized into three domains (Fig. 1a)10. The N-terminal domain (domain I) coordinates a single zinc atom per protein molecule10. Mutations disrupting either the membrane anchor7,16 or zinc binding10 are lethal for RNA replication. Probing the role of NS5A in replication has been hampered by the lack of structural information for this enigmatic multifunctional protein. Herein we report the structure of domain I at 2.5 Å resolution, revealing a novel fold, a new zinc-coordination motif, and a disulfide bond. Molecular surface analysis suggests the location of protein, RNA, and membrane interaction sites.
The structure of domain I (amino acids 36-198) reveals two identical monomers per asymmetric unit packed as a dimer via contacts near the N-terminal ends of the molecules. For ease of discussion, the molecule is divided into two subdomains; the N-terminal subdomain IA and the C-terminal subdomain IB (figures 1b-e). A more schematic view of the fold is shown in Figure 1e. The DALI11 server was unsuccessful at identifying structures related to domain I or either subdomain, indicating this protein represents a novel fold. Subdomain IA consists of an N-terminal extended loop lying adjacent to a 3-stranded anti-parallel β-sheet (strands B1, B2 and B3), with α-helix H2 (designated H2 to allow numbering of the N-terminal membrane anchoring helix H18) at the C-terminus of the third β-strand. These elements comprise the structural scaffold for a four-cysteine zinc atom coordination site at one end of the β-sheet. A view of subdomain IA (aa 36-100) highlighting the cysteine residues involved in zinc coordination is shown in Figure 2a and 2b. The anti-parallel β-sheet, composed of strands B1, B2, and B3, positioning Cys 57, Cys 59, and Cys 80 near the zinc-binding site is essentially as predicted in our previous model.10 The long random coil region positioning Cys 39 and connecting the N-terminus to the β-sheet was incorrectly predicted to be a β-strand, but the arrangement of this region in relation to the other strands matches the original model. The distances of the cysteine side chain sulfur groups to the zinc atom for Cys 39 (2.36 Å), Cys 57 (2.47 Å), Cys 59 (2.42 Å) and Cys 80 (2.45 Å) observed in domain I are close to the ideal 2.35 (+/− 0.09) Å distances for structural metal zinc coordination sites12. Similarly, the side chain geometries are within the acceptable limits of published values12. DALI11 database searches of the subdomain IA region were unable to locate similar metal binding folds. The location of the zinc-binding site in the structure of domain I, combined with previous biochemical characterization, suggests zinc plays a structural role in NS5A fold maintenance. Following the zinc-coordination site, a stretch of random coil exits H2 and passes across the β-sheet and N-terminal strand in an orientation orthogonal to the plane of the sheet. This coiled sequence then enters a tight turn and runs adjacent to the β-sheet away from the zinc atom coordination site before entering a proline rich region that connects subdomain IA to subdomain IB.
Figure 1.
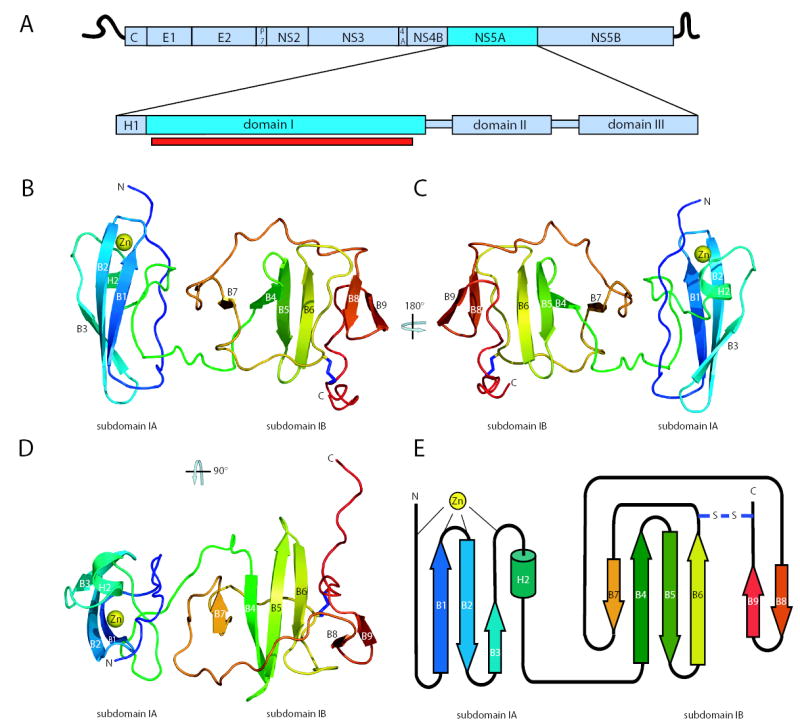
An overview of the NS5A domain I structure. a. Schematic of HCV genome organization and domain structure of the NS5A protein. The portion of domain I in this structure is indicated by the red bar. b. Ribbon diagram of the structure of domain I. The polypeptide chain is colored from N-terminus (blue) to C-terminus (red). The coordinated zinc atom is shown in yellow. The disulfide bond is shown in blue. c. A 180° rotation showing the ‘back’ of domain I. d. A 90° rotation showing the ‘top down’ view of domain I. e. Domain I topology organization model.
Figure 2.
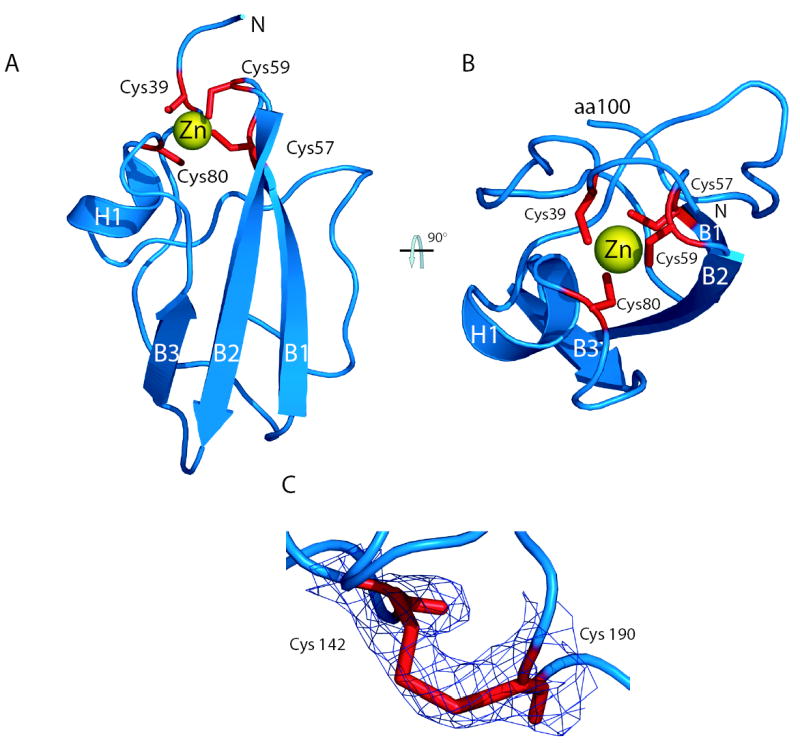
The NS5A zinc-binding motif and disulfide bond. a. A view of subdomain 1A (amino acids 36-100) highlighting the zinc-binding motif. The zinc atom (yellow) is coordinated by four cysteine residues (red). b. A ‘top down’ view of the zinc ion coordination site showing caging of the zinc atom. c. Overlay of experimental electron density map on the model of the disulfide bond at 1σ. Amino acids cysteine 142 and 190 are labeled.
Subdomain IB consists of a four-strand anti-parallel β-sheet (B4, B5, B6, and B7) and a small two-strand anti-parallel β-sheet near the C-terminus (B8 and B9) surrounded by extensive random coil structures (best seen in figure 1d). Perhaps the most surprising observation from model building and refinement of the structure was the presence of a disulfide bond near the C-terminus of domain I. The disulfide bond connects the sidechains of the conserved Cys 142 and Cys 190, resulting in a covalent link between the loop exiting from β-strand B6 to the C-terminal extension of strand B9. Model refinement without the disulfide placed the sidechains of these cysteines in an unfavourable proximity. Refinement with the disulfide led to no problematic geometry for either cysteine, and generated a model that better fit the electron density (Figure 2c). Density corresponding to the disulfide bond is present in both molecules in the asymmetric unit, providing two independent views of this feature. The disulfide bond in the model results in a sulfur to sulfur atom distance of 2.03Å, an ideal value for bond formation13. Although all evidence points to the presence of a disulfide in the crystal, it is not yet clear if this bond exists in NS5A in the context of an HCV infection. Mutagenesis of these cysteine residues produced only a mild defect in RNA replication, indicating the disulfide is not required for the RNA replicase functions of NS5A10. However, it is enticing to imagine the disulfide bond, tethering the C-terminus of domain I and likely altering the arrangement of the C-terminal domain II and III, plays a regulatory role in NS5A function, perhaps serving as a conformation switch to modulate functions of NS5A in and out of the replicase.
The analysis of the molecular surface of domain I is important to furthering our understanding of NS5A. Figure 3a presents two rotational views of the solvent accessible molecular surface of NS5A domain I colored by electrostatic potential. The orientations, left to right, are identical to those shown in ribbon diagrams in figure 1b and c, respectively. Overall these views highlight the strikingly uneven charge distribution within domain I. The N-terminal subdomain IA region has an almost exclusively basic surface, whereas the C-terminal subdomain 1B is predominantly acidic. This unusual charge distribution has interesting implications in the dimeric form of the protein.
Figure 3.
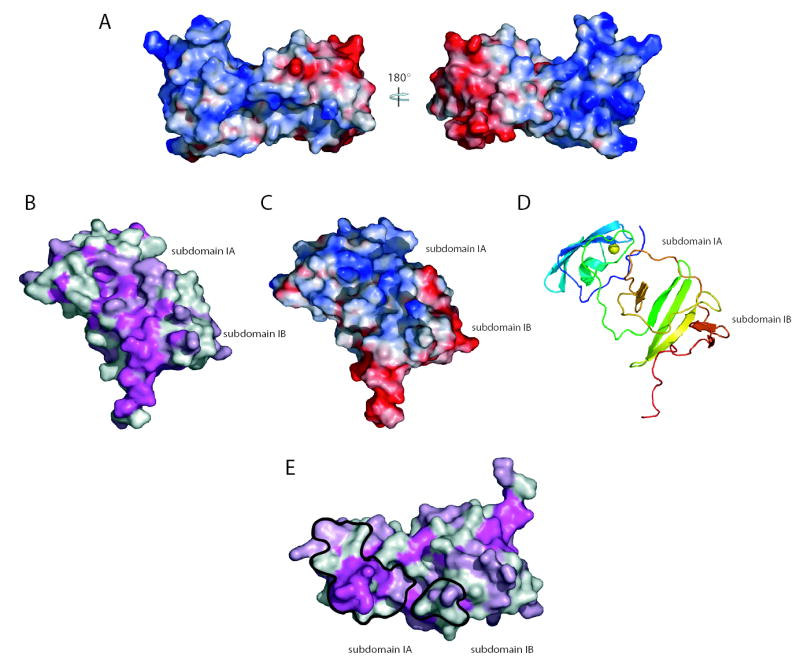
Molecular surfaces of domain I. a. Domain I surface potential, acidic (red), neutral (white), and basic (blue) surfaces are shown. Two rotations of domain I are shown corresponding to the views in 1b and 1c. b. Surface of domain I colored by conservation; magenta ≥ 95% conservation, light pink = 75–95% conservation, and white ≤ 75% conservation. Domain I is oriented to show the most conserved molecular surface. c. A surface potential view of the image in b. d. A ribbon diagram view of the orientation in c and d. e. Conservation plot showing the dimer interface surfaces (black outline).
Analysis of the conservation of surface exposed residues of domain I was performed to define potential sites of interactions. A conservation plot, based on sequence alignments of domain I regions from the 30 HCV genotype reference sequences from the Los Alamos HCV database14 and the related GB virus B is shown in figure 3b. The plot shows the significant overall surface conservation of domain I, and highlights a large patch of conserved residues that may represent a molecular interaction surface. This contiguous surface spans subdomain IA and IB, including a ‘pocket’ generated by the interface between these elements. An electrostatic potential plot of this same surface is also shown, highlighting the complex mixture of charged and hydrophobic residues generating this surface (figure 3c). A ribbon diagram of this orientation of domain I is shown in figure 3d. A large number of proteins have been shown to interact with domain I6. Whether one of the diverse cellular proteins or a viral component of the replicase, including the other domains of NS5A, interacts with this surface remains to be determined.
Another surface of considerable interest was the buried region between monomers to create the dimeric domain I seen in the crystal structure. Figure 3e presents a surface conservation plot highlighting these interaction patches (orientation similar to figure 1d). The dimer interface of 678 Å2 consists of two patches of buried surface area, one located primarily in subdomain IA, and a second located in subdomain IB. The total buried surface area in the domain I dimer is greater than the generally accepted standard of 600 Å2 for protein interfaces and has good shape and electrostatic complementarity15. The contact patch in subdomain IA contains a number of conserved residues, including several residues surrounding the zinc coordination site of domain I. The smaller patch in subdomain IB is of lower sequence conservation. It is important to note that residues of lower conservation involved in these interfaces appear to primarily be involved in mainchain contacts. Homotypic oligomeric interactions of the NS5A protein have been described16. Analytical ultracentrifugation suggests that domain I is monomeric, although it is important to note that these experiments were performed with only a portion of the NS5A protein in the absence of membranes and RNA. It is likely that in the context of the HCV replicase complex, NS5A is present in very high local concentrations that may drive oligomerization. NS5A may, in this respect, be similar to the NS5B protein that is monomeric in solution, yet readily forms 2D arrays on membrane surfaces17. Perhaps the conditions that favor protein-protein interactions in crystallization have captured a glimpse of part of the relevant NS5A oligomerization interactions. While more work is needed to determine the oligomeric state of NS5A in the replicase, the dimeric form presented herein provides a number of interesting features that may have relevance to HCV biology. Figure 4a presents three rotations of the dimer showing the interface between monomers and the large groove between the domain IB regions of the monomers generated by the “claw-like” shape of the dimer.
Figure 4.
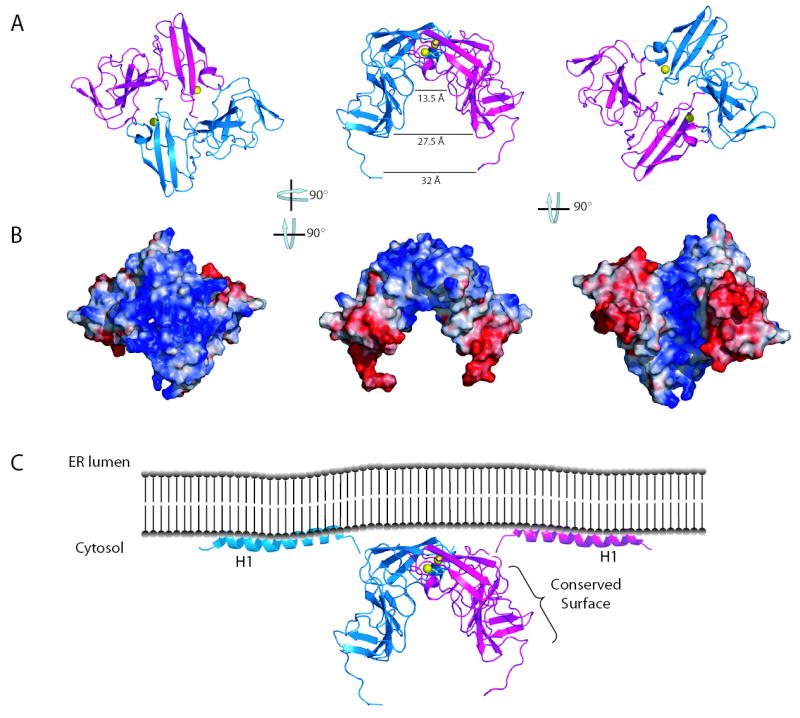
The NS5A domain I dimer reveals potential interaction surfaces. a. Ribbon diagrams of three rotations of the domain I dimer. b. Surface potential plots of the domain I dimer, views correspond to those show in a. Analysis of images in a and b reveal the dimer creates a relatively flat, basic surface near the N-terminus and a large groove between the two subdomain IB regions. c. Model of NS5A position relative to the ER membrane. The location of the conserved surface in figure 3 b is indicated. Helices are from the recent structure of the NS5A N-terminal helix8.
As NS5A likely makes contacts with proteins, RNA, and membranes, an analysis of electrostatic surface potential is important in determining how a dimeric domain I might fit into the replicase (Figre 4b). One interesting feature in these surface potential plots is the basic surface of domain I near the N-termini of the dimer. An NMR structure of the membrane anchor of NS5A has recently been determined8. This anchoring helix is five residues from the N-terminus of the domain I structure suggesting domain I is close to the membrane. The basic nature of the surface of domain I close to the anchor helix is logical, as the protein is likely in close contact with the negatively charged head groups of the membrane. This interaction would position the domain I dimer groove facing away from the membrane where it could interact with RNA. Interactions of NS5A with RNA during protein purification have been described18, although specific binding remains to be demonstrated. This groove is an attractive RNA binding pocket, especially when surface potentials of the groove are plotted. Modeling suggests the groove is of sufficient dimensions to bind to either single or double stranded RNAs. The deep, highly basic portion of the groove has a diameter of 13.5 Å. The groove then expands to diameter of approximately 27.5 Å in a less charged, polar boundary region. An RNA molecule could easily fit in the groove, making both electrostatic contacts with the deep basic groove and other contacts with the groove boundary region. An exposed tryptophan resides (Trp 84) lies on the floor of the basic groove where it could interact with RNA via stacking interactions. The ‘arms’ extending out past this groove are more acidic, perhaps serving as a clamp to prevent RNA from exiting the groove. The positioning of NS5A domain II and III relative to the groove remains unclear, but it is interesting to imagine these domains interact with the RNA positioned by the domain I groove. Furthermore, the dimeric form of domain I places the conserved interaction surface described for the monomer on the outside of the ‘arms’ of the dimer, where they would be available for interactions. A model of the NS5A dimer in relation to the cellular membrane and the N-terminal membrane anchor is presented in figure 4c. The sequence of domain I, highlighting secondary structures and important residues and surfaces has been provided as supplementary material.
The crystal structure of the domain I region of NS5A provides the first molecular view of this important protein, and perhaps a new class of viral replicase proteins. The high-resolution view of domain I has practical applications for anti-viral drug design and provides a rational framework for experimentally addressing NS5A function in the replicase and in the regulation of cellular processes. These experiments may ultimately provide clues to how the complex replicase machinery functions to replicate HCV RNA.
Methods
Protein Preparation
The expression and purification of genotype 1b, Con1 subtype NS5A Domain I-Δh was performed as described previously10. Following initial purification, the C-terminal polyhistidine tag and linker sequence were proteolytically removed from NS5A-Domain I-Δh by on overnight incubation at 25°C in the presence of recombinant light chain enterokinase protease. Once cleavage was complete, the protein was repurified by ion exchange on a HiPrep 16/20 DEAE column using conditions described previously to separate the protease and tag from Domain I-Δh protein. The protein was then exchanged into buffer A (25 mM Tris-HCl pH 8.0, 175 mM NaCl, and 20% (v/v) glycerol) using a HiPrep 26/10 desalting column (Amersham/Pharmacia). Protein was then concentrated to 20–30 mg/ml via Amicon ultra 4 centrifugal concentrators (Millipore). Protein yields were typically between 10 and 12 mg/L of bacterial culture at an estimated 95% purity.
Crystal Growth and Freezing
Crystals of NS5A Domain I-Δh were grown by hanging drop vapor diffusion at 4°C on siliconized cover slips in 24 well Linbro plates. The 1 milliliter of well solution contained 100 mM HEPES pH 7.0, 0.6 M trisodium citrate dihydrate, and 13% (v/v) isopropanol. The drop contained 1.5 microliters of Domain I-Δh protein at 26 mg/ml in buffer A, 1.25 microliters of well solution, and 0.5 microliters of 2 M non-detergent sulfobetaine 201. Crystals of 0.15–0.3 mm in size grew from these conditions in approximately 8 days. For freezing, crystals were transferred from hanging drops to a 2 microliter drop of well solution plus 20% (v/v) glycerol and incubated at 4°C for ten minutes. Crystals were then transferred to a 2 microliter drop of buffer A with a total of 30% (v/v) glycerol and allowed to equilibrate for 10 minutes. Crystals were then harvested and flash frozen in liquid propane.
Data Collection
All data collection was performed at beam line X9A at Brookhaven National Labs National Synchrotron Light Source. Phases were determined from the endogenous zinc present in NS5A. Prior to diffraction data collection, X-Ray fluorescence scans were used to confirm the presence of zinc in the protein crystals and determine the zinc K absorption edge. For zinc multiwavelength anomalous diffraction (MAD), data were collected at the zinc absorption maxima (9660 eV, 1.28345 Å), the maxima inflection point (9657 eV, 1.28385 Å), and at a remote wavelength (9757 eV, 1.27069 Å). Data collection was limited to 2.5 Å resolution by a combination of detector geometry and the presence of an unusually long axis in the crystal.
Data Processing and Model Building
Data were processed and scaled using DENZO/SCALEPACK19. The data for each of the three MAD wavelength datasets were then used to locate anomalous peaks corresponding to the presence of the zinc atom in NS5A. The two zinc sites were found by the program SOLVE 20. An interpretable electron density map was obtained using MLPHARE followed by density modification and phase combination by DM 21. The inflection data set was used for electron density map calculation. Several rounds of iterative model building and refinement were performed using the programs O 22 and CNS 23. The final model contained 96 solvent molecules, two zinc atoms, and two molecules of NS5A consisting of amino acids 36-198 of NS5A, with density not observed for the extreme amino and C-termini. A summary of the final refinement statistics is provided in table 1. PROCHECK 24 revealed no unfavorable (ϕ,Ψ) combinations, and mainchain and side chain structural parameters consistently better than or within the average for structures refined to 2.5 Å. Atomic coordinates have deposited in the Protein Data Bank (PDB ID Code to be supplied at galley proof stage). Secondary structures were assigned using the program DSSP 25. Graphics presented in this manuscript were generated using the programs PyMOL26. APBS was used for calculating surface potentials27. Sequence alignments were performed using ClustalX 28 and plotting of conservation to molecular surfaces was performed using msf_similarity_to_pdb (Dr. David Jeruzalmi, personal communication).
Table 1.
Summary of crystal parameters, data collection, and refinement statistics.
| Crystal Parameters | ||||||
|---|---|---|---|---|---|---|
| Spacegroup | Unit cell lengths (a,b) (Å) | Unit cell lengths (c) (Å) | Unit cell angles (α,β,γ) | Molecules per asymmetric unit | ||
| P4122 | 55.28 | 312.30 | 90° | 2 | ||
|
Data Collection | ||||||
| Wavelength (Å) | Resolution (Å) | Reflections measured/unique | Completenessa (%) | Rsymab (%) | I/σ(I)a | Phasing Powerc (anomalous) |
| λ1 = 1.28345 | 30.0-2.50 | 301,420/17,992 | 100 (99.9) | 6.9 (32) | 23.8 (4.7) | 1.18 |
| 30.0-2.50 | 302,219/17,992 | 100 (99.9) | 4.5 (34) | 22.4 (4.3) | 1.25 | |
| 30.0-2.50 | 314,341/18,007 | 100 (100) | 8.7 (27) | 35.6 (5.7) | 1.21 | |
| figure of merit | acentric | centric | overall | |||
| 0.48 | 0.40 | 0.46 | ||||
|
Refinement against λ3 | ||||||
| Resolution (Å) | Cut-off | Reflections | Completeness (%) | Rcrystd (%) | Rfreee (%) | |
| 30.0-2.50 | |F|/σ|F|>2.0 | 29,570 | 92.5 | 22.1 | 28.6 | |
|
Root Mean Square Deviations | ||||||
| Root mean square deviation molecules C α (Å) | Bond lengths (Å) | Bond angles | Thermal parameters mainchain atoms (Å2) | Thermal parameters sidechain atoms (Å2) | ||
| 0.43 | 0.00625 | 1.34° | 1.28 | 2.15 | ||
Values reported in the format: overall data (last resolution shell)
Rsym =∑|I−<I>|/∑I, where I is observed intensity and <I> is average intensity obtained from multiple observations of symmetry-related reflections.
Phasing power = rms (|FH|/E), where |FH| = heavy atom structure factor amplitude and E = residual lack of closure.
Rcryst =∑|Fobs−Fcalc|/∑|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
Rfree is the same as Rcryst, but is calculated with 10% of the data.
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. Roderick MacKinnon, Dr. Seth Darst, and Hans Mueller for the use of x-ray diffractometers, related equipment, and software at The Rockefeller University. We appreciate access to beamline X9A at the National Synchrotron Light Source (NSLS), Brookhaven National Labs and acknowledge the assistance of the NSLS staff. John-William Carroll provided vital assistance in data collection. Dr. David Jeruzalmi kindly provided the program msf_similarity_to_pdb. We wish to thank Drs. Seth Darst, Matt Evans, Annick Gauthier, Christopher Jones, Brett Lindenbach, Ivo Lorenz, Thomas von Hahn, and Ms. Melissa and Kathryn Tellinghuisen for critical reading of this manuscript. T.T. was supported, in part, by fellowships from the Charles H. Revson Foundation for Biomedical Research and the National Institutes of Health Ruth L. Kirschstein National Research Service Award (5F32 AI51820-03) granted through the National Institute of Allergy and Infectious Disease. J.M. was supported as a Merck Fellow of the Life Sciences Research Foundation. Additional financial support for this work came from grant 5 R01 CA57973-12 from the National Institutes of Health and the Greenberg Medical Research Institute (C.M.R.). T.T., J.M., and C.M.R. conceived these experiments. T.T. generated all reagents, materials, proteins, and crystals used herein with J.M.’s assistance. J.M. and T.T. carried out all data collection. Data processing, model building, and refinement was performed by T.T., with significant input and assistance from J.M. The manuscript was written by T.T. with comments and assistance from the remaining authors.
Footnotes
Supplementary Information accompanies the paper on Nature’s website (http://www.nature.com).
Competing interest statement The authors declare that they have no competing financial interests.
References
- 1.Anonymous World Health Organization - Hepatitis C: global prevalence. Wkly Epidemiol Rec. 1997;72:341–4. [PubMed] [Google Scholar]
- 2.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 3.Lohmann V, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 4.Lohmann V, Korner F, Dobierzewska A, Bartenschlager R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J Virol. 2001;75:1437–1449. doi: 10.1128/JVI.75.3.1437-1449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tellinghuisen TL, Rice CM. Interaction between hepatitis C virus proteins and host cell factors. Current Opinion in Microbiology. 2002;5:419–27. doi: 10.1016/s1369-5274(02)00341-7. [DOI] [PubMed] [Google Scholar]
- 6.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 7.Elazar M, et al. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J Virol. 2003;77:6055–61. doi: 10.1128/JVI.77.10.6055-6061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penin F, et al. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem. 2004;279:40835–43. doi: 10.1074/jbc.M404761200. [DOI] [PubMed] [Google Scholar]
- 9.Brass V, et al. An Amino-terminal Amphipathic alpha -Helix Mediates Membrane Association of the Hepatitis C Virus Nonstructural Protein 5A. J Biol Chem. 2002;277:8130–9. doi: 10.1074/jbc.M111289200. [DOI] [PubMed] [Google Scholar]
- 10.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem. 2004 doi: 10.1074/jbc.M407787200. [DOI] [PubMed] [Google Scholar]
- 11.Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 12.Alberts IL, Nadassy K, Wodak SJ. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 1998;7:1700–16. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engh R, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Cryst A. 1991;47:392–400. [Google Scholar]
- 14.The Hepatitis C Databases. http://gluttony.lanl.gov/content/hcv-db/
- 15.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimitrova M, Imbert I, Kieny MP, Schuster C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol. 2003;77:5401–14. doi: 10.1128/JVI.77.9.5401-5414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang QM, et al. Oligomerization and Cooperative RNA Synthesis Activity of Hepatitis C Virus RNA-Dependent RNA Polymerase. J Virol. 2002;76:3865–72. doi: 10.1128/JVI.76.8.3865-3872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L, et al. Purification and characterization of hepatitis C virus non-structural protein 5A expressed in Escherichia coli. Protein Expr Purif. 2004;37:144–53. doi: 10.1016/j.pep.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski, Z. & Minor, M. in Macromolecular Crystallography, part A, 307–326 (Academic Press (New York), 1997).
- 20.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55 (Pt 4):849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 22.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 (Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54 (Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–67. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch WaSC. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 26.DeLano, W. L. (http;//www.pymol.org, 2002).
- 27.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.