

Abstract
Mouse bone marrow cells transduced with retroviral vectors encoding either of two oncogenic Bcr-Abl isoforms (p210Bcr-Abl and p185Bcr-Abl) induce B cell lympholeukemias when transplanted into lethally irradiated mice. If the activity of the Arf tumor suppressor is compromised, these donor cells initiate a much more highly aggressive and rapidly fatal disease. When mouse bone marrow cells expressing Bcr-Abl are placed in short-term cultures selectively designed to support the outgrowth of pre-B cells, only those lacking one or two Arf alleles can initiate lympholeukemias when inoculated into immunocompetent, syngeneic recipient mice. Although the ABL kinase inhibitor imatinib mesylate (Gleevec) provides highly effective treatment for BCR-ABL-positive chronic myelogenous leukemia, it has proven far less efficacious in the treatment of BCR-ABL-positive acute lymphoblastic leukemias (ALLs), many of which sustain deletions of the INK4A-ARF (CDKN2A) tumor suppressor locus. Mice receiving Arf−/− or Arf+/− p210Bcr-Abl-positive pre-B cells do not achieve remission when maintained on high doses of oral imatinib therapy and rapidly succumb to lympholeukemia. Although cells expressing the Bcr-Abl kinase can proliferate in the absence of IL-7, they remain responsive to this cytokine, which can reduce their sensitivity to imatinib. Treatment of Arf−/−, p210Bcr-Abl-positive pre-B cells with imatinib together with an inhibitor of JAK kinases abrogates this resistance, suggesting that this combination may prove beneficial in the treatment of BCR-ABL-positive acute lymphoblastic leukemia.
Keywords: Arf tumor suppressor, Gleevec, IL-7, JAK kinases, drug resistance
The Philadelphia chromosome (Ph) (1), arising from a balanced translocation involving chromosomes 9 and 22 (2), is the founding genetic lesion and cytogenetic hallmark of chronic myelogenous leukemia (CML) and of a subset of Ph+ acute lymphoblastic leukemias (ALLs). These translocations fuse a breakpoint cluster region (BCR) derived from chromosome 22 to a portion of the c-ABL protooncogene from chromosome 9, thereby leading to formation of alternative BCR-ABL fusion proteins p210BCR-ABL and p185BCR-ABL (hereafter p210 and p185), which are typically detected in CML and Ph+ ALL cells, respectively (3–5). Both proteins encode a constitutive tyrosine-specific protein kinase activity that is essential for cell transformation (4, 6). Although CML occurs in response to the appearance of the t[9;22] in early hematopoietic stem cells, Ph+ ALL results when the translocation affects cells restricted to the B cell lineage (5).
Treatment of CML has been revolutionized by the advent of specific ABL tyrosine kinase inhibitors now used in the front-line management of this disease (7). The first such approved drug, imatinib, when administered as a sole agent to CML patients, provokes remissions in the vast majority without producing serious side effects. Not only is imatinib efficacious, but its use exemplifies the potential for therapies specifically targeted toward activated oncoproteins. However, recent clinical data indicate that ≈5% of patients per year who are maintained on imatinib become resistant to treatment. This drug resistance is most often due to selection for secondary mutations in the BCR-ABL oncoprotein, rather than to “downstream” mutations affecting the signaling pathways subverted by the BCR-ABL kinase (8). Therefore, second-generation kinase inhibitors, such as dasatinib, that effectively block the activity of most mutant forms of BCR-ABL are now being used to treat imatinib-resistant CML (9, 10).
Although CML and Ph+ ALL are triggered by very similar BCR-ABL oncoproteins, durable responses of Ph+ ALL patients to imatinib therapy are uncommon (11), and these patients, both pediatric and adult, receive other conventional combinational chemotherapy and/or bone marrow transplants to stem their disease. At least 30% of Ph+ ALL patients have sustained deletions at chromosome 9p21 that compromise the CDKN2A (hereafter the INK4A-ARF) locus (12, 13), as confirmed by studies of pediatric ALL tumor samples at our own institution. In contrast, a survey of 20 adult CML samples from patients in accelerated phase or blast crisis revealed no examples of INK4A-ARF deletion or ARF promoter methylation (our unpublished data). Therefore, whereas inactivation of INK4A-ARF seems not to contribute to imatinib resistance in CML, it may potentially play such a role in Ph+ ALL.
INK4A-ARF encodes two distinct tumor suppressor genes. The first, p16INK4A, acts to inhibit cyclin D-dependent kinases, thereby regulating transcriptional programs that depend upon the retinoblastoma (RB) protein and its related family members, p130 and p107, for their proper execution. The second protein, p14ARF (p19Arf in the mouse), blocks the E3 ubiquitin ligase activity of HDM2 (Mdm2 in the mouse) to stabilize p53 and so induce a p53-dependent transcriptional program that triggers either cell-cycle arrest or apoptosis in response to oncogenic stress (14). Although the Arf gene is not generally expressed during mouse development or in most normal tissues of adult animals, it is induced by sustained and elevated thresholds of mitogenic signals conveyed by oncoproteins such as Myc, Ras, adenovirus E1A, and v-Abl (15–18). Therefore, Arf activation suppresses the proliferation of incipient cancer cells that have sustained deleterious oncogenic mutations. Conversely, inactivation of Arf facilitates oncogene-induced tumor progression. Interestingly, several lines of evidence indicate that Arf is actively repressed in hematopoietic stem cells (19, 20), which are targeted in CML, but not in pre-B lymphocytes (18, 21–23). Here, we have tested the role of the Arf checkpoint in mouse pre-B cell tumors induced by Bcr-Abl oncoproteins and have studied their response to imatinib treatment in vitro and in vivo.
Results
Bcr-Abl Induces the Arf Checkpoint in Pre-B Cells.
Primary pre-B cells derived from mouse bone marrow progenitors and propagated on stromal cell feeder layers producing IL-7 survive for many weeks in culture but ultimately senesce (24), whereas their Arf-null counterparts can be grown continuously as long as IL-7 is present (25). Abl oncoproteins efficiently kill primary pre-B cells rather than promoting their proliferation; however, rare clonal populations spontaneously arise that are immortal, IL-7-independent, and tumorigenic (18, 26). Resistance to apoptosis results from inactivation of the Ink4a-Arf locus or p53 (18, 25), but the ability to proliferate in an IL-7-independent manner is conferred by Bcr-Abl (26).
To further assess the impact of Bcr-Abl on the Arf checkpoint, we used retroviral vectors encoding GFP to introduce two different Bcr-Abl isoforms into primary pre-B cells derived from the bone marrow of Arf+/+, Arf+/−, and Arf−/− mice. Infected bone marrow cells were propagated on stromal feeder layers producing IL-7 to select for the outgrowth of pre-B cells that expressed B220, CD24, BP-1, and rearranged Ig μ heavy chains, but lacking cell surface IgM (ref. 24 and data not shown). By 7 days after infection, p19Arf was induced in GFP-positive Arf+/+ pre-B cells transduced with either p210 or p185, but not those infected with the control vector encoding GFP alone (Fig. 1A). By this time, >50% of GFP+-Arf+/+ cells could be costained with an Alexa Fluor 647-conjugated monoclonal antibody to p19Arf, which is ≈20-fold more sensitive than previously derived reagents (27) (Fig. 1B).
Fig. 1.

Induction of Arf by Bcr-Abl. (A) Stromally supported pre-B cells (culture day 7) infected with retroviruses encoding p185 or p210 coexpressed p19Arf as detected by immunoblotting, whereas cells infected with a control GFP-expressing vector (Ctl) did not. All pre-B cells expressed p16Ink4a as well as nucleophosmin (NPM) used as a loading control. (B) Fluorescence-activated flow cytometric measurement of cells infected with a vector encoding GFP and p210 were costained with an Alexa Fluor 647-conjugated monoclonal antibody (5-C3-1) to p19Arf (Right) or an isotype-matched control (Left). (C) Pre-B cells of indicated Arf genotypes were removed from stromal support and diluted into liquid medium (day 0), and the levels of p19Arf were determined by immunoblotting at the indicated times thereafter. Actin was used as a loading control.
The Arf genotype only minimally influenced the establishment of these cultures during this initial 7-day period during which stromally supported pre-B cells reached comparable densities of 0.5–1.0 × 106 per ml. However, when Arf+/+, p210+ cells were subsequently removed from stromal support and seeded at 2 × 105 cells per ml into liquid medium lacking IL-7, p19Arf was further induced (Fig. 1C) and the cells died within 6 days, whereas their Arf−/− counterparts continued to expand exponentially (doubling time 18 h). No significant effects of Arf status on the fraction of cells in S phase were observed, but the Arf gene dosage correlated with the apoptotic index (Fig. 6 A and B, which is published as supporting information on the PNAS web site). Moreover, Arf-null pre-B cells expressing p210 efficiently gave rise to clonal cultures derived from single cells plated at limiting dilution, whereas infected Arf+/+ cells did not (Fig. 6C). Age-matched Arf+/− cells expressing less p19Arf protein (Fig. 1C) yielded intermediate phenotypes (Fig. 6 B and C). Therefore, the Arf status did not significantly influence the initiation of stromally supported short-term cultures expressing p210 or p185, but when these cells were shifted to a less favorable culture environment, further Arf induction triggered an efficient apoptotic response. Because p53-null and Arf-null pre-B cells behave similarly (data not shown), we reason that the ability of Bcr-Abl to kill Arf-positive cells is p53-dependent.
When replicate cultures of viable Arf−/− cells expressing p210 were transferred to liquid cultures and diluted to the initiating cell density with fresh medium every 3 days (one passage), they could be expanded exponentially for at least 16 further passages (Fig. 2A). By comparison, Arf+/− pre-B cells that initially exhibited a higher apoptotic index (Fig. 6B) proliferated at slower rates but then adapted, usually between passages 5 and 10, to grow more rapidly (Fig. 2A). Establishment as defined by >90% viability and doubling times of at least 24 h typically occurred by passage 12–16. We used the monoclonal antibody to p19Arf to monitor its expression by immunofluorescence in the four Arf+/− cultures designated A–D in Fig. 2A. This analysis revealed that the frequency of p19Arf-positive cells (Fig. 2B) was proportional to the average population doubling time (Fig. 2A), implying that establishment entailed selection against Arf expression. Loss of p19Arf by passage 12 was confirmed in six additional cultures (E–J) studied by immunoblotting (Fig. 2C). PCR analysis of genomic DNA from the 10 independent Arf+/− cultures at passage 12 revealed concordance between loss of the WT Arf allele and the variably reduced levels of p19Arf expressed in these different populations (Fig. 2D) consistent with Arf gene conversion. In contrast, the linked Ink4a gene was conserved, and its expression increased even as p19Arf levels fell (Fig. 2C). After ionizing irradiation (IR), p53 was robustly induced in all Arf−/− (n = 4) and Arf+/− (n = 10) cultures (Fig. 7, which is published as supporting information on the PNAS web site) as was the expression of the canonical p53-responsive proteins Mdm2 and p21Cip1 (data not shown). Therefore, Arf loss attenuates apoptosis induced by Bcr-Abl in pre-B cells and enables their continuous proliferation in culture.
Fig. 2.

Selection for Arf inactivation during establishment of pre-B cell cultures. (A) Arf−/− cells expressing p210 expand exponentially in liquid cultures, whereas Arf+/−, p210+ cells undergo adaptation. (B) Four Arf+/−, p210+ cultures designated A–D in A were fixed and stained with monoclonal antibody 5-C3-1 to p19Arf (Left) and for DAPI (Right) to visualize all nuclei. Cells with nucleolar p19Arf staining are designated by arrows (Right). (C) Arf+/−, p210+ culture lysates designated E–J were immunoblotted with antibodies to p19Arf and p16Ink4a at passages 4 and 12. The viability of each culture is indicated at the bottom. (D) PCR was used to assay Arf alleles in all 10 Arf+/−, p210+ cultures at passage 12. Loss of the WT allele correlated with loss of p19Arf expression.
Arf Inactivation in a Mouse Bone Marrow Transplantation Model of Ph+ ALL.
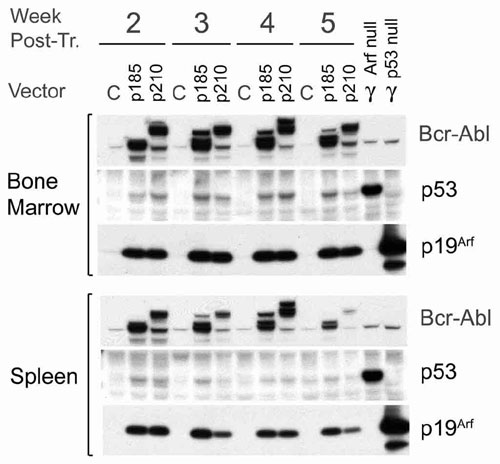
Unconditioned bone marrow cells infected with retroviruses expressing Bcr-Abl induce pre-B cell lymphoid leukemia, but not CML, when used to reconstitute lethally irradiated mice (28). Therefore, bone marrow harvested from Arf+/+ or Arf−/− donor animals was transduced with GFP vectors encoding p185 or p210, and the cells were immediately transplanted after minimal ex vivo manipulation and without culture into cohorts of lethally irradiated mice. Arf loss significantly reduced disease latency (Fig. 3A), and cells recovered from the marrows and spleens of sick animals confirmed the pre-B cell phenotype (B220+, CD24+, and BP-1+) of GFP-marked cells that infiltrated these tissues. In five of five mice transplanted with Arf+/+ cells expressing p185 and in three of four that received Arf+/+ cells expressing p210, p19Arf was induced by Bcr-Abl in peripheral blood leukocytes in clinically well animals by 3 weeks after transplant, revealing early activation of the Arf checkpoint response long before disease was manifested (Fig. 3B). Similarly, p19Arf was detected as early as 2 weeks after transplant in B220+ cells derived from bone marrow and spleen of Bcr-Abl-infected animals; compared with cells transduced with the control vector, those expressing Bcr-Abl also expressed increased levels of p53, which could be induced to higher levels in response to ionizing irradiation (Fig. 8, which is published as supporting information on the PNAS web site). Recipients of Arf+/+ donor cells expressing p210 maintained low levels of GFP+ cells (<1 × 104/μl) in peripheral blood until their clinical deterioration, whereas levels of circulating Arf−/−-GFP+ cells were grossly elevated within 2 weeks of transplant (6–9 × 104/μl). Arf inactivation also increased the severity of leukemia. Spleen weights were 2-fold greater in moribund mice that received Arf-null cells (mean weights 325 mg in Arf−/−-p210+; 410 mg in Arf−/−-p185+; vs. 190 mg in Arf+/+-p210+ or -p185+ mice); bone marrow infiltration with GFP+ cells exceeded 90% in recipients of Arf−/− cells versus 55–76% in recipients of Arf+/+ cells. Therefore, Arf loss shortened the latency and heightened the severity of Bcr-Abl-induced lympholeukemia.
Fig. 3.

Arf-null bone marrow cells expressing p210 or p185 induce accelerated disease after transplant into irradiated recipients. (A) Survival curves after transplantation of bone marrow donor cells expressing p210 (Left) or p185 (Right) into lethally irradiated mice. The genotypes of donor cells were Arf+/+ (open symbols) or Arf−/− (filled symbols). (B) Lysates of peripheral blood leukocytes taken 3 weeks after transplant from mice receiving Arf+/+ donor cells expressing p185, p210, or neither (control) were immunoblotted by using an antibody that detects both Bcr-Abl isoforms (as well as a background band, asterisk). Levels of p19Arf in the same lysates were also determined.
Arf Inactivation Leads to Aggressive Disease in Immunocompetent Mice.
Unlike Bcr-Abl-transduced bone marrow cells injected into irradiated recipients, primary pre-B cells expressing p185 or p210 are only weakly tumorigenic when inoculated into nonirradiated syngeneic mice. We therefore introduced p210 into bone marrow cells from Arf+/+, Arf+/− and Arf−/− mice, cultured the cells on autologous pre-B cell supportive stroma for 7 days, and injected 2 × 106 cells intraperitoneally into cohorts of nonirradiated, syngeneic C57BL/6 recipients. As expected, animals injected with Arf+/+-p210+ cells rarely developed tumors (Fig. 4A). However, animals receiving Arf−/− cells expressing p210 rapidly succumbed to highly aggressive, disseminated lympholeukemias, with a median survival of 14 days (Fig. 4A). These animals had bone marrow infiltration, hepato- and splenomegaly (4- to 5-fold increase in spleen mass with GFP+ lymphoblasts), massive abdominal adenopathy, malignant ascites, and pleural effusions. Animals injected with Arf+/−-p210+ cells developed tumors with delayed kinetics (median survival 33 days) and incomplete penetrance (≈70%) (Fig. 4A) but gave rise to lympholeukemias clinically similar to those of their Arf-null counterparts. Not unexpectedly, isolated GFP+-B220+ tumor cells obtained from Arf+/− mice no longer expressed p19Arf (data not shown).
Fig. 4.

Survival of immunocompetent mice injected i.p. with p210-expressing pre-B cells of various Arf genotypes. (A) Mice received equivalent numbers of p210+ cells of the indicated Arf genotypes. (B) Mice received Arf−/−, p210+ pre-B cells and were treated orally twice per day for the indicated interval (Rx, black bar) with vehicle alone (squares) or imatinib (triangles). (C) A similar experiment was performed with Arf+/−, p210+ donor cells.
In separate experiments, cohorts of mice were injected intravenously with serial log dilutions of donor cells. Mice receiving 2 × 105 p210+, Arf+/+ cells did not develop tumors, whereas animals receiving only 2 × 104 p210+, Arf−/− cells died of lympholeukemia within 3 weeks of injection (Table 1, vehicle cohorts). These results underscore the effects of Arf loss in conferring sensitivity to transformation by Bcr-Abl.
Table 1.
Imatinib response of mice receiving Arf−/−, p210+ pre-B cells
| Treatment | No. of cells injected | No. of mice | Survival, days* | Spleen mass, mg* | GFP+ cells in Blood, 1,000/μl | GFP+ cells in BM, %* |
|---|---|---|---|---|---|---|
| Vehicle | 2 × 105 | 9 | 14.5 ± 0.5 | 361 ± 47 | 26.1–66 | 77 ± 4.8 |
| Imatinib | 2 × 105 | 8 | 20.9 ± 2.3 | 229 ± 31 | 2.8–39.7 | 82 ± 5.5 |
| Vehicle | 2 × 104 | 8 | 17.8 ± 0.5 | 322 ± 51 | 16.5–59.0 | 55 ± 7.6 |
| Imatinib | 2 × 104 | 8 | 23.4 ± 1.9 | 223 ± 55 | 0.2–46.3 | 68 ± 23 |
Animals received the indicated number of cells by i.v. injection. Oral imatinib treatment (100 mg/kg, twice per day) was begun 3 days later and continued until animals became moribund. Normal spleen mass and white blood counts are ≈100 mg and 10,000/μl. BM, bone marrow.
*Mean ± SD.
Arf Inactivation in Bcr-Abl-Driven ALL Limits Response to Imatinib.
Arf-null or p53-null pre-B cells expressing p210 or p185 are highly sensitive to imatinib treatment in vitro (IC50 ≈100 nM) (Fig. 9A, which is published as supporting information on the PNAS web site). The drug inhibits the tyrosine phosphorylation of p185 and p210 as well as that of bona fide Bcr-Abl substrates, including Stat-5 and CrkL, regardless of Arf status (Fig. 9B). However, when mice injected with Arf−/− or Arf+/− cells expressing p210 were treated twice per day by oral gavage with clinically effective doses of imatinib [100 mg/kg capable of inducing durable CML remissions in mouse models (29) and human patients], they developed lympholeukemia and rapidly died, even when maintained on therapy (Fig. 4 B and C). Imatinib treatment decreased the number of circulating lymphoid blasts, reduced spleen size, and modestly extended the animals' survival (Figs. 4 B and C and Table 1). Nonetheless, mice receiving as few as 2 × 104 p210+, Arf −/− cells could not be cured (Table 1). Importantly, tumor cells recovered from bone marrow and pleural effusions of moribund p210+, Arf−/− mice and expanded in culture for 2–5 days showed the same Gleevec sensitivity as the injected donor cells, arguing that drug resistance was not due to mutations in, or overexpression of, Bcr-Abl.
IL-7 Heightens Imatinib Resistance in Arf−/−-p210+ Pre-B Cells.
Although pre-B cells expressing Bcr-Abl exhibit IL-7 independence, the cells remain responsive to this cytokine. Addition of IL-7 to Arf−/−, p210+ pre-B cells counteracted the antiproliferative effects of imatinib treatment at all doses of drug tested (Fig. 5A). Expression of cyclin D2 served as a useful biomarker, because it is essential for IL-7-induced B cell proliferation and for Bcr-Abl-induced transformation in culture (30). Therefore, treatment of Arf−/−, p210+ B cells with increasing concentrations of imatinib lowered cyclin D2 levels and concomitantly prevented S phase entry (Fig. 5B). Both effects were partially overcome in the presence of IL-7. Imatinib treatment selected for cultured cells that expressed higher levels of p210, whether they were treated with IL-7 or not (Fig. 5B), although this induction did not occur in vivo. Based on these findings, we reasoned that inhibitors of IL-7 signaling might complement imatinib treatment in limiting the proliferation of IL-7-stimulated, Arf−/−, p210+ pre-B cells. Using a small molecule inhibitor of JAK kinases, we found that this compound resensitized the cells to imatinib (Fig. 5C).
Fig. 5.

IL-7 desensitizes p210+, Arf −/− pre-B cells to imatinib treatment. (A) p210+, Arf −/− pre-B cells were treated with the indicated doses of imatinib in the absence (open bars) or presence (filled bars) of IL-7. (B) In the absence of IL-7 (−), increasing doses of imatinib select for increased expression of p210 but decreased expression of cyclin D2. Cells expressing low levels of cyclin D2 exit the cell cycle and exhibit a reduced S phase fraction. In the presence of IL-7 (+), the reduction of cyclin D2 and the fraction of S phase cells is attenuated. Arf-null cells lacking p210 (Right) express lower levels of cyclin D2 than untreated p210+ cells. (C) Treatment of Arf−/−, p210+ cells with 1.0 μM imatinib (filled bars) and the indicated concentrations of Jak kinase inhibitor (JAKi) in the presence of IL-7. Error bars represent standard errors.
Discussion
Induction of Arf by a surfeit of potentially oncogenic signals can halt the proliferation of incipient cancer cells or can eliminate them by inducing apoptosis (14). We documented cell-autonomous, tumor-suppressive effects of Arf in mouse models in which the Bcr-Abl oncoprotein induces pre-B cell lympholeukemias reminiscent of Ph+ ALL. In a well tried experimental transplant setting in which Arf+/+ bone marrow progenitors transduced with vectors encoding either p210 or p185 were introduced without ex vivo culture into lethally irradiated mice, recipient animals developed lympholeukemia only after a relatively long latency period. Notably, expression of p19Arf and induction of p53, although absent in the hematopoietic tissues of recipient mice receiving cells transduced with a control GFP vector, occurred very early after transplantation in the bone marrow, spleen, and peripheral blood leukocytes of animals reconstituted with cells expressing Bcr-Abl. Thus, the Arf checkpoint was activated before any overt disease was manifested. When leukemias arose in Arf+/+ animals, GFP-positive tumor cells recovered from these mice generally retained Arf and p53 function and could not be expanded in liquid cultures without stromal support. This indicates, not surprisingly, that genetic events other than mutations in the Arf-p53 pathway can cooperate with Bcr-Abl to contribute to leukemogenesis. However, Bcr-Abl-induced tumors in the Arf−/− background arose very rapidly and were much more aggressive, emphasizing the ability of the Arf checkpoint to halt tumorigenesis at least until other as yet undetermined genetic restraints are lifted.
In a second, more stringent ALL model in which transiently cultured pre-B cells expressing Bcr-Abl were injected into nonirradiated syngeneic recipients, Arf+/+ donor cells rarely induced tumors. In contrast, Bcr-Abl(+), Arf+/− pre-B cells induced lympholeukemias in which the WT Arf allele, but not the closely linked Ink4a gene, was inactivated. The latency of disease was even shorter when Arf−/− cells expressing Bcr-Abl isoforms were introduced into immunocompetent hosts, and many fewer cells were required to induce flagrant disease. Although intraperitoneal administration of cells produced a disseminated disease that included lymphomatous masses at the injection site and overt nodal involvement, i.v. injection of p210+, Arf−/− cells led to a lympholeukemic phenotype restricted to hematopoietic tissues. This model has obvious practical benefits. First, transplantation of genetically manipulated donor cells does not require any preconditioning of the syngeneic recipients, which, unlike irradiated mice, do not require hematopoietic reconstitution, retain their immune function, and resist adventitious infections. Second, the resulting tumor cells can be explanted into culture or serially transplanted into secondary recipients, facilitating their further genetic analysis. Finally, rapidly generated, large cohorts of recipient animals can be easily subjected to therapeutic trials.
Like cells taken from CML patients, pre-B cells expressing Bcr-Abl were sensitive to imatinib treatment regardless of their Arf or p53 status. The drug induces cytostatic effects at lower doses but can trigger apoptosis in cells exposed to higher concentrations for prolonged periods of time. Exit of treated pre-B cells from the cell cycle was associated with a reduction in cyclin D2 levels, which served as a reliable biomarker. Despite the brisk response of these cells in vitro to submicromolar concentrations of imatinib, animals injected with Arf−/− or Arf+/− pre-B cells expressing p210 showed only modest responses to imatinib in vivo. These mice developed lethal disease even in the face of continued twice per day oral dosing with concentrations of drug that are clinically effective (29). Tumor cells recovered from moribund animals exhibited the same IC50 as that of the injected donor cells, implying that drug resistance in vivo was not due to alterations in Bcr-Abl. Rather, drug resistance depended on a tumor cell-extrinsic mechanism. The high-frequency INK4A-ARF inactivation in human Ph+ ALL and the possibility that it contributes to drug resistance in human patients therefore merit consideration.
The manner by which Arf inactivation overrides sensitivity to imatinib in vivo remains a mystery. By attenuating Bcr-Abl-induced apoptosis, loss of Arf facilitates the proliferation of cells expressing the oncoprotein. Indeed, forms of BCR-ABL arising in CML patients have been routinely tested for imatinib sensitivity in IL-3-dependent BAF3 cells in which the Ink4a-Arf locus is silenced by methylation (our unpublished observations). Interestingly, although pre-B cells expressing Bcr-Abl can be propagated in the absence of IL-7, they remain IL-7-responsive, and the addition of IL-7 to cultured cells partially counteracted imatinib's cytostatic effects and restored cyclin D2 expression and S phase entry. The combination of a pan-JAK kinase inhibitor and imatinib circumvented these effects of the cytokine. Could cytokine protection occur in vivo within the microenvironment in which malignant B cells expand? Despite a rationale for the combined use of imatinib and JAK kinase inhibitors in this model, such trials are presently precluded by the high cost of the few commercially available compounds and the general absence of selective inhibitors to specific JAKs. The recent emphasis by pharmaceutical companies in generating targeted therapies should facilitate such efforts.
Methods
Expression Vectors and Retroviral Production.
cDNAs encoding p185 and p210 expressed in mouse stem cell virus–internal ribosome entry site–GFP vectors (MSCV-IRES-GFP) were provided by O. Witte (University of California, Los Angeles). Packaging of replication-incompetent, ecotropic retroviral particles was achieved by transient transfection of Phoenix-Eco packaging cells (G. Nolan, Stanford University, Stanford, CA) by using previously described methods (15).
Culture Conditions.
Marrow was obtained from the long bones of backcrossed C57BL/6 mice harboring targeted deletions of Arf exon 1β (31) or p53 (32). Cells of indicated genotypes were suspended in vector-containing supernatant and plated as described (24) to derive infected pre-B cell cultures on autologous stroma. Alternatively, primary pre-B cell cultures established by means of selection on IL-7-secreting stroma (25) were infected with retroviruses immobilized on Retronectin (Takara Bio, Tokyo). Cultures were maintained in RPMI medium 1640 (Mediatech, Herndon VA) supplemented with 5% FCS, 55 μM 2-mercaptoethanol, 2 mM glutamine, penicillin, and streptomycin in the presence of saturating levels of IL-7-conditioned medium. Where indicated, pre-B cells were transferred to liquid culture in medium with 10% FCS and maintained at 2 × 105 viable cells per ml by dilution into fresh medium every 3 days. Emergent populations were monitored for Arf status by PCR (33). For drug growth inhibitory studies, Arf−/− p210+ cells were plated in triplicate (2 × 104 cells per well in 96-well plates) in the absence or presence of recombinant IL-7 (10 ng/ml, R & D Systems). Medium containing imatinib (Novartis, East Hanover, NJ) and/or a pan-JAK inhibitor (no. 420099; EMD Biosciences, La Jolla, CA) or respective diluents (water or DMSO, respectively) was added to achieve the indicated final concentrations. Cell growth was quantified 72 h later by a methane-thiosulfonate-base assay (CellTiter 96 Aqueous One Solution Reagent; Promega).
Immunoblotting and Immunofluorescence.
Proteins immobilized on poly(vinylidene difluoride) (PVDF) membranes (15) were detected with antibodies to p19Arf (27) p16Ink4a (sc-1207), Abl (sc-23; both Santa Cruz Biotechnology), p53 (Ab-7, EMD Biosciences), cyclin D2 [clone 34-B1-3; (34)], NPM (Invitrogen), and eIF4E and tyrosine-phosphorylated Bcr-Abl, Stat5, and CrkL (Bcr-Abl Pathscan Activity Assay; Cell Signaling Technology, Beverly, MA). Detection of endogenous p19Arf in fixed cells by immunofluorescence was performed as described (35).
Flow Cytometric Analysis of Viability, Apoptosis, Cell-Cycle Distribution, and p19Arf Expression.
Cultured Arf+/−, p210+ cells were harvested at sequential passages and stained at room temperature with Annexin-V APC reagent (Cat. no. 550475; BD Biosciences) as recommended by the manufacturer. Annexin-V/propidium iodide double-negative cells were scored as viable. Cells fixed in 1% paraformaldehyde (PF) (15 min on ice) and sequentially in 70% ethanol overnight at −20°C were assayed by terminal transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) by using a APO-BRDU KIT (BD Biosciences). Measures of DNA content and calculation of S-phase fractions were as described (35). To detect p19Arf by flow cytometry, 5-C3-1 rat anti-p19Arf monoclonal antibody (27) or an isotype-matched control antibody was conjugated to the Alexa Fluor 647 fluorochrome (no. A20186; Molecular Probes, Invitrogen). Cells were washed, fixed with 4% PF (room temperature, 10 min), permeabilized for 30 min with PBS containing 10% FCS and 0.1% Triton X-100, and stained with 1 μg/ml Alexa Fluor 647-conjugated 5-C3-1 or control antibodies before analysis of dual GFP and Alexa Fluor 647 chromophores.
Syngeneic Pre-B Cell Transfer and Imatinib Treatment.
Cohorts of 8- to 12-week-old C57BL/6 male mice were injected intraperitoneally with 2 × 106 viable GFP-marked cells of the indicated genotypes or intravenously with log dilutions of cells (Table 1). Mice were observed daily and killed when moribund (dehydration, ruffled fur, poor mobility, respiratory distress). Imatinib treatment was initiated 3 days after injection by twice daily oral gavage with either imatinib at 100 mg/kg per dose or with a vehicle control and was continued until animals became moribund.
Bone Marrow Transplantation.
For efficient induction of B lineage Ph+ ALL, bone marrow of Arf+/+ or Arf−/− donor mice was harvested and infected in suspension for 3 h with retroviral vectors in the absence of any exogenous cytokines (28). Washed, transduced bone marrow cell suspensions (1 × 106 cells per mouse) were immediately injected into lethally irradiated (11 Gy in two fractions) WT C57BL/6 recipients maintained on antibiotic-containing (Enrofloxacin) water for 3 weeks after transplant. Mice were observed for signs of illness and periodically bled to monitor white blood counts and GFP expression in peripheral blood.
Supplementary Material
Acknowledgments
We thank the members of the C.J.S. and M.F.R. laboratory for helpful suggestions and criticisms, Shelly Wilkerson for treatment and monitoring of animals, David Bertwistle for the preparation of Alexa Fluor 647-conjugated antibodies and development of the FACS assay, and Richard A. Ashmun and Ann Marie Hamilton-Easton for assistance with flow cytometry. This work was supported in part by National Institutes of Health Training Grant T32-CA70089 (to R.T.W.) and Grant CA-71907 (to M.F.R.), Cancer Center Core Grant CA-21765, and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital. C.J.S. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- Ph
Philadelphia chromosome
- CML
chronic myelogenous leukemia
- ALL
acute lymphoblastic leukemia
- BCR
breakpoint cluster region.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Nowell P., Hungerford D. J. Natl. Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 2.Rowley J. D. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 3.Groffen J., Heisterkamp N., de Klein A., Bartram C. R., Grosveld G. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 4.Clark S. S., McLaughlin J., Crist W. M., Champlin R., Witte O. N. Science. 1987;235:85–88. doi: 10.1126/science.3541203. [DOI] [PubMed] [Google Scholar]
- 5.Chan L. C., Karhi K. K., Rayter S. I., Heisterkamp N., Eridani S., Powles R., Lawler S. D., Groffen J., Faulkes J. G., Greaves M. F., et al. Nature. 1987;325:635–637. doi: 10.1038/325635a0. [DOI] [PubMed] [Google Scholar]
- 6.Witte O. N., Dasgupta A., Baltimore D. Nature. 1980;283:826–831. doi: 10.1038/283826a0. [DOI] [PubMed] [Google Scholar]
- 7.Deininger M., Buchdunger E., Druker B. J. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 8.Shah N. P., Nicoll J. M., Nagar B., Gorre M. E., Paquette R. L., Kuriyan J., Sawyers C. L. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 9.Shah N. P., Tran C., Lee F. Y., Chen P., Norris D., Sawyers C. L. Science. 2004;305:319–321. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 10.Shah N. P. Hematology: The American Society of Hematology Education Program Book. Washington, DC: Am. Soc. Hematol.; 2005. pp. 183–187. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong S. A., Look A. T. J. Clin. Oncol. 2005;23:6306–6315. doi: 10.1200/JCO.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 12.Heerema N. A., Harbott J., Galimberti S., Camitta B. M., Gaunon P. S., Janka-Schaub G., Kamps W., Basso G., Pui C.-H., Schrappe M., et al. Leukemia. 2004;18:693–702. doi: 10.1038/sj.leu.2403324. [DOI] [PubMed] [Google Scholar]
- 13.Primo D., Taberbero M. D., Perez J. J., Rasillo A., Sayagues J. M., Espinosa A. B., Lopez-Berges M. C., Garcia-Sanz R., Gutierrez N. C., Hernandez J. M., et al. Leukemia. 2005;19:713–720. doi: 10.1038/sj.leu.2403714. [DOI] [PubMed] [Google Scholar]
- 14.Sherr C. J., McCormick F. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 15.Zindy F., Eischen C. M., Randle D. H., Kamijo T., Cleveland J. L., Sherr C. J., Roussel M. F. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Stanchina E., McCurrach M. E., Zindy F., Shieh S.-Y., Ferbeyre G., Samuelson A. V., Prives C., Roussel M. F., Sherr C. J., Lowe S. W. Genes Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmero I., Pantoja C., Serrano M. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- 18.Radfar A., Unnikrishnan I., Lee H.-W., DePinho R. A., Rosenberg N. Proc. Natl. Acad. Sci. USA. 1998;95:13194–13199. doi: 10.1073/pnas.95.22.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs J. J. L., Kieboom K., Marino S., DePinho R. A., van Lohuizen M. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 20.Valk-Lingbeek M. E., Bruggeman S. W., van Lohuizen M. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Eischen C. M., Weber J. D., Roussel M. F., Sherr C. J., Cleveland J. L. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitt C. A., McCurrach M. E., De Stanchina E., Lowe S. W. Genes Dev. 1999;13:2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobs J. J. L., Scheijen B., Vonchen J.-W., Kieboom K., Berns A., van Lohuizen M. Genes Dev. 1999;13:2678–2690. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitlock C. A., Witte O. N. Methods Enzymol. 1987;150:275–286. doi: 10.1016/0076-6879(87)50085-4. [DOI] [PubMed] [Google Scholar]
- 25.Randle D. H., Zindy F., Sherr C. J., Roussel M. F. Proc. Natl. Acad. Sci. USA. 2001;98:9654–9659. doi: 10.1073/pnas.171217498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaughlin J., Chianese E., Witte O. N. Proc. Natl. Acad. Sci. USA. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertwistle D., Zindy F., Sherr C. J., Roussel M. F. Hybridoma Hybridomics. 2005;23:293–300. doi: 10.1089/hyb.2004.23.293. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y., Liu Y., Pelletier S., Buchdunger E., Warmuth M., Fabbro D., Hallek M., Van Etten R. A., Li S. Nat. Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 29.Wolff N. C., Ilaria R. L. Blood. 2001;98:2808–2816. doi: 10.1182/blood.v98.9.2808. [DOI] [PubMed] [Google Scholar]
- 30.Jena N., Deng M., Sicinska E., Sicinski P., Daley G. Q. Cancer Res. 2002;62:535–541. [PubMed] [Google Scholar]
- 31.Kamijo T., Zindy F., Roussel M. F., Quelle D. E., Downing J. R., Ashmun R. A., Grosveld G., Sherr C. J. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 32.Jacks T., Remington L., Williams B. O., Schmitt E. M., Halachmi S., Bronson R. T., Weinberg R. A. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 33.McKeller R. N., Fowler J. L., Cunningham J. J., Warner N., Smeyne R. J., Zindy F., Skapek S. X. Proc. Natl. Acad. Sci. USA. 2002;99:3848–3853. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallance S. J., Lee H.-M., Roussel M. F., Shurtleff S. A., Kato J.-Y., Strom D. K., Sherr C. J. Hybridoma. 1994;13:37–44. doi: 10.1089/hyb.1994.13.37. [DOI] [PubMed] [Google Scholar]
- 35.den Besten W., Kuo M.-L., Williams R. T., Sherr C. J. Cell Cycle. 2005;4:1593–1598. doi: 10.4161/cc.4.11.2174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}