

Abstract
Glioblastoma multiforme is an intracranial tumor that has very poor prognosis. Patients usually succumb to their disease 6 to 12 months after they are diagnosed despite very aggressive treatment modalities. We tested the efficacy of a potent differentiation and proliferation factor for the professional antigen-presenting dendritic cells (DCs), i.e., Flt3L, for its potential role as a novel therapy for gliomas. We investigated the ability of recombinant adenoviral vectors encoding human soluble Flt3L (hsFlt3L) to improve the survival of Lewis rats bearing intracranial syngeneic CNS-1 gliomas. We show that RAdhsFlt3L can improve survival in a dose-dependent manner. Seventy percent of rats survive when treated with 8 × 107 pfu RAdhsFlt3L ( P < 0.0005). In addition we demonstrate in both naïve Lewis rats and C57BL/6 mice the presence of increased numbers of cells bearing DC markers (OX62 and MHCII, in rats, or CD11C, 33D1, MHCII, and F4/80, but not DEC205, in mice) in sites of brain delivery of RAdhsFlt3L. These results show that expression of hsFlt3L in the brain leads to the presence of cells displaying DC markers. We demonstrate that treatment with hsFlt3L leads to inhibition of tumor growth and significantly increased life span of animals implanted with syngeneic CNS-1 glioma cells. Animals that had survived for long periods, i.e., 6 months, had eliminated the implanted tumors after neuropathological analysis; on the other hand, some of the 3-month survivors still appeared to harbor brain tumors. Our results have profound implications for immune-mediated brain tumor therapy and also suggest the ability to recruit DC-like cells within the brain parenchyma in response to the local expression of Flt3L from adenoviral vectors.
Keywords: adenovirus, brain, HSV1-TK, Flt3L, immunology, tumor
Introduction
Malignant brain gliomas are the most common subtype of primary brain tumors. These aggressive, highly invasive, and neurologically destructive tumors are among the deadliest of human cancers [1]. In its most aggressive manifestation, glioblastoma multiforme median survival ranges from 6 to 12 months, despite aggressive therapeutic interventions. This statistical fact has changed very little over several decades of technological advances in neuro-surgery, radiation therapy, and clinical trials with novel therapeutic modalities [2–8].
A number of important clinical trials investigating the efficacy of gene therapy for the treatment of glioma have been carried out. The most widely used approach has been suicide therapy using a prodrug activation system, i.e., the herpes simplex type 1–thymidine kinase gene in combination with the prodrug ganciclovir (HSV1-TK/GCV); this is 100% efficient in most preclinical models. In human patients bearing malignant glioma, both replication-defective retroviruses and adenoviruses, and replication-competent adenoviruses and HSV viruses, have been used [3,9–13]. These studies are important as they have shown that the HSV1-TK/GCV is safe and have determined the doses of vector that are tolerated without adverse side effects. However, the bystander effect anticipated by this suicide gene therapy approach in clinical trials has been less than expected, suggesting insufficient vector distribution and/or low transduction efficiency. Nevertheless, investigators have demonstrated a statistically significant increase in the long-term survival of patients treated with first-generation recombinant adenovirus vectors expressing HSV1-TK driven by the human cytomegalovirus promoter (hCMV) [3], in double-blind, randomized controlled clinical trials of adenovirus vs retrovirus, both expressing HSV1-TK, and treatment with ganciclovir. This has prompted the movement of the trials with adenovirus forward into Phase II and III clinical trials.
A potential explanation for this apparent discrepancy between the experimental and the clinical trials centers around the animal models used in the preclinical experiments. The experimental animal models consist of clonal glioma cell lines implanted into the brain, with particular gene therapies being delivered directly into the growing tumors. Usually, this is done at a time when tumors are small, and this may need to be reconsidered in the future. The use of human-derived tumor models tends to replicate tumor biology somewhat better, although the need to use them in immune-deficient animals precludes their implementation for immunotherapy studies. Additionally, novel transgenic mouse models have been developed; their use may provide new experimental approaches to the gene therapy of brain tumors, but this avenue remains to be further explored [14]. The advantage of the CNS-1 model is that, by being syngeneic, immunotherapeutic approaches can indeed be tested.
In this article we investigate the efficacy of human soluble fms-like tyrosine kinase 3 ligand (hsFlt3L) gene therapy in a syngeneic intracranial model of glioma. The rationale for using hsFlt3L is its unique ability to increase the number of dendritic cells (DC) from circulating precursors [15,16]. DCs are the primary professional afferent antigen-presenting cells (APCs). Although the brain contains cell types that can present antigen to activated T cells (such as endothelial cells, pericytes, perivascular macrophages, and microglial cells) there is consensus that the naïve brain lacks DCs, although there are reports of DC-like cells within the brain ventricles, meninges, and choroid plexus and within the inflamed brain [17]. The brain has classically been thought of as an immunoprivileged site. It is now appreciated that this immune privilege is an active process regulated at the tissue level by several factors, including the presence of the blood–brain barrier, a lack of conventional lymphatics, and an immunosuppressive microenvironment and also by the lack of resident professional APCs [18] able to take up antigen and transport it to the lymph nodes to present antigen to naïve T lymphocytes. We thus hypothesized that increasing the presence of these cells in the brain could be therapeutically advantageous and thus used hsFlt3L to promote the infiltration and/or differentiation of DCs into the CNS and potentially to increase the capacity of the brain to promote antigen presentation.
To this end, we used the intracranial, syngeneic CNS-1/Lewis glioma model [19–22]. In this model, hsFlt3-L-mediated gene therapy elicited a 70% (P < 0.0005) survival of tumor-bearing animals, and this increased survival was monitored for up to 6 months. This is the first demonstration that hsFlt3L, a cytokine used to stimulate anti-tumor responses in tumors in other organs, may have an important role for treating intracranial brain tumors.
Results
Recombinant Adenovirus (RAd) Expressing Human Soluble Flt3L: Molecular Characterization and Expression in Vitro
We first analyzed putative Flt3L-encoding RAds by restriction enzyme analysis of the adenovirus genome digested with HindIII. We compared the restriction pattern generated to the restriction pattern generated by the HindIII digestion of the adenoviral vector (pJM17; Figs. 1A and 1B, L1) and the shuttle vector pAL119-Flt3L (Figs. 1A, 1B, and 1C, L2). Restriction digestion of the shuttle vector generated three bands: first a large band corresponding to the remaining pAL119 vector, a second band 1496 bp in length corresponding to the hsFlt3L transgene and poly(A) sequences, and a band of 250 bp corresponding to the hCMV sequence (Figs. 1A, 1B, and 1C, L2). The bands corresponding to the hCMV promoter and the transgene sequence and the SV40 poly(A) sequence are also present in the HindIII restriction pattern of RAdhsFlt3L (Figs. 1B and 1C, L3). We used the HindIII restriction pattern of pJM17 to compare it with the HindIII digest of RAdFlt3L to confirm that no recombination had occurred during cotransfection between the viral vector, the shuttle vector, and the E1 region of the trans-complementing 293 cell line. We confirmed the 1496-bp band, which corresponds to the hCMV promoter and hsFlt3L transgene sequences, by hybridizing with a DIG-labeled Flt3L-specific probe (1496 bp) generated by the restriction digestion of the shuttle vector with HindIII and extraction of the DNA band corresponding to 1496 bp. The probe produced a positive signal in lane L2 (pAL119-Flt3L) and lane L3 (RAdhsFlt3L), which corresponded to the transgene sequence (Fig. 1C).
FIG. 1.
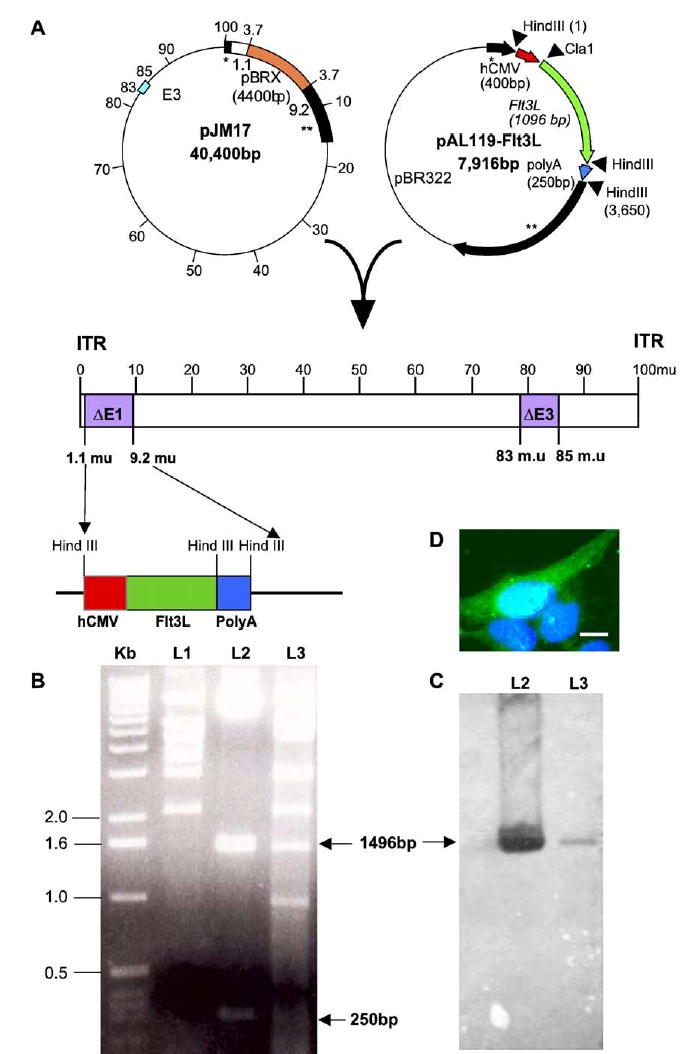
Characterization of RAdhsFlt3L. (A) Genomic organization of RAdhsFlt3L. The adenoviral plasmid pJM17 and shuttle vector pAL119 were cotransfected into 293 cells to generate the recombinant adenovirus. The expression cassette, which consists of the hCMV promoter driving the expression of hsFlt3L, is situated in the E1-deleted region of the adenovirus genome. (B) Gel electrophoretic separation of the HindIII-digested pJM17 (L1), pAL119Flt3L shuttle vector (L2), and putative RAdhsFlt3L viral clone (L3). (C) The Southern blot used a 1496-bp DIG-labeled probe directed against the hCMV promoter and hsFlt3L transgene sequences to detect the presence of the transgene, which is indicated by the corresponding band. (D) Expression of hsFlt3L in COS7 cells as assessed using immunofluorescence staining.
To characterize expression of hsFlt3L from RAd-hsFlt3L, we infected COS7 and HeLa cells with an increasing multiplicity of infection ranging from 0 to 500. We incubated the cells with the virus for 48 h at 37°C, with 5% CO2. After 48 h we washed, fixed, and permeabilized the cells and determined protein expression by immunofluorescence cytochemistry. We detected hsFlt3L expression using a rat anti-Flt3L primary antibody (provided by Immunex) and a goat anti-rat FITC secondary antibody. Cells were counterstained with DAPI to label nuclei (Fig. 1D).
Time Course of Expression of hsFLT3L within the CNS of Lewis Rats
We injected male Lewis rats with saline, RAd35 expressing β-galactosidase (1 × 107 iu), or RAdhsFlt3L (1 × 107 iu), into the striatum. We measured adenovirally expressed hsFlt3L at 7, 14, and 30 days post-RAd injection (Fig. 2A). Fig. 2A illustrates that no hsFlt3L immunoreactivity was detected in RAd35-or saline-(data not shown) injected animals. To quantitate the levels of the respective protein products from both RAd35 and RAdhsFlt3L delivered into the striatum, we used an ELISA specific for hsFlt3L and we measured the enzymatic activity for β-galactosidase. While endogenous rat Flt3L protein expression was low and remained constant across all three groups (approximately 20 pg/ml, Table 1), hsFlt3L was detected at much higher levels (7 ng/ml) only in RAdhsFlt3L-injected animals (Fig. 2B). β-Galactosidase enzymatic activity was detected only in brains in which RAd35 was injected (Fig. 2C). The levels of Flt3L expressed were stable for up to a month post-brain delivery.
FIG. 2.
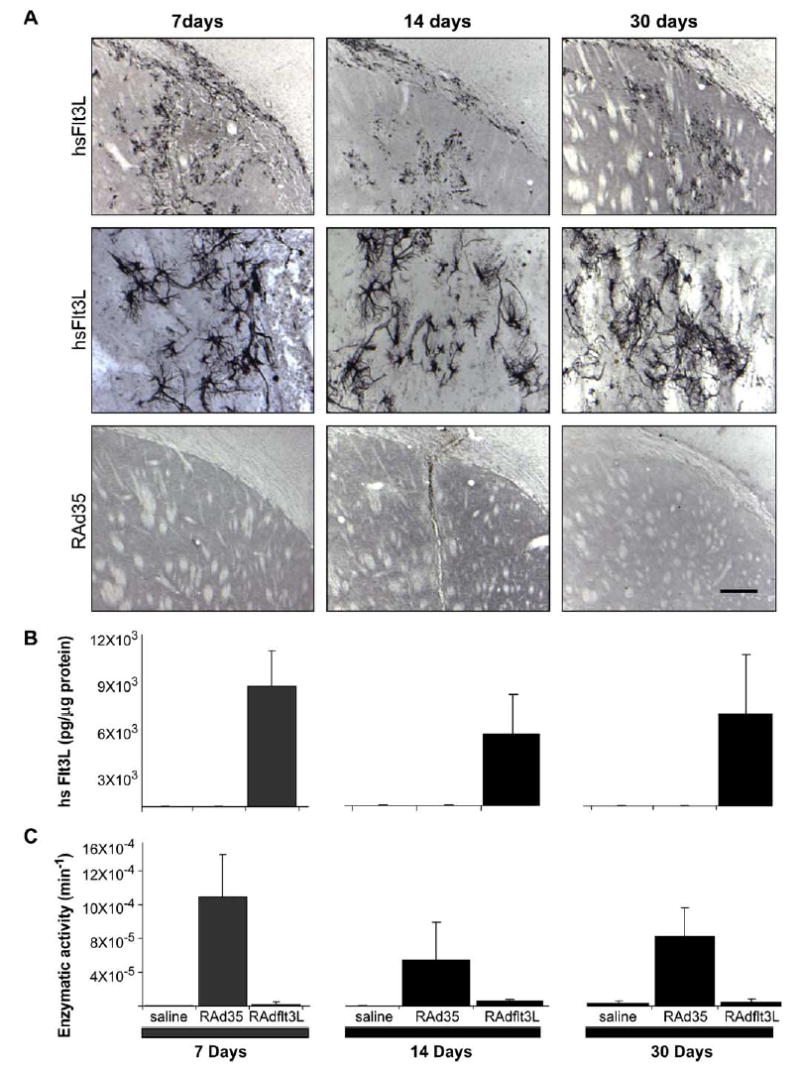
Expression and activity of RAd35 and RAdhsFlt3L. (A) Immunohistochemistry for the presence of hsFlt3L in brains injected with RAd35 or RAdhsFlt3L; in the middle panels, hsFlt3L-immunoreactive cells can be clearly identified. In these experiments hsFlt3L immunoreactivity was detected by immunohistochemistry methods using a specific anti-hsFlt3L polyclonal rabbit antibody generated by us using specific unique peptides as immunogens. (B) hsFlt3L ELISA (pg/μg protein) of brain homogenates from Lewis rats injected with saline, RAd35, or RadhsFlt3L. (C) β-Galactosidase enzymatic activity assay of brain homogenates from Lewis rats injected with saline, RAd35, or RadhsFlt3L. For B and C, values are expressed as means ± SEM (n = 3). The scale bar, shown by the bottom right panel in A, equals 200 μm for the top and lower panels, and 60 μm for the middle panels.
TABLE 1.
Endogenous rat soluble Flt3L ELISA
| Treatment | 7 days | 14 days | 30 days |
|---|---|---|---|
| Saline | 17.75 ± 1.53 | 18.36 ± 2.24 | 15.24 ± 2.02 |
| RAd35 | 14.17 ± 4.43 | 13.43 ± 2.55 | 12.36 ± 3.01 |
| RAdhsFlt3L | 16.43 ± 2.73 | 16.58 ± 5.72 | 25.53 ± 2.3 |
Levels of endogenous, rat soluble Flt3L in brain homogenates from saline-, RAd35-, and RAdhsFlt3L-injected Lewis rats’ brains. Values are expressed as means ± SEM (n = 3).
In Vivo Effects of RAdhsFlt3L Delivered into the CNS of Lewis Rats
We injected male Lewis rats with RAdhsFlt3L (1 × 107 iu), into the striatum; we used injection of RAd35 (1 × 107 iu) expressing β-galactosidase and saline as controls. We characterized brain inflammation following intrastriatal injection 48 h, 7 days, and 14 days postsurgery using the following markers of inflammatory activation in the brain: ED1 (activated macrophages and microglial cells), MHCI, MHCII, CD8 (CD8+ T cells), CD4 (CD4+ T cells), and OX62 (dendritic and γδT cells). We examined potential tissue toxicity associated with the delivery of RAds and the expression of hsFlt3L immunohistochemically by examining for immunoreactivity to GFAP (astrocytes), NeuN (neurons), and MBP (myelin basic protein) for myelin integrity.
The most dramatic changes in brain inflammation were characterized by the upregulation of ED1, MHCI, and CD8 immunoreactivity. We detected monocyte, macrophage, and microglial activation by the expression of ED1. The ED1 response to the injection of saline was negligible compared to the injection of RAd35 or RAdhsFlt3L, at 48 h and 7 days post-viral injection. Although the increase in ED1 immunoreactivity was significant for both RAd35 and RAdhsFlt3L, the effect of hsFlt3L was three to four times that of RAd35. There was a decrease in ED1 activation over time; the effect of RAd35 disappeared by day 14, while the activation induced by RAdhsFlt3L remained elevated (Fig. 3A).
FIG. 3.
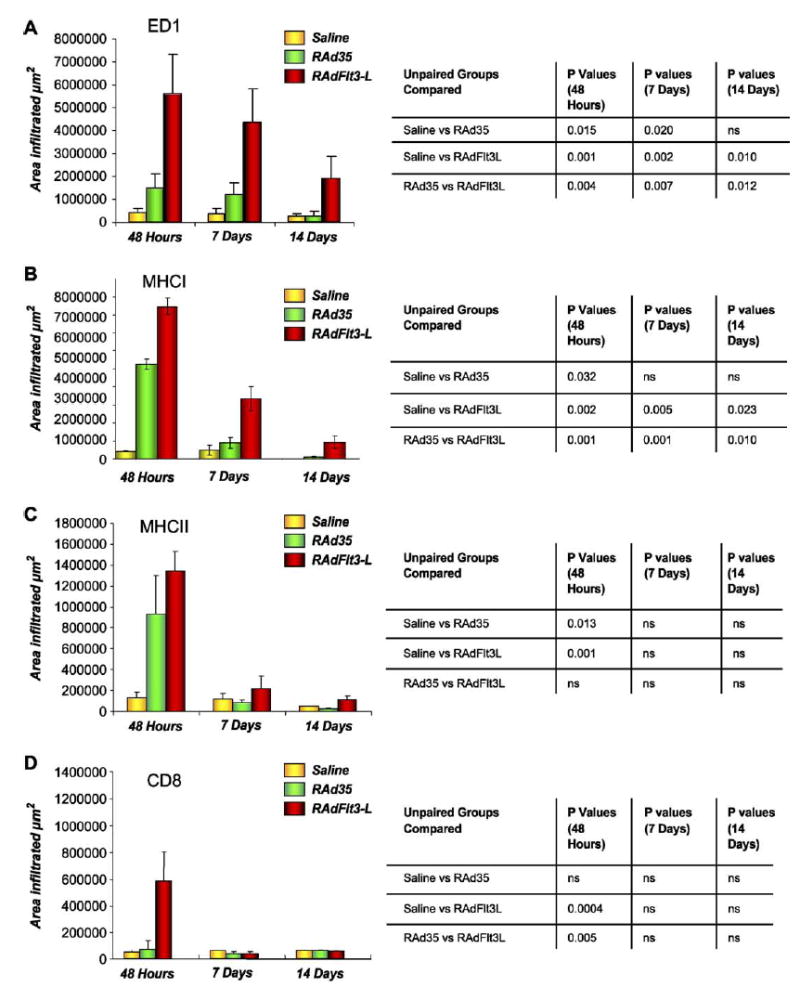
Inflammatory effects of RAdhsFlt3L delivered into the brain of Lewis rats. (A) ED1 quantification. (B) MHCI quantification. (C) MHCII quantification. (D) CD8 quantification. Quantification of the area occupied by ED1, MHCI, MHCII, and CD8 immunoreactivity, 48 h, 7 days, and 14 days post-intrastriatal injection was performed using a semiautomatic Quantimet imaging system. Serial sections were studied and infiltration was determined using the sections displaying the largest amounts of infiltration per brain, per treatment. Error bars show the SEM value from four animals in each experimental group. ED1, MHCI, MHCII, and CD8 immunoreactivities observed in each experimental group were compared to one another at each time point using an unpaired Student t test; the results are presented in the tables next to each bar graph (ns, not significant).
We detected MHCI upregulation on many cells, including invading inflammatory-and tissue-specific cells. MHCI molecules, however, are widely expressed on other cell types as well, i.e., resident microglia and infiltrating monocytes/microglia. MHCI upregulation was highest at 48 h postinjection and decreased thereafter (Fig. 3B). MHCI expression in brains treated with RAdhsFlt3L was significantly higher than that observed in RAd35-or saline-treated brains at 48 h ( P < 0.05) (Fig. 3B). MHCI expression remained higher in the RAdhsFlt3L treatment group at each time point studied ( P < 0.005) (Fig. 3B).
MCHII expression, as predicted, appeared to be restricted mainly to immune infiltrating cells within the ipsilateral striatum. The greatest MCHII expression was seen in RAdhsFlt3L-treated brains at 48 h compared to controls ( P < 0.05) (Fig. 3C). There was a dense infiltration of MHCII-positive cells in the injection site and perivascular cuffing in areas adjacent to it. Although MHCII expression remained elevated in RAdhsFlt3L-injected brains compared to those injected with saline, the peak in increased expression was at 48 h and, compared to ED1 and MHCI, appeared to be to a large extent virus-specific more than transgene-specific, i.e., there are no significant differences between the RAd35-and the RAdhsFlt3L-treated animals (Fig. 3C).
Other markers examined include CD8, CD4, and OX62. The CD8 infiltration was not as intense or as widely distributed in the CNS as the ED1 immunoreactivity. The highest levels of CD8 immunoreactivity (marker for cytotoxic T cells and NK cells) were detected at 48 h for each treatment group, with statistically much higher infiltration in the brains of animals treated with RAdhsFlt3L compared to RAd35 or saline controls ( P < 0.05) (Fig. 3D). Interestingly, CD8-immunoreactive cells were back to normal values by day 7 postinjection.
The CD4 staining followed a pattern very similar to that of the CD8 immunoreactivity. The anti-CD4 monoclonal antibody used identifies expression of CD4 on activated macrophages and microglia (in rats CD4 is expressed within the monocyte lineage), as well as CD4 helper T cells. At 48 h cells resembling microglia and CD4-positive T cells were detectable in the striatum, with the greatest expression in RAdhsFlt3L-treated brains. The CD4 expression was greatly reduced by 7 days and essentially restricted to small spherical cells with typical T cell characteristics. By 14 days very little staining was detectable in all groups (results not shown).
A large number of OX62-positive cells associated with blood vessels and the CNS parenchyma were detectable only at 7 days post-intrastriatal injection (Fig. 4). A greater number of these cells were present in the brains of animals treated with RAdhsFlt3L compared with RAd35 or saline, which had no OX62-positive cells. These cells displayed an elongated, amoeboid morphology, with the occasional dendritic process seen protruding from one end of the cellular body. OX62-positive cells were not detectable by 14 days. The increased number of OX62-immunoreactive cells, together with the upregulation of MHC expression and enhanced inflammation in RAdhsFlt3L-treated brains, is indicative of a proinflammatory effect of hsFlt3L within the CNS.
FIG. 4.
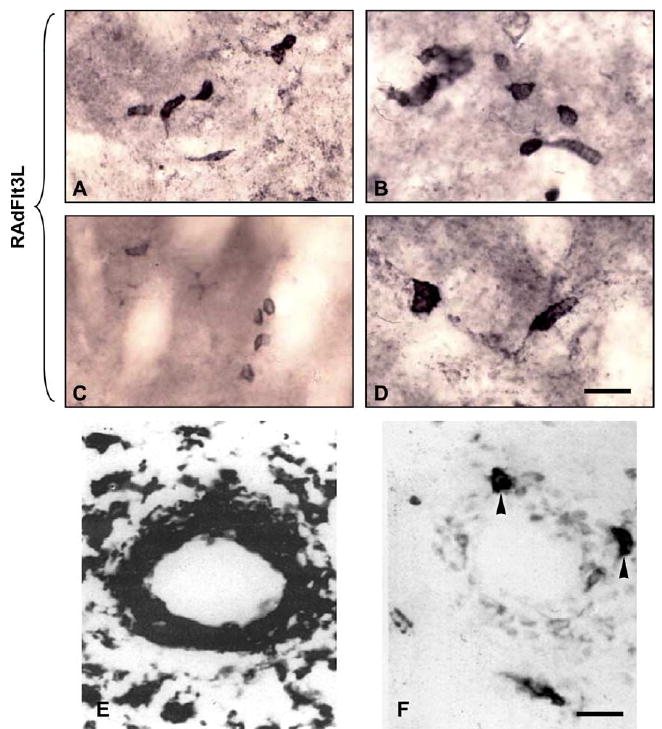
(A–D) High-magnification images of OX62-immunoreactive cells within the brains of Lewis rats treated with RAdhsFlt3L. Images were taken at either 20 ×2.5 or 100 × 2.5 magnification. (E) OX6 (MHCII) immunoreactivity around the perivascular cuffs of a blood vessel in the CNS parenchyma. (F) Serial section of the same blood vessel showing OX62-positive cells that are also MHCII positive. The scale bar for A–D (shown in D) equals 20 μm; the scale bar for E and F (shown in F) equals 20 μm.
We assessed possible tissue toxicity associated with adenoviral vector delivery and hsFlt3L expression within the brain by immunostaining for GFAP, NeuN, and MBP (results not shown). Administration of both RAd35 and RAdhsFlt3L caused astrocyte activation (GFAP upregulation) throughout the ipsilateral striatum. This widespread glial activation was not seen in brains injected with saline. We detected greater GFAP immunoreactivity in the brains treated with RAdhsFlt3L compared to RAd35 or saline, and this peaked at 7 days postinjection and may be related to the activation caused not only by the mechanical stimulation but also by the injection of recombinant adenovirus. GFAP immunoreactivity decreased by day 14. Similar to GFAP staining, we detected a small area of neuronal loss (assessed with NeuN staining) due to the mechanical injury that was observed for each treatment and time point (results not shown). No differences were detected between treatment groups. Also there was no change in myelin integrity at any time point studied with either of the treatments (results not shown).
Biological Activity of hsFlt3L in Vivo in C57BL/6 Mice
Although following the delivery of hsFlt3L into the brains of Lewis rats provides evidence for the presence of cells expressing DC markers, as shown by the detection of OX62-positive cells, there are more markers available for the characterization of DC in mice. Because of the importance of characterizing the presence of DCs in the brain in response to Flt3L, we decided to determine the expression of DC markers in the brains of C57BL/6 mice following the injection of 1 × 107 iu of RAdhsFlt3L (Fig. 5A) or RAd35 (Fig. 5B). We determined the presence of cells expressing DC markers immunohistochemically 7 days post-intrastriatal injection using the following markers: MHC II, F4/80 (macrophages and microglia), 33D1, CD11c (two markers of myeloid DCs), and DEC205 (a marker of lymphoid-derived DCs). While injection of RAd35 expressing β-galactosidase only caused minor upregulation of some of the inflammatory markers (Fig. 5B), injection of RAdhsFlt3L was followed by an upregulation of MHCII immunoreactivity (Fig. 5A), which was detected throughout the ipsilateral striatum, corpus callosum, and cortex. The macrophage and microglial marker F4/80 was upregulated to a lesser extent than MHCII. We also detected immunoreactivity for 33D1 and CD11c, but expression was again lower than for either MHCII or F4/80. Expression of MHCII, F4/ 80, 33D1, and CD11c could be present in either infiltrating and/or resident cells within the brain. The marker of lymphoid-derived DCs, DEC205, was not detected within cells located within the brain parenchyma, but was only rarely seen surrounding blood vessels. All these results taken together indicate that hsFlt3L induces a very high expression of MHCII on brain cells, as well as infiltration of myeloid-, but not lymphoid-, derived DCs (Fig. 5A).
FIG. 5.
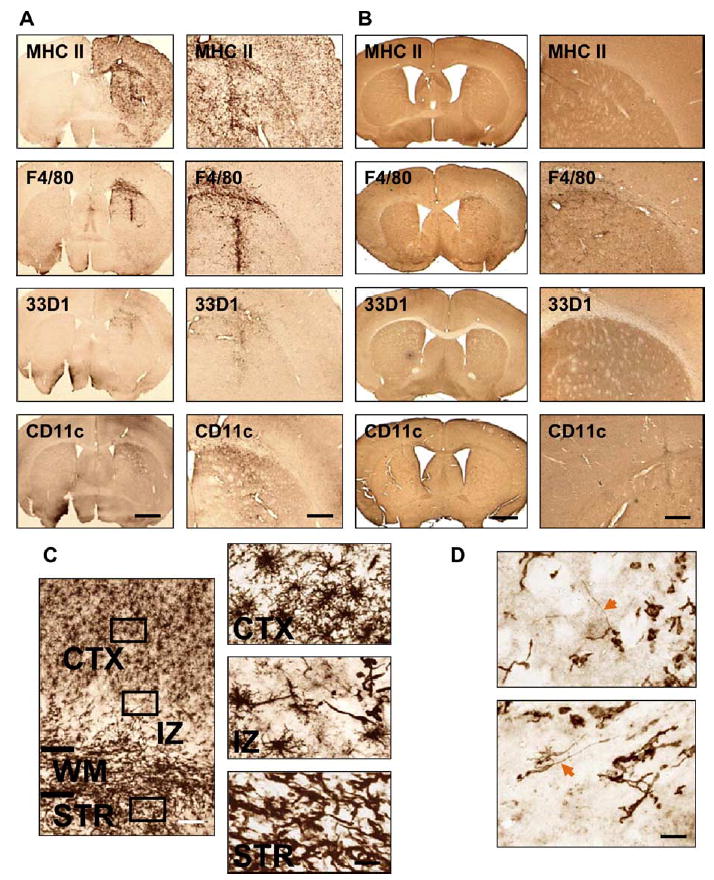
Effects of RAdhsFlt3L in vivo in the brain of C57BL/6 mice. (A) Expression of myeloid DC markers in the brains of RAdhsFlt3L-treated C57BL/6 mouse brains 7 days post-intrastriatal injection (n = 4). (B) Expression of myeloid DC markers in brains of RAd35-treated mice 7 days post-intrastriatal injection (n = 3). (C) High-magnification demonstration of MHCII immunoreactivity. Three distinct cellular morphologies can be seen in three different zones of the brain section, suggesting differentiation of dendritic cells (STR, striatum; CTX, cortex; IZ, intermediate zone). (D) High-magnification illustration of 33D1-positive cells. These cells have unique long processes protruding from the cellular body. The scale bar for the left-hand side panels of A and B, shown on the last panel, equals 0.5 mm; the scale bar for the right-hand side panels of A and B, shown on the last panel, equals 0.15 mm. The scale bar for the left-hand side image in C, equals 100 μm; the one at the bottom right of the right-hand side panels of C equals 20 μm. The scale bar for D equals 20 μm.
Examination of cells expressing MHCII at high magnification identified the presence of cells displaying three different morphologies (Fig. 5C). Closest to the injection site itself, cells with an elongated amoeboid morphology can be seen infiltrating the striatum, while in the cortex, which represents the region farthest away from the injection site, the cells resemble typical ramified microglia. There is an interphase/intermediate zone between the striatum and cortex, where cells have an intermediate morphology, not exclusively amoeboid or exclusively ramified.
Similarly, examination of cells expressing a DC-specific antigen recognized by antibody 33D1 at high magnification identifies a cell type with a cellular morphology consistent with that of differentiated DCs, i.e., long processes extending from a central cell body (Fig. 5D).
RAdhsFlt3L Improves the Survival of Lewis Rats Bearing Intracranial Syngeneic Gliomas in a Dose–Dependent Manner
To assess the anti-tumor activity of RAdhsFlt3L, we injected rats bearing intracranial CNS-1 gliomas with increasing doses (1 × 107 or 8 × 107 iu) of either RAdhsFlt3L or RAd35. We used RAd35 and saline as negative controls. In our syngeneic microscopic glioma model the survival rates for rats treated with RAdhsFlt3L were better than the survival rates of rats treated with either RAd35 or saline (Fig. 6). At a dose of 1 × 107 iu, 20% of the rats survived long term when treated with RAdhsFlt3L ( P < 0.005). Increasing the dose of virus from 1 × 107 to 8 × 107 iu elicited a dose-dependent increase in the number of rats surviving when treated with RAdhsFlt3L to 60% ( P < 0.0005). Increasing the dose of RAd35 failed to improve the survival of rats bearing microscopic tumors, with the rats succumbing to the tumors between 18 and 20 days.
FIG. 6.
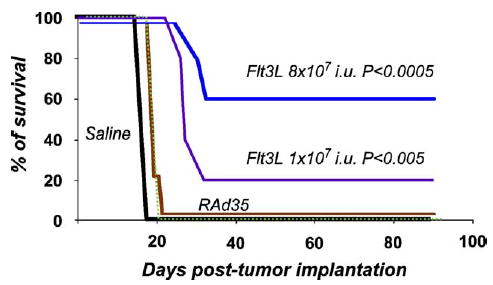
Survival of animals injected with 1 × 107 and 8 × 107 iu virus of RAd35 and RAdhsFlt3L. Animals with microscopic tumors were treated on day 3 with either RAdhsFlt3L or RAd35 (1 × 107 or 8 × 107 iu) or saline included as a control. hsFlt3L significantly increased, in a dose-dependent manner, the survival of animals with microscopic tumors.
We perfused/fixed rats surviving treatment with RAdhsFlt3L for 6 months and performed immunohistochemical and neuropathological analyses. In 3-month survivors, ~60% of the animals had a tumor mass remaining in the ipsilateral hemisphere (Figs. 7 and 8). This suggests that treatment with RAdhsFlt3L somehow inhibits tumor growth, possibly due to the stimulation of a local inflammatory or systemic immune anti-tumor response. However, rats that survived for up to 6 months did not show evidence of tumor (Figs. 7 and 8). Animals treated with RAd35 did not survive beyond 20 days and thus were unavailable for direct time point comparison with the RAdhsFlt3L-treated, tumor-bearing Lewis rats.
FIG. 7.
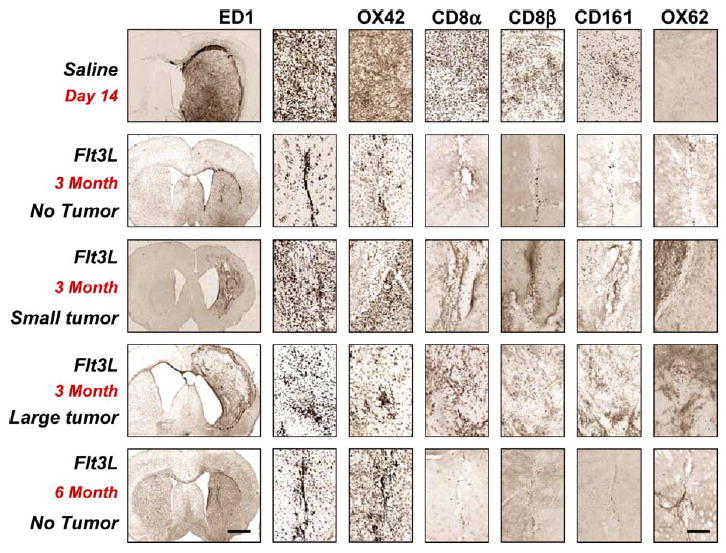
Immunohistochemical analysis of brains from animals bearing microscopic tumors treated with either hsFlt3L or saline. Animals that are completely tumor free are infiltrated with activated monocytes and macrophages mainly along the needle tract. Animals that have small tumors have greater inflammation as characterized by increased ED1 and CD8 immunoreactivity, which is concentrated around islands of growing tumor cells. Characterization of the CD8-immunoreactive cells shows that the majority of the infiltrating cells are CD8β positive (cytotoxic T cells), although there is also infiltration of NK cells. OX62-positive cells were seen only in animals surviving hsFlt3L treatment long term. The scale bar for the low magnification left-hand side panels, shown at the bottom left-hand side panel, equals 1 mm; the scale bar for all other panels, shown at the bottom right-hand side, equals 100 μm.
FIG. 8.
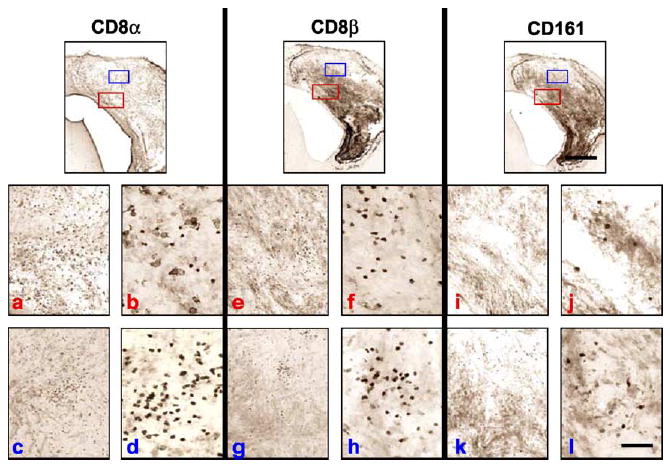
Comparison of the CD8α, CD8β, and CD161 immunoreactivities in serial sections of Lewis rat brains bearing microscopic tumors surviving treatment with RAdhsFlt3L 3 months post-CNS-1 cell implantation. CD8β-positive cells are present in greater numbers than CD161-positive cells in serial brain sections, suggesting a greater role for CD8β CTLs in inhibiting tumor growth. However, if the numbers of CD8β-positive and CD161-positive cells are combined, the total number of cells does not account for the total number of cells represented by the CD8α staining, which means there is a third cell population, most probably CD8α positive. Red letters (a, b, e, f, i, j) indicate low- (a, e, i) and subsequent high- (b, f, j) magnification images taken from the red boxed areas in each ICC staining condition shown in the top row. Blue letters (c, d, g, h, k, l) indicate low- (c, g, k) and subsequent high- (d, h, l) magnification images taken from the blue boxed areas in each ICC staining condition shown in the top row. Two areas are illustrated to highlight the heterogeneity encountered within the tumors of long-term survivors. Such heterogeneity was found only in these animals. The scale bar for the low magnification images, shown on the right-hand side panel, equals 0.5 mm; the scale bar for all other images, shown on the lower right panel, equals 50 μm.
We analyzed brains of rats surviving treatment with RAdhsFlt3L histopathologically to assess the distribution of inflammatory and immune markers, as well as the integrity of myelin fibers. We characterized brain inflammation using the markers ED1, OX42, CD8α, CD8β, CD161, and OX62 (Figs. 7 and 8). Rats injected with saline died between 18 and 22 days. At 2 weeks these animals had large solid tumors, which filled the entire striatum and protruded into the cerebral ventricles. Rats that survived treatment but still had a tumor at 3 months had irregular tumors, which appeared to be expanding into the cortex (Figs. 7 and 8). Rats that were completely tumor free had a scar in the injection site, with enlarged ventricles, an indicator that there had been striatal tissue loss. Activated monocytes/macrophages and microglia (ED1 and OX42) represented the largest inflammatory or immune cell population infiltrating any of the tumors (Fig. 8); they remained as an indicator of scar tissue in animals that had eliminated the tumors.
Other inflammatory markers investigated follow a similar pattern of staining, although there was much less infiltration of other immune cells; CD8β and CD161, which are markers for CTLs and NK cells, respectively, both infiltrated tumors, albeit to a much lesser degree than monocyte-derived cells (Figs. 7 and 8). We detected higher numbers of CD8β cells infiltrating the tumors compared to the numbers of CD161-immunoreactive cells, suggesting a greater role for CTLs in the control of tumor growth (Figs. 7 and 8). Interestingly the expression of the myeloid lineage dendritic cell marker OX62 was present only in rat brains surviving treatment with RAdhsFlt3L up to 3 and 6 months.
Discussion
We have previously demonstrated that the CNS-1 glioma model in Lewis rats is advantageous since the CNS-1 cells are syngeneic in this strain of rats [20]. Because many glioma models have been established either in allogeneic host strains or in animals with various immune deficiencies, testing immune-stimulatory or proinflammatory approaches has been difficult.
Using the CNS-1 glioma model we tested the gene therapy efficacy of a cytokine, i.e., hsFlt3L, which exerts a powerful differentiation factor for monocyte-derived cells to become DCs. The brain, in its naive or unstimulated state, is believed to be naive of bona fide DCs although some reports have detected DCs in the inflamed brain [17]. Also, the brain contains a large number of local tissue macrophages called microglial cells that are downregulated in the normal brain, but can be activated by various stimuli. We hypothesized that hsFlt3L would have a proinflammatory and/or immune-stimulatory effect that could induce the elimination of brain gliomas. RAdhsFlt3L was indeed effective in inducing brain inflammation and in providing a powerful anti-tumor effect in the CNS-1 syngeneic glioma model. DCs are not detected in the naive, noninflamed brain. However, they have been detected in the brain under conditions of inflammation, caused by various mechanisms. Two hypotheses have been brought forward to account for the appearance of cells displaying DC markers and function in the CNS during inflammation. One hypothesis proposes that brain DCs are derived from circulating myeloid DC precursors that differentiate into myeloid DCs within the brain parenchyma. Alternatively, it has been proposed that brain microglial cells can, during brain inflammation, display bona fide DC markers and behave like myeloid-derived DCs. Alternatively, it is possible that cells displaying markers and functioning as DCs in the CNS are derived from both sources [23–34]. The ultimate origin and function of DCs in the CNS remain to be determined. Importantly, no lymphoid-derived DCs have been found within the brain under any circumstances.
RAdhsFlt3L also induced a dose-dependent brain inflammation. There was an increase in activated macrophages and microglia (ED1 staining) and an important upregulation of both MHCI and MHCII. Upregulation of ED1 and MHCI followed similar time courses, with an early peak and progressive reduction up to day 14. This suggests that MHCI is being expressed mainly on infiltrating macrophages and/or activated microglia or other resident brain cells. The peak in MHCII expression was narrower, centered on 2 days postinjection, while remaining somewhat elevated on days 7 and 14. Influx of CD8+ cells was strictly limited to a very narrow peak at 2 days postinjection that returned to normal at 7 days. These data appear to indicate that expression of MHCII may be localized to infiltrating cells. However, immunohistochemical detection of MHCII in the brains of mice stained activated microglial cells, suggesting that both endogenous and infiltrating cells upregulate expression of MHCII in response to hsFlt3L. Cells expressing DC antigens, like OX62 in the rat and CD11c and 33D1, were also detected, although in much smaller numbers than the other inflammatory cells and markers. While DCs seem the likely candidate for hsFlt3L effects it is also possible that the expression of hsFlt3L triggers induction of downstream mediators, i.e., chemokines, which could be responsible for tumor regression unrelated to DC maturation.
Interestingly, the kinetics of brain immune responses differ between mice and rats. Although in rats, brain inflammation peaks earlier than in mice, in general, in mice inflammation tends to lasts longer. Similar data have also been shown both in systems examining immune responses to viral vector injections in the CNS and in immune responses to spinal cord injury [35–37]. The basis of this difference has not been elucidated and is likely dependent on mouse/rat strain.
While our data suggest a greater role for CTLs than other cell types in controlling tumor growth, further examination of which cells are involved is necessary for definitive conclusions. Of specific interest are the effects of NK cells, which are known to be involved in tumor eradication upon systemic Flt3L treatment [38]. Likewise, anti-tumor antibodies or cytokine mediation of tumor cell death via FasL or TNF-α may warrant further exploration.
Concurrent with its proinflammatory effect, we also detected a dose-dependent increase in the survival of glioma-bearing Lewis rats. This therapeutic response to hsFlt3L treatment is not unique to tumors growing in the CNS. Similar results with hsFlt3L treatment using daily administration of the protein have been obtained with tumors growing in other organs [15]. Maraskovsky et al. [15] also showed that the beneficial effects of hsFlt3L treatment not only are dose dependent, they also depend on the length of time hsFlt3L treatment is administered. Studies in mouse tumor models have demonstrated that hsFlt3L treatment leads to the generation of protective anti-tumor immune responses. It is thought that the dramatic increase in DC numbers in hsFlt3L-treated animals leads to amplified uptake of tumor cell antigens and an anti-tumor immune response [38–40]. The immunohistopathological data from long-term survivors in our experiment show that the tumors are infiltrated with both CD8β-(a marker for CTLs) and CD161-(a marker for NK cells) positive cells (both cell types also express CD8α) and are consistent with the data of Maliszewski et al. [53]. Although infiltration of CTLs and NK cells is evident, the presence and density of immune cells is not necessarily correlated directly with their function. Even if several immune cell types are found in the brain tumors under different treatment conditions, their numbers alone are unlikely to determine their activity. HsFlt3L or HSV1-TK/ GCV may have conferred onto the immune cells the cytotoxic activity that stimulates their anti-tumor effects. There is also an undefined third CD8α-positive cell population. Although we have not characterized this population of cells, it could perhaps represent lymphoid-derived DCs. Although no demyelination was observed up to 3 months in the animals that survived, it is still a possibility that with continuous expression of Flt3L in the CNS, an autoimmune reaction might occur. For clinical implementation, Flt3L expression would need to be under the control of an inducible promoter system.
Although we have used hsFlt3L in these studies, it is unlikely that immune infiltrates observed in hsFlt3L-treated animals were due to the putative inflammatory effects of the hsFlt3L molecule in rats, since no such effects were seen at longer time points analyzed. Also, no effects were observed in animals treated with the control RAd expressing β-galactosidase, an Escherichia coli protein with no homologue in rat or mouse or with any other foreign protein expressed in the CNS using RAds. In addition, the degree of conservation between human and rat or mouse Flt3L is ~70%.
Interestingly, this cytokine gene-based approach, compared to the conditional cytotoxic gene therapy studies by Dewey et al. [20] using HSV1-TK + GCV, does not lead to the complete elimination of the tumor at 3 months. In animals treated with RAdTK+GCV, surviving animals at 3 months do not show any evidence of the presence of remaining tumors. Control animals die in less than 30 days. Herein, however, of all the rats surviving treatment with RAdhsFlt3L and studied at 3 months, 33.3% were completely tumor free, while the remaining 66.6% had irregular tumors growing within the striatum and the overlying neocortex. We interpret this as the treatment providing a veritable struggle between the tumor and the immune system, the outcome of which is not 100% predictable. Thus at 3 months some animals are already cured (no tumor), some may be cured at a late time point (small tumor), and some will die (large tumor). This is important because the increased survival provided by our novel treatment will allow us to model the tumor/immune system struggle, an important area of the immune-stimulatory anti-tumor strategies that has not been able to be modeled by any of the commonly used tumor models.
Phenomenologically, it appears that the tumor slowed down its growth rate in hsFlt3L-treated animals. Some of the tumor-bearing animals surviving 3 months were found to be tumor free by 6 months since at this longer time point all of the surviving animals were tumor free, and no animals died between the 3-and the 6-month time points. These results are compatible with an ongoing “battle” between the tumor and the immune system and could be interpreted as providing evidence for the equilibrium phase of the cancer immunoediting hypothesis [41], which, following stimulation of an inflammatory and possibly an immune response due to overexpression of hsFlt3L, eventually leads to elimination of the tumor in many animals, but slower, continued growth in others; e.g., one animal succumbed to the tumor after 151 days in the macroscopic model of glioma. The fact that hsFlt3L significantly inhibited CNS-1 tumor growth and/or completely eliminated the tumors makes this an attractive approach to inhibit tumor recurrences after surgery.
The combined data presented here have important implications for the implementation of immunotherapy for brain tumors. Glioma patients are highly immunocompromised, and if immunotherapy is to be successful for the treatment of brain gliomas, it will have to be combined with other conventional treatment strategies such as surgery and/or radiotherapy and chemotherapy. It has been reported that surgical debulking of the tumor leads to restoration of the immune competence of the host [42]. We demonstrate for the first time that adenoviral-mediated expression of hsFlt3L within tumor-bearing rats leads to either elimination of the tumor mass (in the 6 months survivors) or significant prolongation of the life span of these animals.
Materials and Methods
Construction of RAdhsFlt3L (expressing fms-like tyrosine kinase ligand)
RAds used for this study are first-generation replication-defective recombinant adenovirus type 5 vectors expressing the transgenes under the transcriptional control of the human cytomegalovirus intermediate early promoter within the E1 region. RAd35 (an adenovirus encoding LacZ under the control of the hCMV promoter) was originally described by Wilkinson and Akrigg [43] and has been used previously [20,44–48]. RAdhsFlt3L was generated by cloning the Flt3L cDNA (provided by Immunex, Seattle, WA, USA) into the unique BamHI cloning site of the pAL119 shuttle vector. The shuttle vector was then cotransfected with the E1-deleted adenoviral vector plasmid PJM17 (Microbix Biosystems, Toronto, ON, Canada) into the human embryonic kidney HEK 293 cell line. The presence of the transgenes within the RAds was tested by restriction analysis of the viral DNA and by analysis of protein expression using immunocytochemistry [49] (Fig. 1). The methods for adenoviral generation, production, characterization, and scale up and viral vector purification have been previously described [44,49]. Titrations were carried out in triplicate and in parallel for all viruses by end-point dilution, cytopathic effect assay, with centrifugation of infected 96-well plates as described in detail by Nyberg-Hoffman et al. [50]. The titer determined was 6.55 × 1011 iu/ml for RAdhsFlt3-L and 1.64 × 1011 iu for RAd35. All viral preparations were screened for the presence of replication-competent adenovirus (RCA) [51] and for LPS contamination, using the Limulus amebocyte gel clot assay (Biowhittaker, UK) [52]. Virus preparations used were free from RCA and LPS contamination. All relevant adenoviral methods and quality control procedures are described in detail in Southgate et al. [49]. Viruses were diluted in sterile saline for injection.
Animal model
Male Lewis rats (225–250 g in body weight) were anesthetized with halothane and placed in a stereotaxic frame that had been modified for use with inhalational anesthesia [48]. Animals were injected in the left striatum (1 mm forward from bregma, 3 mm lateral and ventral 4 mm from the dura) with 5 × 103 CNS-1 cells [20]. The cells were administered in a volume of 3 μl using a 10-μl Hamilton syringe. For each injection a small pocket was created before the deposition of cells in the striatum. This involved moving the needle 0.2 mm below the stated coordinates and holding for 1 min, before moving up to the stated coordinates and slowly injecting all the cells over a period of 3 min. The needle was left in place for a further 5 min before being removed.
Viruses were injected into the tumor 3 days post-CNS-1 cell implantation, using the same anterior and lateral coordinates. Doses of 1 × 107 and 8 × 107 iuRAdhsFlt3L were injected intratumorally. RAd35 and saline were used as controls. Animals were monitored daily, and rats showing morbidity were perfused-fixed with heparinized Tyrode followed by 4% paraformaldehyde in PBS. The brains were removed for histological analysis. Animals surviving treatment were allowed to survive for up to 3 and 6 months to assess the efficiency and side effects of the treatment within the brain.
ELISA for Flt3L
ELISA for rodent or human soluble Flt3L (R&D Systems, Minneapolis, MN, USA) were conducted exactly as described by the manufacturer’s instructions.
β-Galactosidase enzymatic activity assay
Blocks of brain tissue around the injection site were homogenized and a single-cell suspension was created by multiple freeze–thaw cycles. Cellular debris was pelleted and supernatant stored on ice until use. β-Galactosidase activity was measured by conversion of o-nitrophenyl-β-d-galactopyranoside in 10 mM MgCl2/0.45 M 2-mercaptoethanol. After careful monitoring of color conversion at 37°C, the enzymatic reaction was stopped with 1 M Na2CO3. A protein standard curve was generated by the Lowry method using BSA. A nitrophenol standard curve was generated using o-nitrophenol. Enzymatic activity was calculated as follows: enzymatic activity/min = [o-nitrophenol]/(time × [protein]).
Brain immunohistochemistry
Forty-micrometer-thick coronal sections were cut through the striatum using a Vibratome. Free-floating immunohistochemistry was performed to detect inflammatory and immune cell markers. Endogenous peroxidase was inactivated with 0.3% hydrogen peroxide, and sections were blocked with 10% horse serum (Life Technologies, Paisley, Scotland) before being incubated overnight with primary antibody diluted in PBS containing 1% horse serum and 0.5% Triton X-100. The primary antibodies and the dilutions at which they were used were ED1, 1:1000 (activated macrophages/microglial cells; Serotec), anti-CD8, 1:500 (cytotoxic T lymphocytes and NK cells; Serotec), anti-CD8β,1:2000 (β chain of MHCI-restricted T cells; Becton–Dickinson), anti-CD161, 1:2000 (receptor on NK cells; Becton–Dickinson), anti OX62, 1:20 (dendritic cells; Serotec), anti-myelin basic protein, 1:2000 (Dako), and anti-β-galactosidase, 1:1000 (provided by Dr. R. Goya, University of La Plata, Argentina). Primary antibody specific to hsFlt3L (1:1000) was generated by injection of an antigenic peptide (CETVFHRVSQDGLDL) into New Zealand White rabbits (New England Peptides, Gardner, MA, USA). The antigenic peptide was coupled to KLH via the cysteine residue. Rabbits were pre-bled and immunized 14 and 28 days later with antibodies collected on days 35 and 42. Antibody specificity was verified by ELISA to the antigenic peptide and by Western blotting to the purified hsFlt3L protein (results not shown). All other primary antibodies were mouse monoclonal anti-rat, except for anti-β-galactosidase, which was a rabbit polyclonal. Secondary antibodies were biotinylated rabbit anti-mouse or biotinylated swine anti-rabbit (Dako), diluted 1:200 in 0.5% Triton X-100 with 1% horse serum, and were detected using the Vectastain Elite ABC horseradish peroxidase method (Vector Laboratories). After being developed with diaminobenzidine and glucose oxidase, sections were mounted on gelatinized glass slides and dehydrated through graded ethanol solutions before coverslipping.
Microscopy
Tissues were observed using an Olympus (AH2-RFL) microscope and images captured by standard photographic techniques. Low-and high-magnification images presented were photographed at 1 × 4 or 10 × 2.5, respectively. Morphological analysis involved higher magnifications, 40 × 2.5 or 60 × 2.5 or 10 × 2.5. Images of whole brain sections were taken at 1 × 2.5.
Statistical analysis
Survival data were analyzed by Kaplan–Meier estimator analysis and compared using the log-rank tests performed using the SPSS software and PRISM software.
Acknowledgments
Gene therapy projects for neurological diseases are funded by National Institutes of Health/National Institute of Neurological Disorders & Stroke Grant 1R01 NS44556.01; National Institute of Diabetes, Digestive and Kidney Diseases Grant 1 RO3 TW006273-01 to M.G.C.; National Institutes of Health/National Institute of Neurological Disorders & Stroke Grants NS 42893.01, U54 NS045309-01, and 1R21 NS047298-01 and Bram and Elaine Goldsmith Chair in Gene Therapeutics to P.R.L.; and The Linda Tallen & David Paul Kane Annual Fellowship to M.G.C and P.R.L. We thank Professors Hans Lassmann (Vienna) and Georg Kreutzberg (Munich) for their help in identifying the phenotype of the MHCII-and 33D1-expressing cells. We also thank the generous funding our institute receives from the Board of Governors at Cedars–Sinai Medical Center and the encouragement and support of each and every one of its members. We also acknowledge the unparalleled support and academic leadership of Dr. Shlomo Melmed. We are grateful to Mr. Richard Katzman for his superb administrative and organizational skills and to Mr. Danny Malaniak for his encouragement, support, and commitment. We also acknowledge Dr. C. A. Gerdes, for useful advice and assistance with Adobe PhotoShop.
References
- 1.Castro MG, et al. Current and future strategies for the treatment of malignant brain tumors. Pharmacol Ther. 2003;98:71 – 108. doi: 10.1016/s0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 2.Puumalainen A, et al. Beta-galactosidase gene transfer to human malignant glioma in vivo using replication-deficient retroviruses and adenoviruses. Hum Gene Ther. 1998;9:1769 – 1774. doi: 10.1089/hum.1998.9.12-1769. [DOI] [PubMed] [Google Scholar]
- 3.Sandmair AM, et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197 – 2205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- 4.Liau LM, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90:1115 – 1124. doi: 10.3171/jns.1999.90.6.1115. [DOI] [PubMed] [Google Scholar]
- 5.Post DE, Fulci G, Chiocca EA, VanMeir EG. Replicative oncolytic herpes simplex viruses in combination cancer therapies. Curr Gene Ther. 2004;4:41 – 51. doi: 10.2174/1566523044577988. [DOI] [PubMed] [Google Scholar]
- 6.Fulci G, Chiocca EA. Oncolytic viruses for the therapy of brain tumors and other solid malignancies: a review. Front Biosci. 2003;1:346 – 360. doi: 10.2741/976. [DOI] [PubMed] [Google Scholar]
- 7.Ehtesham M, et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia. 2004;6:287 – 293. doi: 10.1593/neo.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen specific cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2003;64:4973 – 4979. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 9.Izquierdo M, et al. Human malignant brain tumor response to herpes simplex thymidine kinase (HSVtk)/ganciclovir gene therapy. Gene Ther. 1996;3:491 – 495. [PubMed] [Google Scholar]
- 10.Ram Z, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354 – 1361. doi: 10.1038/nm1297-1354. [DOI] [PubMed] [Google Scholar]
- 11.Trask TW, et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195 – 203. doi: 10.1006/mthe.2000.0030. [DOI] [PubMed] [Google Scholar]
- 12.Markert JM, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867 – 874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 13.Rampling R, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7 doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 14.Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120 – 129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- 15.Maraskovsky E, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953 – 1962. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulendran B, et al. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J Immunol. 1997;159:2222 – 2231. [PubMed] [Google Scholar]
- 17.Serot JM, Foliguet B, Bene MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport. 1997;8:1995 – 1998. doi: 10.1097/00001756-199705260-00039. [DOI] [PubMed] [Google Scholar]
- 18.Lowenstein PR. Dendritic cells and immune responses in the central nervous system. Trends Immunol. 2002;23:70. doi: 10.1016/s1471-4906(01)02151-2. [DOI] [PubMed] [Google Scholar]
- 19.Kruse CA, Molleston MC, Parks EP, Schiltz PM, Kleinschmidt-DeMasters BK, Hickey WF. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol. 1994;22:191 – 200. doi: 10.1007/BF01052919. [DOI] [PubMed] [Google Scholar]
- 20.Dewey RA, et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat Med. 1999;5:1256 – 1263. doi: 10.1038/15207. [DOI] [PubMed] [Google Scholar]
- 21.Cowsill C, et al. Central nervous system toxicity of two adenoviral vectors encoding variants of the herpes simplex virus type 1 thymidine kinase: reduced cytotoxicity of a truncated HSV1-TK. Gene Ther. 2000;7:679 – 685. doi: 10.1038/sj.gt.3301147. [DOI] [PubMed] [Google Scholar]
- 22.Bigliari, A., et al. (2004). Effects of ectopic decorin in modulating intracranial glioma progression in vivo, in a rat syngeneic model. Mol. Ther. (in press). [DOI] [PMC free article] [PubMed]
- 23.Lowenstein PR. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 2002;23:23 – 30. doi: 10.1016/s1471-4906(01)02063-4. [DOI] [PubMed] [Google Scholar]
- 24.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991 – 2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164 – 174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21 :141 – 147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 27.Fischer HG, Reichmann G. Brain dendritic cells and macrophages/ microglia in central nervous system inflammation. J Immunol. 2001;166:2717 – 2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- 28.Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J Immunol. 2000;164:4826 – 4834. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- 29.Suter T, et al. The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol. 2003;33:2998 – 3006. doi: 10.1002/eji.200323611. [DOI] [PubMed] [Google Scholar]
- 30.Kivisakk P, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627 – 638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 31.Pashenkov M, Teleshova N, Link H. Inflammation in the central nervous system: the role for dendritic cells. Brain Pathol. 2003;13:23 – 33. doi: 10.1111/j.1750-3639.2003.tb00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stevenson PG, Austyn JM, Hawke S. Uncoupling of virus-induced inflammation and anti-viral immunity in the brain parenchyma. J Gen Virol. 2002;83:1735 – 1743. doi: 10.1099/0022-1317-83-7-1735. [DOI] [PubMed] [Google Scholar]
- 33.Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of CD8+T cells into the central nervous system by professional antigen-presenting cells. Am J Pathol. 1999;154:481 – 494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599 – 608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 35.Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 2003;462:223 – 240. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- 36.Zirger JM, et al. Immune system regulation of transgene expression in the brain. 2 Differential increase of mRNAs encoding for interferon-regulated, chemokine, and T-cell genes during either innate acute or chronic systemic adaptive immune responses to first generation adenoviral viral vectors. Mol Ther. 2004;9(Suppl 1):1019. [Google Scholar]
- 37.Barcia C, et al. Immune system regulation of transgene expression in the brain. 1 Systemically activated immune responses silence adenoviral vector-encoded transgene expression in the brain through both noncytolytic and cytotoxic mechanisms. Mol Ther. 2004;9(Suppl 1):459. [Google Scholar]
- 38.Fernandez NC, et al. Dendritic cells directly trigger NK cell functions: crosstalk relevant in innate anti-tumor immune responses in vivo. Nat Med. 1999;5:405 – 411. doi: 10.1038/7403. [DOI] [PubMed] [Google Scholar]
- 39.Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. 1997;3:625 – 631. doi: 10.1038/nm0697-625. [DOI] [PubMed] [Google Scholar]
- 40.Lynch DH. Induction of dendritic cells (DC) by Flt3 Ligand (FL) promotes the generation of tumor-specific immune responses in vivo. Crit Rev Immunol. 1998;18:99 – 107. doi: 10.1615/critrevimmunol.v18.i1-2.110. [DOI] [PubMed] [Google Scholar]
- 41.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991 – 998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 42.Mukherjee S, et al. The immune anti-tumor effects of GM-CSF and B7-1 gene transfection are enhanced by surgical debulking of tumor. Cancer Gene Ther. 2001;8:580 – 588. doi: 10.1038/sj.cgt.7700347. [DOI] [PubMed] [Google Scholar]
- 43.Wilkinson GW, Akrigg A. Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res. 1992;20:2233 – 2239. doi: 10.1093/nar/20.9.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shering AF, et al. Cell type-specific expression in brain cell cultures from a short human cytomegalovirus major immediate early promoter depends on whether it is inserted into herpesvirus or adenovirus vectors. J Gen Virol. 1997;78:445 – 459. doi: 10.1099/0022-1317-78-2-445. [DOI] [PubMed] [Google Scholar]
- 45.Gerdes CA, Castro MG, Lowenstein PR. Strong promoters are the key to highly efficient, noninflammatory and noncytotoxic adenoviral-mediated transgene delivery into the brain in vivo. Mol Ther. 2000;2:330 – 338. doi: 10.1006/mthe.2000.0140. [DOI] [PubMed] [Google Scholar]
- 46.Morelli AE, et al. Neuronal and glial cell type-specific promoters within adenovirus recombinants restrict the expression of the apoptosis-inducing molecule Fas ligand to predetermined brain cell types, and abolish peripheral liver toxicity. J Gen Virol. 1999;80:571 – 583. doi: 10.1099/0022-1317-80-3-571. [DOI] [PubMed] [Google Scholar]
- 47.Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc Natl Acad Sci USA. 2000;97:7482 – 7487. doi: 10.1073/pnas.120474397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas CE, Birkett D, Anozie I, Castro MG, Lowenstein PR. Acute direct adenoviral vector cytotoxicity and chronic, but not acute, inflammatory responses correlate with decreased vector-mediated transgene expression in the brain. Mol Ther. 2001;3:36 – 46. doi: 10.1006/mthe.2000.0224. [DOI] [PubMed] [Google Scholar]
- 49.Southgate, T., and Kingston, P. (2000). Gene transfer into neural cells in vivo using adenoviral vectors. In Current Protocols in Neuroscience (C. R. Gerfen, R. McKay, M. A. Rogawski, D. R. Sibley, P. Skolnick Eds.), pp. 4.23.1 – 4.23.40. Wiley, New York.
- 50.Nyberg-Hoffman C, Aguilar-Cordova E. Instability of adenoviral vectors during transport and its implication for clinical studies. Nat Med. 1999;5:955 – 957. doi: 10.1038/11400. [DOI] [PubMed] [Google Scholar]
- 51.Dion LD, Fang J, Garver RI., Jr Supernatant rescue assay vs. polymerase chain reaction for detection of wild type adenovirus contaminating recombinant adenovirus stocks. J Virol Methods. 1996;56:99 – 107. doi: 10.1016/0166-0934(95)01973-1. [DOI] [PubMed] [Google Scholar]
- 52.Cotten M, Baker A, Saltik M, Wagner E, Buschle M. Lipopolysaccharide is a frequent contaminant of plasmid DNA preparations and can be toxic to primary human cells in the presence of adenovirus. Gene Ther. 1994;1:239 – 246. [PubMed] [Google Scholar]
- 53.Maliszewski C, et al. Dendritic cells in models of tumor immunity. Role Flt3 ligand. Pathol Biol (Paris) 2001;49:481 – 483. doi: 10.1016/s0369-8114(01)00172-9. [DOI] [PubMed] [Google Scholar]