

Abstract
The degradation of the extracellular matrix is regulated by matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). Matrix components of the basement membrane play critical roles in the development and maintenance of the neuromuscular junction (NMJ), yet almost nothing is known about the regulation of MMP and TIMP expression in either the presynaptic or postsynaptic compartments. Here, we demonstrate that TIMP-2 is expressed by both spinal motor neurons and skeletal muscle. To determine whether motor function is altered in the absence of TIMP-2, motor behavior was assessed using a battery of tests (e.g., RotaRod, balance beam, hindlimb extension, grip strength, loaded grid and gait analysis). TIMP-2−/− mice fall off the RotaRod significantly faster than wild-type littermates. In addition, hindlimb extension is reduced and gait is both splayed and lengthened in TIMP-2−/− mice. Motor dysfunction is more pronounced during early postnatal development. A preliminary analysis revealed NMJ alterations in TIMP-2−/− mice. Juvenile TIMP-2−/− mice have increased nerve branching and acetylcholine receptor expression. Adult TIMP-2−/− endplates are enlarged and more complex. This suggests a role for TIMP-2 in NMJ sculpting during development. In contrast to the increased NMJ nerve branching, cerebellar Purkinje cells have decreased neurite outgrowth. Thus, the TIMP-2−/− motor phenotype is likely due to both peripheral and central defects. The tissue specificity of the nerve branching phenotype suggests the involvement of different MMPs and/or extracellular matrix molecules underlying the TIMP-2−/− motor phenotype.
Keywords: cerebellum, knockout, muscle, neuromuscular junction, TIMP
Introduction
Extracellular matrix (ECM) proteins of the synaptic basal lamina play a critical role in the maturation and maintenance of the neuromuscular junction (NMJ). Agrin binds to laminin-2 (α2β1γ1) and laminin-4 (α2β2γ1), the two predominant laminin isoforms expressed by muscle fibers. In addition, agrin binds to N-CAM, heparan sulfate proteoglycans, and αvβ1 and α7β1 integrins expressed on the myotube surface (reviewed in Sanes and Lichtman, 2001). In vivo time-lapse imaging has demonstrated the highly dynamic nature of NMJ maturation with axons extending and retracting over 24 hour intervals (Walsh and Lichtman, 2003). This dynamic process likely requires alterations in basal lamina composition of the synaptic cleft and the perisynaptic ECM.
The metzincin family of proteases are the main mediators of ECM degradation. The metzincin family includes matrixin/matrix metalloproteinase (MMP), ADAM (A Disintegrin And Metalloproteinase domain) and ADAM-TS (ADAM proteases containing a thrombospondin [TS] domain) members (Nagase and Woessner, 1999). Inasmuch as MMPs function in the turnover of a broad-spectrum of ECM (Nagase, 1997) and non-ECM proteins (McCawley and Matrisian, 2001), MMP activity must be tightly regulated. Many of the synaptic basal lamina constituents are substrates for MMPs, including agrin (VanSaun and Werle, 2000), laminin and perlecan (d’Ortho et al., 1997), integrin (Ratnikov et al., 2002) and β-dystroglycan (Yamada et al., 2001). MMP activity is regulated by tissue inhibitor of matrix metalloproteinases (TIMPs). TIMPs are 20–30 kDa secreted proteins that form tight, but relatively low selectivity complexes with the active forms of MMPs. Of the four known TIMPs, TIMP-2 is abundantly expressed in the central nervous system (CNS) and expression is maintained into adulthood (Fager and Jaworski, 2000; Young et al., 2002). In contrast to the sustained CNS expression, TIMP-2 mRNA expression in muscle is down-regulated at postnatal day 21 (P21) (Young et al., 2002). To characterize the role of TIMP-2 in NMJ maturation, we examined TIMP-2 deficient mice.
Two laboratories independently generated mice with altered TIMP-2 expression. No TIMP-2 mRNA was detected in mice generated by Soloway and colleagues, indicating a null mutation (Wang et al., 2000). These mice will be referred to as TIMP-2 knockout. In contrast, mice generated by Birkedal-Hansen and colleagues possess a 14 kDa protein that has a 300-fold lower MMP-inhibitory activity than that of the wild-type 22 kDa protein (Caterina et al., 2000). These hypomorphs will be referred to as TIMP-2 knock-down mice. Thus far, the only phenotype reported for both TIMP-2−/− mice is the impairment of proMMP-2 activation (Caterina et al., 2000; Wang et al., 2000). This reflects the unique ability of TIMP-2 to regulate both the activation of latent proMMPs to the proteolytically active form and the inhibition of MMP activity. Here, we report the regulation of TIMP-2 mRNA and protein expression in mouse CNS and muscle throughout postnatal development. In addition, we report the presence of both behavioral and histological alterations in TIMP-2 deficient mice. This observation substantiates the need for more careful examination of mice previously reported to lack a phenotype with behavioral methodologies.
Methods
Animals
Mice bearing a targeted disruption of the TIMP-2 gene have been described elsewhere (Caterina et al., 2000; Wang et al., 2000). Heterozygous breeding was performed to generate mice of all three genotypes such that littermates would serve as controls. TIMP-2 knock-down mice were maintained as homozygous recessive; therefore, littermate controls were not available. Offspring carrying the TIMP-2 mutant gene were identified by PCR analysis of ear punch DNA. Genomic DNA was prepared using a DNA extraction kit (Stratagene; Cedar Creek, TX) following manufacturer’s instructions. Products were amplified using HotStarTaq master mix (Qiagen; Valenica, CA) and either a forward oligo for neomycin (5′-GCCCAGTCATAGCCGAATAGCC-3′) or a forward oligo specific for TIMP-2 (PT2: 5′-CCCCTCCACCGTTCCTCTCTTTTC-3′) in conjunction with a reverse oligo specific for TIMP-2 (PT1: 5′-TCACCCAGCCAGCACCTCACC-3′). After an initial activation of the HotStar Taq at 94 °C for 6 min, 40 cycles of amplification were performed (94 °C for 30 sec, 60 ° for 30 sec and 72 °C for 1 min 15 sec [at a ramp rate of 12 °C, 0.8 °C/sec]). PCR products (750 bp for the mutant gene and 250 bp for wild-type) were separated on a 1.8% agarose gel. Procedures that involved animals were in accordance with the institutional guidelines of the Animal Care and Use Committee at the University of Vermont.
Northern Blot Analysis
Northern blot analysis was performed as previously described (Jaworski et al., 1994). RNA (25 μg total RNA for nervous system, 2.5 μg poly A+ RNA for muscle) was electrophoresed on a denaturing gel, transferred to Zeta-Probe membrane (Bio Rad; Hercules, CA) by capillary transfer and hybridized with a 32P-labeled cDNA to full length rat TIMP-2. To determine equal loading of RNA per lane, the TIMP-2 probe was removed in 0.1X SSC/0.1% SDS at 95 °C for 30 min and the membrane hybridized with the non-developmentally regulated gene cyclophilin (Lenoir et al., 1986).
Western Blot Analysis
Western blot analysis was performed as previously described (Jaworski and Fager, 2000) with minor modifications. Tissue was homogenized in Triton Lysis Buffer (20 mM Tris pH 7.4, 137 mM NaCl, 25 mM β-glycerolphosphate pH 7.4, 2 mM sodium pyrophosphate, 2 mM EDTA pH 7.4, 1% Triton X-100, 10% glycerol) containing a cocktail of protease inhibitors (1 mM EDTA, 1 mM phenyl-methylsulfonyl fluoride, 5 mM N-ethylmaleamide, 5 mM caproic acid, 5 μg/ml leupeptin and 5 μg/ml pepstatin A). Lysates were spun at 14,000 rpm for 15 min at 4 °C, and the supernatant used for western blot analysis. Proteins (20 μg for nervous system, 25 μg for muscle) were fractionated by SDS-PAGE on a 12% gel and transferred to Immobilon-P membranes (Schleicher & Schuell; Keene, NH). Membranes were blocked with 5% nonfat dry milk (NFDM) in Tris-buffer saline (TBS; 10 mM Tris, pH 7.5, 150 mM NaCl) with 0.4% Tween-20 (TBST) followed by overnight incubation at 4 °C with rabbit anti-human TIMP-2 (1,500X; Chemicon, Temecula, CA) diluted in TBST containing 3% BSA. After three washes with TBST, membranes were incubated with HRP-conjugated donkey anti-sheep (3,000X; Jackson ImmunoResearch; West Grove, PA) in 5% NFDM in TBST for 2 hrs at room temp. After washing as before, immunocomplexes were visualized by enhanced chemiluminescence according to manufacturer’s instructions (PerkinElmer Life Sciences; Boston, MA). Afterwards, the blots were stripped with 0.2 M glycine, 0.5M NaCl, pH 2.5 twice for 10 min, washed with TBST, and blocked as before. To determine equal loading of protein per lane, the blots were incubated with goat anti-human actin (5,000X; Santa Cruz Biotechnology; Santa Cruz, CA) and HRP-conjugated donkey anti-goat (5,000X; Jackson ImmunoResearch) as described above.
Fluorescence Microscopy
Immunohistochemistry was performed as previously described (Jaworski et al., 1999). Tissue sections (40 μm) were incubated in sheep anti-human TIMP-2 (200X; Biogenesis; Kingston, NH), rabbit anti-bovine neurofilament-200 (250X; Chemicon), rabbit anti-rat calbindin (1000X); Sigma; St. Louis, MO) or mouse anti-neuronal nuclei (NeuN) monoclonal antibody (500X; Chemicon) at 4 °C overnight. After 3 washes with PBS, the sections were incubated in the appropriate Cy 3-conjugated secondary antibody (500X; Jackson ImmunoResearch) for 2 hrs at room temp. After 3 washes as before, the tissue was mounted onto gelatin coated glass slides with Citifluor (Electron Microscopy Sciences; Hatfield, PA), examined with a Nikon E800 microscope (Micro Video Instruments; Avon, MA) and images captured with a Spot RT digital camera (Diagnostic Instruments, Sterling Heights, MI).
Muscle immunohistochemistry was performed as described (Son and Thompson, 1995). Muscle was removed from the perfused animal, rinsed 3 times (10 min each) in PBS, and stained with rhodamine-conjugated α-bungarotoxin (Btx) (10 μg/ml; Molecular Probes; Eugene, OR) for 15 min. After rinsing as before, the muscle was incubated in methanol for 5 min at −20 °C. The muscle was rinsed as before and blocked in PBS with 0.2 % Triton X-100, 2.0 % BSA, and 0.1 % sodium azide for 1 hr. The muscle was incubated with rabbit anti-rat neurofilament-145 (500X; Chemicon), diluted in blocking buffer, overnight at room temperature. After rinsing 3 times with blocking buffer, the muscle was incubated in Cy-2 conjugated goat anti-rabbit (100X; Jackson ImmunoResearch), diluted in block, for 4 hrs. After washing with PBS 3 times, the muscle was mounted onto gelatin coated glass slides with Citifluor and examined with a Nikon E800 microscope and images captured with a Spot RT digital camera. Endplate size was determined by encircling the total endplate area using software provided with the Spot digital camera.
Motor Behavioral Tests
TIMP-2−/− mice were examined using a subset of the SHIRPA protocol for behavioral and functional phenotypic assessment (Rogers et al., 1997). The tertiary screen of this protocol revealed increased anxiety in TIMP-2−/− mice as indicated by reduced prepulse inhibition and fear-potentiated startle (Jaworski et al., 2005).
RotaRod
The effect of TIMP-2 deletion on motor function was tested with a RotaRod (Dunham and Miya, 1957) using three fixed speed rotations (4, 20, 40 rpm). Animals were placed on the RotaRod rotating at 4 rpm for a maximum of 200 secs or until they fell off (latency to fall). Regardless of completion or fall, each animal was allowed to rest for 2 mins between each trial. Each testing session consisted of 10 trials. Animals were tested at 20 rpm and 40 rpm on consecutive days. With increasing speeds of rotation, animals tended to grip the rod and passively spin. Thus, at 40 rpm both the animal’s latency to fall and latency to passive rotation (the time it takes the mouse to make one complete revolution while griping) were recorded.
Balance beam
Balance was determined by measuring the time (up to 90 sec for P21 mice and 120 sec for adult mice) that mice were able to stay on a round wood beam. Three beams of decreasing diameter were used.
Hindlimb extension
Mice were suspended by the tail and the extent of hindlimb extension was observed. A score of 0 corresponds to the absence of hindlimb extension or the mouse gripping its tail (Fig. 5A). A score of 1 corresponds to an extension reflex of only one hindlimb or extension of both hindlimbs without splayed toes (Fig. 5B). A score of 2 corresponds to a normal extension reflex in both hindlimbs, including splaying of toes (Fig. 5C). Only hindlimbs extended > 90 ° were scored.
Figure 5. Hindlimb extension is reduced in TIMP-2−/− mice.
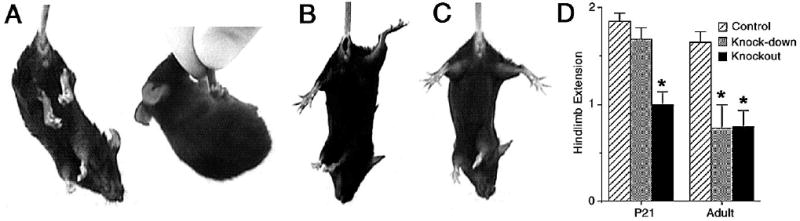
A-C) Photomicrographs showing representative hindlimb extension reflex scores of 0 (A), 1 (B) and 2 (C) in P21 mice. D) Hindlimb extension is reduced in P21 (F2, 28 = 17.17, p = 0.000) and adult (F2, 28 = 10.29, p = 0.000) knockout mice, but only adult knock-down mice. * p < 0.001 by t-test.
Trapeze
Grip strength was assessed by measuring the time (up to 30 sec) mice were able to hang from a non-slippery wire (2 mm diameter) with their forearms.
Loaded grid
Mice were allowed to grip a weight (5g for P21, 10g for adult) and then suspended by their tail. The time that the mouse was able to carry the weight (up to 30 sec) was recorded.
Gait analysis
Peripheral nerve function was assessed based on measurements made from walking tracks (de Medinaceli et al., 1982). Mice were manually restrained and their hindpaws dipped in non-toxic ink. Animals were then allowed to walk on white paper within a restricted area (70 x 400 mm). Stride length (mean distance between consecutive left or right paw prints), gait base of support (mean distance perpendicular to parallel left and right paw prints), and intrastep distance (mean distance between alternate left and right paw prints) were measured (Fig. 7A).
Figure 7. Gait is altered in TIMP-2−/− mice.

A) Representative footprints of P21 wild-type and knockout mice demonstrating the three parameters measured. B) Stride length is increased in P21, but not adult knockout mice and is unaltered in knock-down mice at either age (P21: F3, 20 = 3.27, p = 0.043; Adult: F3, 23 = 2.44, p = 0.094). C) The gait of both TIMP-2−/− genotypes is increased in both P21 and adult mice (P21: F3, 20 = 10.21, p = 0.000; Adult: F3, 23 = 3.38, p = 0.038). D) Intrastep distance is increased in P21, but not adult knockout mice and is unaltered in knock-down mice at either age (P21: F3, 20 = 2.95, p = 0.058; Adult: F3, 23 = 2.04, p = 0.141). * p < 0.05 relative to wild-type, # p < 0.05 relative to heterozygous by t-test.
Statistics
Data are presented as mean ± s.e.m. Variance between subjects within a litter was determined prior to pooling data from multiple litters. Significant interactions identified by ANOVA were then assessed with t-tests. Criterion for statistical significance is p < 0.05
Results
TIMP-2 expression is differentially regulated during murine CNS and muscle development
To investigate the role of TIMP-2 in NMJ maturation and maintenance, the expression of TIMP-2 mRNA and protein was examined (Fig. 1). Northern blot analysis of CNS (including brain and spinal cord) reveals that TIMP-2 mRNA expression increases throughout postnatal development (Fig. 1A). In the human, two TIMP-2 mRNA transcripts are generated by alternative polyadenylation (Hammani et al., 1996). In the murine CNS, the 1.0 kb transcript is constitutively expressed while the 3.5 kb transcript predominates and is developmentally up-regulated. This confirms the regulation of TIMP-2 mRNA previously reported during rat (Fager and Jaworski, 2000) and mouse (Young et al., 2002; Nuttall et al., 2004) brain development. To examine expression specifically in the spinal cord, immunohistochemistry was performed (Fig. 1B). TIMP-2 is expressed throughout the gray matter with expression detected in both the dorsal and ventral horns (Fig. 1Ba). In the ventral horn, TIMP-2 is expressed by motor neurons (Fig. 1Bb). Northern blot analysis of skeletal muscle (all muscles of the lower extremity) reveals that TIMP-2 mRNA expression differs from that in the CNS (Fig. 1C). In contrast to the CNS where the 3.5 kb TIMP-2 transcript is expressed at a greater level, in skeletal muscle the 1.0 kb transcript is enriched. Unlike the CNS, expression of both the 1.0 and 3.5 kb transcripts is down-regulated with muscle development. This down-regulation confirms a previous quantitative PCR analysis of TIMP-2 in muscle (Young et al., 2002). Although TIMP-2 mRNA expression decreases during muscle development, TIMP-2 protein expression is unchanged (Fig. 1D). Because the muscle samples included slow-twitch muscles (e.g., soleus), fast-twitch (e.g., extensor digitorum longus) and muscles containing both fiber types (e.g., gastrocnemius), it is not known whether TIMP-2 is differentially expressed in a particular muscle type.
Figure 1. Developmental regulation of TIMP-2 expression in murine CNS and skeletal muscle.
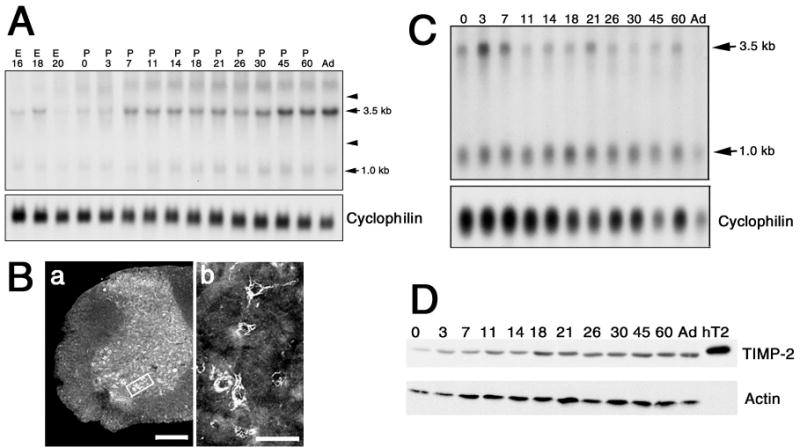
A) Northern blot analysis with 25 μg total RNA of CNS (brain and spinal cord) at the indicated embryonic (E) and postnatal (P) time points. The housekeeping gene cyclophilin was used to verify equal loading of RNA per lane. While the 1.0 kb TIMP-2 transcript is constitutively expressed, the 3.5 kb transcript increases throughout development. Arrows indicate the positions of 28S and 18S rRNA. B) Immunohistochemistry reveals that TIMP-2 is expressed throughout the adult spinal cord gray matter (a) and is expressed by ventral horn motor neurons (b, area indicated by box in a). Scale bar = 250 μm (a), 50 μm (b). C) Northern blot analysis with 2.5 μg poly A+ RNA from lower extremity muscles at the indicated postnatal ages. TIMP-2 RNA expression decreases throughout development. D) Western blot analysis (25 μg) of crude protein homogenates from lower extremity muscles at the indicated postnatal ages. In contrast to TIMP-2 mRNA, TIMP-2 protein expression remains high after P21. Recombinant human TIMP-2 (hT2) was used as a positive control. Actin was used to verify equal loading of protein per lane.
Motor behavior is altered in TIMP-2 deficient mice
Inasmuch as TIMP-2 is expressed in both motor neurons and muscle, we sought to determine whether motor function was affected in TIMP-2 deficient mice. Motor function was assessed on a RotaRod (Dunham and Miya, 1957) using a fixed speed testing strategy (as described in Methods). Both male and female animals were tested. Since ANOVA showed no gender by genotype difference, data from male and female animals were analyzed together. Although postnatal day 90 (P90) TIMP-2 knockout mice are able to maintain motor activity on the rod comparable to wild-type and heterozygous littermate control animals when the rod rotated at 4 rpm, they fall off much faster (decreased latency to fall) at 20 and 40 rpm (Fig. 2A). In addition, knockout mice more quickly grip the rod and spin rather than attempting locomotion than controls (decreased latency to passive rotation). Latency to passive rotation represents the time at which the mouse makes one complete revolution while gripping the rod. Notice that in knockout mice the latency to passive rotation is similar to the latency to fall, indicating that once the animal passively spins it can no longer maintain motor activity on the rod and falls off. In contrast, heterozygous mice take longer before they passively rotate and even resume motor activity afterwards. TIMP-2 knock-down mice are maintained as homozygous recessive; thus, littermate controls are not available. Knock-down mice show decreased latency to fall, but only at 40 rpm. To determine whether the decreased latency observed at 40 rpm was a result of fatigue, individual trial data was analyzed (Fig. 2B). Wild-type and heterozygous mice quickly attain the maximal testing duration within three trials. At all trials, knockout mice fall off significantly faster than knock-down, heterozygous or wild-type mice. The decreased motor function of knockout mice is not likely due to fatigue since their duration on the rod increases, not decreases, with each trial. Whether this increase is due to motor learning or increased neurotransmission efficacy over time is yet to be determined.
Figure 2. TIMP-2−/− mice have altered locomotor function.

P90 wild-type, heterozygous and TIMP-2 knockout (KO) mice littermates, and TIMP-2 knockdown (KD) mice were tested at 4, 20 and 40 rpm on consecutive days on a motorized rotating rod. A) Knock-down mice fall off faster than heterozygous or wild-type mice only at 40 rpm. Knockout mice fall off faster at 20 and 40 rpm. In addition, knockout mice spend more time passively rotating. Latency to passive rotation is the time at which a mouse first makes a complete revolution while holding onto the rod rather than actively moving. 4 rpm: F3, 133 = 1.36, p = 0.257; 20 rpm: F3, 133 = 19.67, p = 0.000; 40 rpm: F3, 133 = 29.99, p = 0.000; passive F2, 72 = 13.53, p = 0.000 B) TIMP-2 knockout mice are different from the other three genotypes at all 40 rpm test trials. * p < 0.05 by t-test relative to wild-type.
To determine whether the motor dysfunction displayed by TIMP-2−/− mice was developmentally regulated, animals were tested at P21, P26, P30 and P45 (Fig. 3). Knockout mice could not be tested prior to P21. When P10 pups were placed on the rod they immediately fell off without attempting locomotion. From P10 to P21, knockout mice are extremely hyperexcitable and sensitive to touch. Attempts to touch the pup results in them jumping out of the cage. Mice are kept in 6 inch deep rat cages for this reason. This hyperexcitability diminishes as development proceeds. In addition, the knockout mice tremble when at rest and “hop/leap” when locomotion is initiated. Once locomotion commences, the mouse’s gait is splayed (discussed below). Even at P21, as soon as the paws of knockout mice touch the rod they often leap off, even at 4 rpm. These motor deficits are not visibly apparent in knock-down mice. At P21, the three genotypes do not show differences in latency to fall at any speed. However, knockout mice have significantly reduced latency to passive rotation and often spend the entire time on the rod passively rotating. As development proceeds, the knockout mice attempt locomotion and do not simply spin (latency to passive rotation increases). However, their ability to maintain motor function is diminished; therefore, there is a concomitant decrease in the latency to fall. By P45, knock-down mice show reduced time on the rod at 40 rpm and knockout mice at 20 and 40 rpm comparable to that observed in P90 animals.
Figure 3. Motor dysfunction in TIMP-2−/− mice is developmentally regulated.
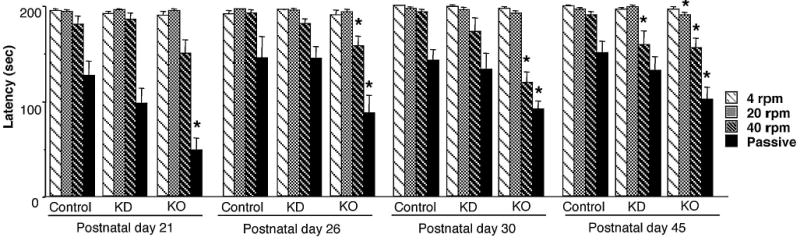
Early in development knockout mice spend most of the time on the RotaRod passively spinning. Thus, only passive rotation is different between the genotypes. However, as the knockout mice attempt to move on the rod, their time on the rod decreases. At P45, both knockout and knockdown mice differ from control mice at 40 rpm, similar to that observed at P90. P21: 4 rpm F2, 37, < 1.0; 20 rpm F2, 37 < 1.0; 40 rpm F2, 37 = 4.13, p = 0.024; passive F2, 23 = 11.30, p = 0.000. P26: 4 rpm F2, 38< 1.0; 20 rpm F2, 38< 1.0; 40 rpm F2, 38 = 4.29, p = 0.021; passive F2, 29 = 3.62, p = 0.040. P30: 4 rpm F2, 54 = 1.75, p = 0.184; 20 rpm F2, 54 = 2.10, p = 0.133; 40 rpm F2, 54 = 17.35, p = 0.000; passive F2, 43 = 6.21, p = 0.004. P45: 4 rpm F2, 59< 1.0; 20 rpm F2, 59 = 6.44, p = 0.003; 40 rpm F2, 59 = 3.86, p = 0.026; passive F2, 37 = 4.20, p = 0.023. * p < 0.05 by t-test.
Because the RotaRod is a complex motor task that involves coordination, balance and motor learning, additional tests were performed to assess individual aspects of motor function. Mice were tested at P21, the period of maximal dyskinesia, and P90 (adult), when RotaRod function was tested. When t-test showed that wild-type and heterozygous mice did not differ data was pooled as control.
Balance, as measured by the amount of time a mouse stays on a small stationary beam, is reduced in P21 knockout, but not knock-down mice (Fig. 4). Neither TIMP-2 genotype shows reduced balance as adults. Although total time on the beam was comparable between adult control and TIMP-2−/− mice, behavior while on the beam was strikingly different. The beam was suspended only 10 cm high because knockout mice show signs of increased anxiety (Jaworski et al., 2005). Control mice spent ~90% of their time walking on the beam while knockout mice froze in place and spent less than 10% of their time in locomotion. Nonetheless, adult TIMP-2−/−mice were able to maintain their balance.
Figure 4. Balance is reduced in juvenile TIMP-2 knockout mice.

Balance of both TIMP-2−/− genotypes is normal at P21 on the large balance beam (9 mm; F2, 28 = 0.33, p = 0.723). However, knockout mice had reduced time on the medium (7.5 mm; F2, 28 = 10.01, p = 0.000) and small (6 mm; F2, 28 = 3.52, p = 0.043) beams. Balance of adult TIMP-2−/− mice is normal on all three beam sizes (large: 18 mm; F2, 27 = 1.32, p = 0.283; medium: 10 mm; F2, 27 = 1.57, p = 0.226; small: 7.5 mm; F2, 27 = 1.89, p = 0.170). * p < 0.05 by t-test.
Motor neuron disease was assessed by testing hindlimb extension. Normally, an extension reflex of the hindlimb is observed when a mouse is suspended in the air by its tail (Fig. 5B, C). However, in mice with motor neuron disease, hindlimb retraction is more commonly observed (Fig. 5A). Both P21 and adult knockout mice show reduced hindlimb extension (Fig. 5D). At P21, knockout mice often swing their body to grab their tail with their fore and hindpaws rather than being suspended upside-down (Fig. 5A). Hindlimb extension is reduced in adult, but not P21, knock-down mice.
Strength was assessed using two tests. Grip strength was measured as the amount of time a mouse is able to remain suspended from a trapeze by its forearms. Muscle strength was assessed using the loaded grid test, which measures the amount of time a mouse is able to hold a weight with its forearms when suspended by the tail. Grip strength is not reduced in either TIMP-2−/− genotype at P21, but is reduced in both genotypes in adults (Fig. 6A). This result is not surprising based on RotaRod observations. At P21, TIMP-2−/− spent most of their time passively spinning while holding onto the RotaRod, suggesting grip strength was unaffected. When adult mice could no longer maintain locomotion they were unable to hold onto the RotaRod, suggesting weakness. However, muscle strength is unaltered in either P21 or adult TIMP-2−/− mice (Fig. 6B). The increased grip time for adult TIMP-2−/− mice is deceiving because they tended to grip the weight with both their fore and hindpaws and; thus, were able to hold the weight longer.
Figure 6. Strength is altered in adult TIMP-2−/− mice.
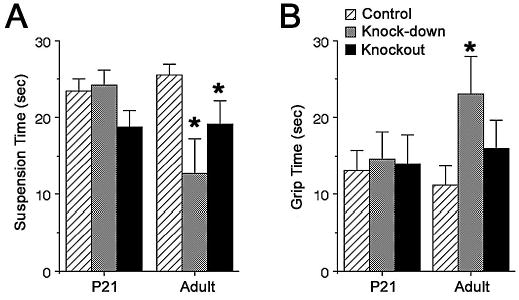
Forearm grip strength on the trapeze (A, F2, 28 = 1.98, p = 0.157) and muscle strength on the loaded grid test (B, F2, 28 = 0.06, p = 0.939) are normal in TIMP-2−/− mice at P21. Forearm grip strength is reduced in adult TIMP-2−/− mice (F2, 26 = 3.58, p = 0.042), while muscle strength is increased in adult knock-down mice (F2, 24 = 2.08, p = 0.017). * p < 0.05 by t-test.
Altered motor activity in knockout mice was suspected based on observing their walking pattern as juveniles (P14 – P21). To quantitate these observations the stride length, gait width, and intrastep distance between right and left paw prints were measured (Fig. 7A). Stride length is increased in P21, but not adult knockout mice (Fig. 7B). Even heterozygous mice show increased stride length at P21. Stride length in knock-down mice is unaltered. The most significant difference in walking pattern is gait width, which is increased in both TIMP-2−/− genotypes of P21 and adult mice (Fig. 7C). Gait width is also increased in P21 heterozygous mice. Like stride length, intrastep distance is increased only in P21 knockout mice (Fig. 7D).
TIMP-2 knockout mice display histological alterations
To determine whether the observed motor dysfunction was associated with histological defects in the peripheral and/or central nervous system, the NMJ and cerebellum were analyzed immunohistochemically. Since TIMP-2 knock-down mice did not show significantly decreased motor function they were not analyzed. Motor neuron morphology was visualized with an antibody to neurofilament-145 while the distribution of acetylcholine receptors (AChRs) was examined using rhodamine-conjugated α-bungarotoxin (α-Btx) (Fig. 8A). NMJ organization is strikingly different in knockout mice. At P7 and P14, wild-type mice possess a discrete endplate band consisting of well aligned AChR clusters. In contrast, the endplate region of knockout mice is much broader with increased neuronal branching. At birth, all endplates are polyneuronally innervated, but over the first two postnatal weeks all but one of the nerve terminals are eliminated from the NMJ (Redfern, 1970; Brown et al., 1976; Balice-Gordon and Lichtman, 1993). At P14, the endplates of both wild-type and knockout mice are appropriately monoinnervated suggesting that the timing of synapse elimination is unaffected by the absence of TIMP-2.
Figure 8. Neuromuscular junction organization in TIMP-2 knockout mice is altered.

A) Immunohistochemistry with α-Btx (red) and neurofilament-145 (green) reveals the organization of the neuromuscular junction in the soleus muscle of P7 and P14 wild-type (WT) and TIMP-2 knockout (KO) mice. Both prior to (a, c) and subsequent to (b, d) the period of synapse elimination the endplate region is more diffuse in knockout mice. Examination at higher magnification shows that the endplates in both knockout and wild-types muscles are innervated by a single nerve terminal. Scale bar = 100 μm, 50 μm for inset. B) α-Btx labeling was used to quantitate endplate area (in μm2) in at least 40 endplates of three adult wild-type (a-c) and knockout (d-f) spinotrapezius muscle. The area of AChR distribution is larger in TIMP-2 knockout muscles (p < 0.05). Scale bar = 25 μm. C) Cerebellar histogenesis is altered in TIMP-2 knockout mice. Cresyl violet stained sections of P7 cerebellar cortex demonstrates the dramatic reduction in cerebellar size in knockout mice (b) relative to wild-type littermates (a). The reduced size is due to a reduction in Purkinje cell processes, as revealed by neurofilament immunolabeling (c, d), and in the number of Purkinje cells, as revealed by calbindin immunolabeling (e, f). The knockout cerebellar cortex remains reduced in size at P14 (g, h). NeuN, a neuron-specific nuclear protein, reveals a reduction of granule cells in knockout mice (i, j). Similar to P7, calbindin labeling of Purkinje cells remains reduced in P14 knockout mice (k, l). MOL - molecular, PcL - Purkinje cell, GL - granule cell layers; WM - white mater. Scale bar = 25 μm, Purkinje cell inset = 37.5 μm.
In addition to synapse elimination, the postnatal endplate cytoarchitecture matures as a simple plaque of AChRs change into a pretzel-like array of branches (Sanes and Lichtman, 2001). Individual endplates were more closely examined to determine whether AChR distribution was altered in TIMP-2 knockout mice. The endplates of adult TIMP-2 knockout muscle are significantly larger than wild-type littermates (Fig. 8B). In addition to being enlarged, the endplates of TIMP-2 knockout muscles are much more complexly convoluted. Therefore, the actual area of AChR expression is likely underrepresented by simply measuring circumferential area.
Since the RotaRod is a complex motor task that tests not only muscle function but balance, and balance was reduced in P21 knockout mice, we asked whether the motor deficit could be due to alterations in cerebellar cytoarchitecture. The P7 knockout cerebellar cortex is thinner than wild-type mice as revealed by Nissl stain (Fig. 8Ca,b). This reduction in size could result from decreased cell number, smaller cell size, or decreased process outgrowth. Neurofilament staining demonstrates that both the length and complexity of Purkinje cell processes are reduced in knockout mice (Fig. 8Cc,d). Immunoreactivity for calbindin, a Purkinje cell-specific calcium binding protein, is reduced, suggesting decreased Purkinje cell genesis in knockout mice (Fig. 8Ce, f). In addition, ectopic calbindin immunolabeling is detected within the internal granule cell layer in knockout mice (Fig. 8Cf). These histologic alterations persist at P14. While the overall thickness of the cerebellar cortex of knockout mice is reduced, the external granule cell layer is increased, suggesting delayed granule cell genesis and/or migration (Fig. 8Cg, h). Immunolabeling with the neuron-specific nuclear protein NeuN is reduced in knockout mice, suggesting reduced granule cell number (Fig. 8Ci, j). As observed at P7, calbindin immunoreactivity is reduced at P14 (Fig. 8Ck,l). The histologic alterations observed at P7 and P14 are no longer apparent at P21 (data not shown). Taken together these data suggest that cerebellar corticogenesis is affected in TIMP-2 knockout mice and indicates that the increased NMJ nerve branching is a specific phenotype not present in all neuronal types. Whether the TIMP-2 null motor phenotype is due to the neuromuscular and/or cerebellar defects warrants further investigation.
Discussion
Here, we report a motor phenotype in TIMP-2 knockout mice. Grossly, the knockout mice have a dyskinesia, including trembling prior to locomotion, excessive jumping upon moving, and a splayed, shortened gait. It is not surprising that this phenotype was not previously detected because it is developmentally regulated. The hyperkinesia is most pronounced during the second to third postnatal week. By P30 knockout mice can no longer be visually distinguished from littermate controls. However, behavioral testing (e.g., RotaRod, hindlimb extension, grip strength) demonstrated that knockout mice indeed have altered motor function even as adults. It is recognized that normal rodent neonates have a tremor that is most apparent during the first few postnatal days and gradually subsides over the next week (Pranzatelli, 1990). Therefore, we are cautious not to over interpret this tremor as a NMJ dysfunction. However, the tremor in TIMP-2 knockout mice is not most pronounced in the first postnatal week, but from ~P14 - P21 and sufficiently obvious that knockout mice can be distinguished from their wild-type littermates. Furthermore, behavioral and histological alterations, which may underlie the motor phenotype, have been detected in knockout mice.
The basis for the TIMP-2 null phenotype is likely complex and involves central (e.g., cortical, cerebellar, bulbar) and peripheral (e.g., NMJ) deficits. The most striking histological phenotype detected is an alteration in nerve branching. Interestingly, this phenotype displays tissue specificity - cerebellar reduction and NMJ augmentation. This should not be unexpected since the repertoire of MMPs and ECM substrates are likely different in the central and peripheral nervous systems. Similar to the cerebellar cortex, nerurite length is reduced in the neonatal cerebral cortex (Pérez-Martínez and Jaworski, 2005). Reduced neurite length is no longer apparent in either cortice at P21, suggesting that either MMP-mediated proteolysis is diminished after neuronal migration and synaptogenesis or other TIMPs compensate for the loss of TIMP-2. Increased intramuscular nerve branching is observed in mice deficient for agrin, rapsyn, MuSK (Gautam et al., 1999), choline acetyltransferase (Brandon et al., 2003) and mice deficient in ß1 integrin within muscle (Schwander et al., 2004). Since paralysis promotes sprouting (reviewed in Tam and Gordon, 2003), the increased branching in these mice may be a response to reduced or absent synaptic transmission. In contrast to these mice, TIMP-2 knockout mice are not paralyzed. Cell adhesion molecules, cytokines, and growth factors also regulate axonal sprouting (reviewed in English, 2003). Both agrin and ß2-laminin arrest neurite outgrowth and promote motor nerve terminal differentiation. Inasmuch as agrin (VanSaun and Werle, 2000; Werle and VanSaun, 2003) and laminin (d’Ortho et al., 1997) are MMP substrates, their proteolysis could produce uncontrolled sprouting upon contact with the muscle. Interestingly, the knock-down mice displayed no visible motor deficits and only reduced latency at 40 rpm, suggesting that the mutant TIMP-2 protein present in these mice provides sufficient MMP inhibition required for adequate NMJ maturation. The TIMP-2 null phenotype described here is also similar to that described for the over-expression of glial cell line-derived neurotrophic factor (GDNF) (Nguyen et al., 1998). In addition to tremor, mice injected with GDNF as neonates (Keller-Peck et al., 2001) have tail kinks reminiscent of TIMP-2 knockout mice. The kinks in TIMP-2 knockout mice can be so pronounced they curl into a “pigtail”. Although the tail kinks may be a consequence of excess collagenolysis, it is also plausible that the kinks are derived from unbalanced muscle tone because a greater number of muscle fibers on one side of the tail are activated.
Although both MMPs and TIMPs are expressed in muscle, little is known about their role in skeletal muscle development and function (Carmeli et al., 2004). Proteases have been suggested as mediators of NMJ synapse elimination ( O’Brien et al., 1984; Champaneria et al., 1992). In one model, postsynaptic cells indiscriminately release proteases, but presynaptic activity causes release of protease inhibitors that protects the most active nerve terminals from removal (Liu et al., 1994; Liu et al., 1994). In support of this hypothesis, treatment of muscles with the protease inhibitors leupeptin (Connold et al., 1986) or hirudin (Zoubine et al., 1996) results in a delay of synapse elimination. One would predict that mice would display hastened synapse elimination in the absence of TIMP-2. However, the juvenile TIMP-2 null phenotype appears in contradiction to this hypothesis. Instead, the hyperkinesia of knockout mice is more suggestive of the persistence of polyneuronal innervation. Yet, P14 knockout NMJs appear appropriately monoinnervated. Whether motor unit size and/or electrophysiological properties of TIMP-2 knockout muscle are altered is yet to be determined. Once fully differentiated, MMPs/TIMPs play a role in the maintenance of basal lamina homeostasis during muscle activity. MMP expression is up-regulated by muscle contraction (Ahtikoski et al., 2003), electrical stimulation (Haas et al., 2000) or vasodilation (Van Gieson and Skalak, 2001). MMP activity is likely up-regulated in the absence of TIMP-2. Because slow-twitch fibers contain more collagen (Zimmerman et al., 1993), they should be less susceptible to the damaging MMP effects. Inasmuch as high intensity exercise up-regulates MMP-2 activity in skeletal muscles containing a high percentage of fast-twitch fibers (Carmeli et al., 2005), the knockout mice probably could not maintain locomotion on the RotaRod at 40 rpm due to structural destabilization of fast-twitch muscle. It will be interesting to determine whether fast-twitch muscle shows greater cytoarchitectural alterations than slow-twitch muscle.
MMPs are thought to play a role in axonal growth (Vaillant et al., 1999; Hayashita-Kinoh et al., 2001; Vaillant et al., 2003) and neuronal migration (Vaillant et al., 1999; Vaillant et al., 2003; Ayoub et al., 2005) during cerebellar neurogenesis. To date, only one study has examined TIMP expression within the cerebellum (Vaillant et al., 1999). This study suggested that TIMP-1 participates in the development of olivocerebellar connectivity, while TIMP-3 plays a role in cerebellar plasticity by facilitating Purkinje cell arborization and synaptogenesis. The spatial and temporal expression pattern of TIMP-2 suggested a role in granule cell proliferation and migration. Our observations of reduced granule cell labeling with NeuN and increased external granule cell layer thickness support this hypothesis. Given that TIMP-2 is expressed on Purkinje cell somata (Vaillant et al., 1999), it also likely plays a role in Purkinje cell differentiation. Although TIMP-3 was the only TIMP to label Purkinje cell dendrites (Vaillant et al., 1999), it is clear that neurite outgrowth is reduced in the absence of TIMP-2, suggesting it may also play a role in Purkinje cell dendritic arborization.
Further studies are required to elucidate the molecular basis for the altered nerve branching in TIMP-2 knockout mice. However, what is clear is the need to examine genetically modified mice at all developmental stages for the presence of a phenotype. Moreover, mice that were previously reported to lack a phenotype should also be examined for behavioral and/or histological alterations that may not be readily apparent.
Acknowledgments
This work was supported by NIH Grant NS35874, Grant NS045225 co-funded by NINDS and NCRR, American Heart Association Grant 9950039N and UVM College of Medicine (DMJ) and NIH Grant P20 RR16435 from the COBRE Program of the National Center for Research Resources (WAF). We thank Karl Zurn of Med Associates (Georgia, VT) for the use of the RotaRod, Gentian Lluri for muscle immunohistochemistry and Jason Boone for measuring endplate size.
References
- Ahtikoski AM, Koskinen SO, Virtanen P, Kovanen V, Risteli J, Takala TE. Synthesis and degradation of type IV collagen in rat skeletal muscle during immobilization in shortened and lengthened positions. Acta Physiol Scand. 2003;177:473–481. doi: 10.1046/j.1365-201X.2003.01061.x. [DOI] [PubMed] [Google Scholar]
- Ayoub AE, Cai TQ, Kaplan RA, Luo J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J Comp Neurol. 2005;481:403–415. doi: 10.1002/cne.20375. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during synapse elimination at developing neuromuscular junctions in living mice. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. 2003;23:539–549. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Jansen JKS, Van Essen D. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J Physiol (Lond) 1976;261:387–422. doi: 10.1113/jphysiol.1976.sp011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli E, Moas M, Lennon S, Powers SK. 2005. High intensity exercise increases expression of matrix metalloproteinases in fast skeletal muscle fibers. Exp Phsyiol E-pub. [DOI] [PubMed]
- Carmeli E, Moas M, Reznick AZ, Coleman R. Matrix metalloproteinases and skeletal muscle: a brief review. Muscle Nerve. 2004;29:191–197. doi: 10.1002/mus.10529. [DOI] [PubMed] [Google Scholar]
- Caterina JJ, Yamada S, Caterina NCM, Longenecker G, Holmbäck K, Shi J, Yermovsky AE, Engler JA, Birkedal-Hansen H. Inactivating mutation of the mouse Tissue Inhibitor of Metalloproteinases-2 (Timp-2) gene alters proMMP-2 activation. J Biol Chem. 2000;275:26416–26422. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- Champaneria S, Swenarchuk LE, Anderson MJ. Increases in pericellular proteolysis at developing neuromuscular junctions in culture. Dev Biol. 1992;149:261–277. doi: 10.1016/0012-1606(92)90283-m. [DOI] [PubMed] [Google Scholar]
- Connold AL, Evers JV, Vrbova G. Effect of low calcium and protease inhibitors on synapse elimination during postnatal development in the rat soleus muscle. Brain Res. 1986;393:99–107. doi: 10.1016/0165-3806(86)90069-6. [DOI] [PubMed] [Google Scholar]
- d’Ortho MP, Will H, Atkinson S, Butler G, Messent A, Gavrilovic J, Smith B, Timpl R, Zardi L, Murphy G. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur J Biochem. 1997;250:751–757. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- de Medinaceli L, Freed WJ, Wyatt RJ. An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol. 1982;77:634–643. doi: 10.1016/0014-4886(82)90234-5. [DOI] [PubMed] [Google Scholar]
- Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharmacol Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- English AW. Cytokines, growth factors and sprouting at the neuromuscular junction. J Neurocytol. 2003;32:943–960. doi: 10.1023/B:NEUR.0000020634.59639.cf. [DOI] [PubMed] [Google Scholar]
- Fager N, Jaworski DM. Differential spatial distribution and temporal regulation of tissue inhibitor of metalloproteinase mRNA expression during rat central nervous system development. Mech Dev. 2000;98:105–109. doi: 10.1016/s0925-4773(00)00437-8. [DOI] [PubMed] [Google Scholar]
- Gautam M, DeChiara TM, Glass DJ, Yancopoulos GD, Sanes JR. Distinct phenotypes of mutant mice lacking agrin, MuSK, or rapsyn. Dev Brain Res. 1999;114:171–178. doi: 10.1016/s0165-3806(99)00013-9. [DOI] [PubMed] [Google Scholar]
- Haas TL, Milkiewicz M, Davis SJ, Zhou AL, Egginton S, Brown MD, Madri JA, Hudlicka O. Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2000;279:H1540–1547. doi: 10.1152/ajpheart.2000.279.4.H1540. [DOI] [PubMed] [Google Scholar]
- Hammani K, Blakis A, Morsette D, Bowcock AM, Schmutte C, Henriet P, DeClerck YA. Structure and characterization of the human tissue inhibitor of metalloproteinase-2 gene. J Biol Chem. 1996;271:25498–25505. doi: 10.1074/jbc.271.41.25498. [DOI] [PubMed] [Google Scholar]
- Hayashita-Kinoh H, HKinoh H, Okada A, Komori K, Yoshifumi I, Chiba T, Kajita M, Yana I, Seiki M. Membrane-type 5 matrix metalloproteinase is expressed in differentiated neurons and regulates axonal growth. Cell Growth Differ. 2001;12:573–580. [PubMed] [Google Scholar]
- Jaworski DM, Boone J, Caterina J, Soloway P, Falls WA. 2005. Prepulse inhibition and fear-potentiated startle are altered in Tissue Inhibitor of Metalloproteinase-2 (TIMP-2) knockout mice. Brain Res in press. [DOI] [PubMed]
- Jaworski DM, Fager N. Regulation of tissue inhibitor of metalloproteinase-3 (TIMP-3) mRNA expression during rat CNS development. J Neurosci Res. 2000;61:396–408. doi: 10.1002/1097-4547(20000815)61:4<396::AID-JNR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jaworski DM, Kelly GM, Hockfield S. BEHAB, a new member of the proteoglycan tandem repeat family of hyaluronan-binding proteins that is restricted to brain. J Cell Biol. 1994;125:495–509. doi: 10.1083/jcb.125.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski DM, Kelly GM, Hockfield S. Intracranial injury acutely induces the expression of the secreted isoform of the CNS-specific hyaluronan-binding protein BEHAB/brevican. Exp Neurol. 1999;157:327–337. doi: 10.1006/exnr.1999.7062. [DOI] [PubMed] [Google Scholar]
- Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD. Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci. 2001;21:6136–6146. doi: 10.1523/JNEUROSCI.21-16-06136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir D, Battenberg E, Kiel M, Bloom FE, Milner RJ. The brain-specific gene 1B236 is expressed postnatally in the developing rat brain. J Neurosci. 1986;6:522–530. doi: 10.1523/JNEUROSCI.06-02-00522.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fields RD, Festoff BW, Nelson PG. Proteolytic action of thrombin is required for electrical activity-dependent synapse reduction. Proc Natl Acad Sci. 1994;91:10300–10304. doi: 10.1073/pnas.91.22.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fields RD, Fitzgerald S, Festoff BW, Nelson PG. Proteolytic activity, synapse elimination, and the Hebb synapse. J Neurobiol. 1994;25:325–335. doi: 10.1002/neu.480250312. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- Nagase H. Activation mechanisms of matrix metalloproteinases. BiolChem. 1997;378:151–160. [PubMed] [Google Scholar]
- Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Lett. 2004;563:129–134. doi: 10.1016/S0014-5793(04)00281-9. [DOI] [PubMed] [Google Scholar]
- O’Brien RAD, Ostberg AJC, Vrbova G. Protease inhibitors reduce the loss of nerve terminals induced by activity and calcium in developing rat soleus muscles in vitro. Neuroscience. 1984;12:637–646. doi: 10.1016/0306-4522(84)90079-4. [DOI] [PubMed] [Google Scholar]
- Pérez-Martínez L, Jaworski DM. Tissue Inhibitor of Metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J Neurosci. 2005;25:4917–4929. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranzatelli MR. Antimyoclonic effect of MK-801: a possible role for NMDA receptors in developmental myoclonus of the neonatal rat. Clin Neuropharmacol. 1990;13:329–338. [PubMed] [Google Scholar]
- Ratnikov BI, Rozanov DV, Postnova TI, Baciu PG, Zhang H, DiScipio RG, Chestukhina GG, Smith JW, Deryugina EI, Strongin AY. An alternative processing of integrin alpha(v) subunit in tumor cells by membrane type-1 matrix metalloproteinase. J Biol Chem. 2002;277:7377–7385. doi: 10.1074/jbc.M109580200. [DOI] [PubMed] [Google Scholar]
- Redfern PA. Neuromuscular transmission in newborn rats. J Physiol (Lond) 1970;209:701–709. doi: 10.1113/jphysiol.1970.sp009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Schwander M, Shirasaki R, Pfaff SL, Muller U. β1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J Neurosci. 2004;24:8181–8191. doi: 10.1523/JNEUROSCI.1345-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995;14:125–132. doi: 10.1016/0896-6273(95)90246-5. [DOI] [PubMed] [Google Scholar]
- Tam SL, Gordon T. Mechanisms controlling axonal sprouting at the neuromuscular junction. J Neurocytol. 2003;32:961–974. doi: 10.1023/B:NEUR.0000020635.41233.0f. [DOI] [PubMed] [Google Scholar]
- Vaillant C, Didier-Bazès M, Hutter A, Belin M-F, Thomasset N. Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J Neurosci. 1999;19:4994–5004. doi: 10.1523/JNEUROSCI.19-12-04994.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant C, Meissirel C, Mutin M, Belin MF, Lund LR, Thomasset N. MMP-9 deficiency affects axonal outgrowth, migration, and apoptosis in the developing cerebellum. Mol Cell Neurosci. 2003;24:395–408. doi: 10.1016/s1044-7431(03)00196-9. [DOI] [PubMed] [Google Scholar]
- Van Gieson EJ, Skalak TC. Chronic vasodilation induces matrix metalloproteinase 9 (MMP-9) expression during microvascular remodeling in rat skeletal muscle. Microcirculation. 2001;8:25–31. [PubMed] [Google Scholar]
- VanSaun M, Werle MJ. Matrix metalloproteinase-3 removes agrin from synaptic basal lamina. J Neurobiol. 2000;43:140–149. [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275:26411–26415. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle MJ, VanSaun M. Activity dependent removal of agrin from synaptic basal lamina by matrix metalloproteinase 3. J Neurocytol. 2003;32:905–913. doi: 10.1023/B:NEUR.0000020631.69804.f5. [DOI] [PubMed] [Google Scholar]
- Yamada H, Saito F, Fukuta-Ohi H, Zhong D, Hase A, Arai K, Okuyama A, Maekawa R, Shimizu T, Matsumura K. Processing of β-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum Mol Genet. 2001;15:1563–1569. doi: 10.1093/hmg/10.15.1563. [DOI] [PubMed] [Google Scholar]
- Young DA, Phillips BW, Lundy C, Nuttall RK, Hogan A, Schultz GA, Leco KJ, Clark IM, Edwards DR. Identification of an initiator-like element essential for the expression of the tissue inhibitor of metalloproteinases-4 (Timp-4) gene. Biochem J. 2002;364:89–99. doi: 10.1042/bj3640089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman SD, McCormick RJ, Vadlamudi RK, Thomas DP. Age and training alter collagen characteristics in fast- and slow-twitch rat limb muscle. J Appl Physiol. 1993;75:1670–1674. doi: 10.1152/jappl.1993.75.4.1670. [DOI] [PubMed] [Google Scholar]
- Zoubine MN, Ma JY, Smirnova IV, Citron BA, Festoff BW. A molecular mechanism for synapse elimination: novel inhibition of locally generated thrombin delays synapse loss in neonatal mouse muscle. Dev Biol. 1996;179:447–457. doi: 10.1006/dbio.1996.0274. [DOI] [PubMed] [Google Scholar]