

Abstract
Incorrect folding of proteins in living cells may lead to malfunctioning of the cell machinery. To prevent such cellular disasters from happening, all cells contain molecular chaperones that assist nonnative proteins in folding into the correct native structure. One of the most studied chaperone complexes is the GroEL-GroES complex. The GroEL part has a “double-barrel” structure, which consists of two cylindrical chambers joined at the bottom in a symmetrical fashion. The hydrophobic rim of one of the GroEL chambers captures nonnative proteins. The GroES part acts as a lid that temporarily closes the filled chamber during the folding process. Several capture-folding-release cycles are required before the nonnative protein reaches its native state. Here we report molecular simulations that suggest that translocation of the nonnative protein through the equatorial plane of the complex boosts the efficiency of the chaperonin action. If the target protein is correctly folded after translocation, it is released. However, if it is still nonnative, it is likely to remain trapped in the second chamber, which then closes to start a reverse translocation process. This shuttling back and forth continues until the protein is correctly folded. Our model provides a natural explanation for the prevalence of double-barreled chaperonins. Moreover, we argue that internal folding is both more efficient and safer than a scenario where partially refolded proteins escape from the complex before being recaptured.
INTRODUCTION
Partially folded proteins are dangerous substances to have in a cell. Not only do they fail to perform their biological function, but they also tend to interact with other biomolecules in ways that disrupt the normal activity of the cell. For this reason, both prokaryotic and eukaryotic cells have developed “chaperone” protein complexes that capture and refold partially folded proteins, thereby preventing them from indulging in cellular mischief (1–3).
An important class of chaperone complexes is the cage chaperones or “chaperonins”. They can trap a partially folded protein in their cavity and perform a number of folding cycles until the native state of the protein is reached. Interestingly, a single chaperonin complex is able to assist the folding of a variety of proteins with quite different amino acid sequences. Hence, the chaperonin is able to distinguish partly folded states from the native state, independent of the amino acid sequence. Sigler et al. (4) proposed the following scenario for the action of the GroEL/GroES chaperonin complex: initially, the GroEL is in an “open-barrel” state, exposing a hydrophobic inner surface to which the target proteins can bind. The next step is an ATP-dependent capping of the GroEL barrel by the GroES cap. The nonnative protein now becomes trapped inside the folding cavity, the inner surface of which is mainly hydrophilic. The protein is allowed to fold during the time it takes the ATP to hydrolyze. Upon binding of ATP to the trans ring, the cap is released. At this stage, the correctly folded protein is thought to be released, since it no longer has properties that allow it to bind to the hydrophobic inner rim of the GroEL ring.
As mentioned above, the GroEL complex has a symmetric double-barrel structure, and after the first barrel (cis ring) releases its protein, the other barrel (trans ring) is ready to receive the next partially folded protein. The mechanism by which the chaperonin assists a partially folded protein to reach its native state has been the focus of much experimental and theoretical research (5,6). Vaart et al. (7) performed detailed molecular simulations and observed that unfolding of Rhodanese upon binding is induced by a conformational change in the apical domain of GroEL. Jewett et al. (8) proposed a model for the chaperonin cavity whereby the internal wall of the folding cage is designed to have a weak attractive interaction with the hydrophobic residues of the protein. The adsorption on the inner surface lowers the barrier to go from a folding intermediate state to the native state.
In the existing models, the fact that the GroEL-GroES complex has a double-barrel structure has no special significance. Here we report molecular simulations that lead us to propose an alternative scenario for the GroEL-GroES complex, namely that the central step in the folding process is a translocation of partially folded proteins from one barrel of the GroEL complex to the other. Such a translocation may be possible because there is a protein-coded channel that connects the two barrels of the chaperonin complex. Crystallographic studies indicate that the chaperonin complex has a well-defined structure except for a fairly large (∼30-Å) “hole” between the two barrels (4). However, the hole in the x-ray structure does not correspond to a region free of amino acids. Rather, it indicates that the amino acids in this region are disordered and highly mobile. Low-resolution small-angle neutron scattering experiments (9) and cryoelectron microscopy (10,11) suggest that there is, in fact, a high density of disordered amino acids near the central hole. Yet this does not preclude translocation through this region, as it is well known (12) that disordered protein filaments near a translocation pore may enhance the selectivity of the translocation process.
Intrachaperonin translocation should be both safer and more efficient than a scenario where partially folded proteins are released from the complex and then recaptured. The enhanced safety is obvious: as long as a partially folded protein is trapped inside the complex it cannot create havoc in the cell. The enhanced efficiency is related to the fact that the probability that the other barrel of the GroEL complex captures a released protein is only ∼30% (13). Of course, the partially folded protein may be captured by another GroEL complex, but on its way there, the partially folded protein is unchaperoned and therefore dangerous. Note that translocation can work both ways. Hence, a protein that partially unfolds during translocation need not complete the translocation to the other cavity. An alternative is that a folding nucleus forms in the original cavity and the partially translocated protein is reeled back into the original cavity to form a more stable (possibly native) conformation.
Finally, proteins that are close to the native state at the beginning of a cycle can fold into the native state without ever leaving the folding cavity. The presence of the translocation channel strongly reduces the probability that nonnative proteins will escape into the environment. Below, we describe molecular simulations that show how the confinement of a (nonnative) protein in a small hydrophilic cage is enough to induce rapid translocation to the other (open) barrel. The translocation process will break any preexisting compact structures. As the translocation proceeds, the protein binds to the open barrel. This folding provides the thermodynamic driving force for translocation. After translocation, proteins not yet in their native state will bind to the surface of the open barrel, which is predominantly hydrophobic. There are examples where translocation facilitates folding. For instance, using a model similar to ours, Morrissey et al. (14) showed that a protein that is extruded gradually from the ribosome folds faster than it does from its fully denatured state.
MODEL
Modeling of the GroEL complex is facilitated by the fact that its action is not sequence-specific (5,8). This suggests that the action of this chaperone can be represented by a simple model, provided that the model can describe protein folding and the interaction of polypeptides with heterogeneous protein surfaces. We consider a lattice protein and a model chaperonin in a cubic simulation box with hard walls. The lateral size of the simulation box is three times the contour length of the protein (64 amino acids). In the middle of the simulation box we placed a cubic chaperonin cage (Fig.1) of 5 × 5 × 5 lattice units. It is connected by a hole (3 × 3 cross shape) to the open barrel, modeled as a 2 × 5 × 5 box surrounded by a repulsive outer layer 3 × 7 × 7 to avoid binding on the outside of the rim. The volume of this cage is ∼1.95 times larger than the volume of protein in a compact (but not necessarily native) state. This would correspond to the ratio between the volume of a typical globular protein with a radius of gyration of ∼27.5 Å, and the volume of the closed chamber in the GroEL-GroES complex (∼170 Å3).
FIGURE 1.

Lattice model for GroEL-GroES complex. (a) The closed GroEL-GroES compartment is modeled as a cage (red) connected by a hole (blue) to the open barrel, modeled as a box surrounded by a repulsive outer layer to avoid binding on the outside of the rim. The attractive internal lateral surface was represented by the strongly attractive and hydrophobic amino acid Phe (average pair interaction with the other amino acids, API = – 0.23 kT (see Eq. 6)), whereas a repulsive and hydrophilic back surface was made of Arg (API = 0.38 kT). The absolute system size is not a crucial physical parameter; in fact, what really matters is the ratio between the accessible volume inside the cage and the volume of the protein. For our model this value is ∼1.95, which is typical for the values found in experiments. (b) Space-filling representation of the x-ray structure of the GroEL-GroES-ADP complex (4). Colors represent the type of surface: all hydrophobic amino acids (A, V, L, I, M, F, P, and Y) are in yellow, and the polar ones (S, T, H, C, N, Q, K, R, D, and E) are in red. (c) Intermediate conformation during the extrusion process from the hydrophilic cage. If the inner surface of the closed GroEL-GroES compartment was made of the mildly hydrophobic Tyr (API = −0.16 kT), extrusion did not take place.
We model the protein as a linear heteropolymer living on a lattice. The amino acids of the chain have nearest-neighbor interactions. The conformational energy E of the system is given by the following expression:
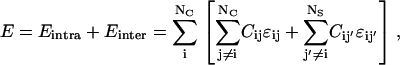 |
(1) |
where Eintra is the total interaction among the amino acids in the protein and Einter is the binding energy between the protein and the walls of the cage. The indices i and j run over the residues of the protein, whereas j′ runs over the elements of the cage, C is the contact matrix, defined as
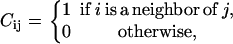 |
(2) |
and ɛ is the interaction matrix. For ɛ, we use the 20 × 20 matrix derived from the method of Betancourt and Thirumalai (15) from the matrix ɛ′ determined by Miyazawa and Jernigan (16) on the basis of the observed frequency of contacts between each pair of amino acids. The matrix ɛ′ has some inconsistency in reproducing the hydrophobic and hydrophilic nature of the amino acids because it is not straightforward to estimate the effective number of interactions between water molecules and the residues of a real protein in the native state. Betancourt and Thirumalai proposed rescaling all the values in the matrix with respect to the interaction with the amino acid Thr in the following way:
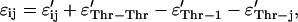 |
where ɛ′ is the interaction matrix calculated by Miyazawa and Jernigan (16). The choice of Thr is justified because it gives the best correlation between experimental hydrophobicities and the self-interaction term ɛThr–Thr/2. Although these interaction energies are, strictly speaking, neither energies nor free energies, they do provide a reasonable representation of the heterogeneity in the interactions between different amino acids.
The chaperonin cage is modeled as a rigid object and hence Eq. 1 does not include the interactions between the amino acids that form the cage.
RESULTS
To explore the relative importance of translocation and intrachamber folding, we performed molecular simulations on a lattice model for the GroEL-GroES complex (Fig. 1). The building blocks of both the polypeptide and the chaperone cage are amino acids with interaction parameters, as described previously (15,16). Although this lattice model provides only a crude representation of a protein in a chaperone cage, it retains important characteristics, such as the amino acid heterogeneity and the folding ability of the amino acid chain. In our simulations, we used a nonnative conformation of a 64-residue lattice protein (referred to as P-64) in one barrel of the GroEL complex. The P-64 sequence was designed (17–19) to have a well-defined native state (see Appendix). The absolute size of the GroEL system was not considered as an essential physical parameter. However, what does matter is the ratio between the accessible volume inside the cage and the volume of the protein. For our model, this value is ∼1.95, which is typical for values that have been determined experimentally. In the simulations, we computed the free-energy barrier that the protein must overcome to perform the successive steps in the chaperonin-assisted protein-folding process (Fig. 2). In the initial configuration (1), the chaperonin barrel is open and exposes a hydrophobic rim for binding partially folded proteins. Using free-energy calculations (20), we verified that the rim of the open barrel binds intermediate conformations strongly, but not the protein in its native state (Fig. 3). After the nonnative protein is captured, the GroEL-GroES complex closes (i.e., the barrel gets capped) and we move to the actual translocation process (Fig. 2, 2 and 3). The extrusion takes place through the flexible region in the bottom of the hydrophilic cage (see Fig. 1 a). To model a hydrophilic cage, the walls of the cavity were coated with Arg. The free energy was then computed as a function of the degree of translocation and the degree of folding. The results demonstrate that the most favorable state is one where the chain has folded outside of the folding cage (Fig. 4). Initially, the chain is stretched across the hole, but as soon as a sufficient number of amino acids are outside the cage, they fold into a compact structure. This process takes place in the trans chamber, where the folding free energy (Fig. 3) shows a clear minimum corresponding to the protein in the native state. The early stages of extrusion cost free energy, as the protein must locally unfold to initiate the extrusion. This implies that the hydrophilic cage will preferentially expel nonnative conformations. The gain in free energy as a result of folding facilitates the extrusion process. Interestingly, the free-energy barrier for extrusion for the native state of P-64 (∼10 KBT) is considerably larger than for a partially folded state (∼4 KBT). However, partial extrusion can still help the folding process: Fig. 5 shows that the folding efficiency is higher in cages where translocation is allowed than in those where it is inhibited. The number of complete translocations decreases as the initial configuration becomes more similar to the native state, as a consequence of the increase in the free energy barrier. In our simulations, translocation of highly nonnative proteins was always much faster than folding inside the chaperonin cage. In fact, on the timescale of a translocation-plus-folding event, we never observed complete folding inside the cis ring. This is shown in Fig. S3 (Supplementary Material), where we compared the rate of intracavity folding with the rate of intercavity translocation for a protein that is initially in a completely nonnative state (fraction of initial native contacts equal to 0). To test whether repulsion inside the chaperonin cage is important for the extrusion process, the previous calculations were repeated with a different (strongly hydrophobic) Phe coating of the internal walls of the cavity. Fig. S4 shows the free energy profile for extrusion from such a hydrophobic cage, suggesting that the driving force for extrusion has now been reversed, i.e., rather than expelling the protein, the cage sucks it in. The attraction is strong enough to cause partial adsorption and unfolding of the chain inside the cage. This is not the case for a moderately hydrophobic surface (modeled by Tyr – average interaction energy Ia = −0.16), where the native state is not disrupted by the adsorption on the inner walls (see Fig. S5). However, the attraction is strong enough to inhibit the translocation process. Hence, for translocation to occur, the protein should initially be confined in a hydrophilic cavity. This offers a rationale for the strong hydrophilic nature of the closed cavity. When the protein enters the open barrel of the chaperonin complex (Fig. 2, 4), it need not end up in its native state, as the surface of the open barrel traps nonnative proteins. In this way, the folding cycle can start again, with the capping of the second cavity and the opening of the first. The results we have just presented were obtained at a temperature below the folding temperature. However we also explored the behavior of the system at higher temperatures (data not shown). The main effect of the temperature is to increase the effect of entropy: this favors translocation of the chains toward the open barrel.
FIGURE 2.

Folding process of the double-cage chaperonin. The first step is the trapping of nonnative proteins in the open cage. The second step is encapsulation upon binding of the GroES cap, and change of the internal wall from hydrophobic (red) to hydrophilic (blue) (Fig. 1 a). At this point, the extrusion process starts (Fig. 1 b) with two possible outcomes: 1), the protein does not fold in the native state; or 2), it does. If the former is the case, then the protein goes through another round of extrusion.
FIGURE 3.

Plots of the free energy, F(Q) at T = TF/2, as a function of the number of native contacts Q (Eq. 7) for the conformation that have at least one amino acid inside the open cage (circles), and for the one free in solution (crosses). The open cage is covered with the most attractive amino acid, Phe, with API = −0.23 kT in the rim area, whereas for the hydrophilic back surface we used Arg. For chain conformation close to the native state (Q > 45), there is not free-energy gain in the trapping, indicating that those states can easily diffuse away. However, for the nonnative states, there is a strong preference (up to 5 kT) to bind to the rim.
FIGURE 4.

Plots of the free-energy landscape 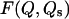 as a function of the number of native contacts Q and of the number of residues inside the cage Qs, computed at T = TF/2, where TF is the temperature at which there is equilibrium between the unfolded and the native states. The states with the lowest free energy correspond to conformation of the chain folded outside the cage (Qs = 0 and Q = 81), demonstrating a preference for the extrusion-plus-folding process. The arrows represent trajectories for the folding-plus-translocation (dashed arrow) and for the intracage folding (solid arrows). Although both translocation and intracage refolding are possible, the first scenario leads to a greater reduction in free energy. The cage is covered with the most repulsive amino acid, Arg, with an average repulsive strength per contact of 0.38. The nonsampled region corresponds to conformations of the chain too compact to exist across the hole.
as a function of the number of native contacts Q and of the number of residues inside the cage Qs, computed at T = TF/2, where TF is the temperature at which there is equilibrium between the unfolded and the native states. The states with the lowest free energy correspond to conformation of the chain folded outside the cage (Qs = 0 and Q = 81), demonstrating a preference for the extrusion-plus-folding process. The arrows represent trajectories for the folding-plus-translocation (dashed arrow) and for the intracage folding (solid arrows). Although both translocation and intracage refolding are possible, the first scenario leads to a greater reduction in free energy. The cage is covered with the most repulsive amino acid, Arg, with an average repulsive strength per contact of 0.38. The nonsampled region corresponds to conformations of the chain too compact to exist across the hole.
FIGURE 5.

Plot of the fraction of successful folding events as a function of the percentage of native contacts in the initial conformation. We compared the folding efficiency in the presence (⋄) and absence (□) of translocation. The picture shows that translocation results in a strong increase in the folding efficiency of highly nonnative states. For conformations that are closer to the native state, the role of translocation becomes less important. Complete translocation becomes less likely as the percentage of native contacts in the initial (prefolding) state increases. This observation is in agreement with the experimental evidence that successful folding can take place in a single GroEL barrel. In these experiments, the GroEL barrel will trap a wide range of proteins at varying degrees of folding. Intrabarrel conversion to the native state should be easy for proteins that already have a fair number of native contacts.
DISCUSSION
This folding scenario has one attractive feature: it offers a natural explanation of the double-barrel structure of the chaperonin, making plausible the idea that nonnative proteins are shuttled back and forth between the barrels until they reach the native state and can escape from the hydrophobic rim of the open barrel. Moreover, as the barrier for translocation is higher for native states than for nonnative states, native states that happen to be trapped in the GroEL-GroES complex stand a good chance of surviving until the barrel opens again. We argue that the large amount of available experimental data is compatible with the translocation scenario that we propose. Weissman et al. (21,22) studied chaperonin-assisted protein folding by using a single-ring mutant (SR1) of the GroEL. This variant is characterized by four mutations in the equatorial region that reduce the binding affinity between the two rings. As a consequence, the individual barrels of the GroEL complex do not dimerize. Interestingly, SR1 does not release the GroES cap under otherwise normal folding conditions (high ATP concentration and 5 mM KCl), and hence the cavity cannot efficiently release the substrate protein. In fact, experiments by Hayer-Hartl et al. (23) indicate that the SR1 complex only releases some 20–30% of the captured proteins. Nevertheless, the folding efficiency of SR1 is comparable to that of the wild-type complex. Analogous experiments on the wild-type GroEL-GroES complex that was kept in the encapsulation state through exposure to ADP or to a nonhydrolizable ATP analog (24) also indicated that a considerable fraction of all substrate proteins was still trapped in the cis cavity, even after several minutes. What these experiments clearly show is that folding can take place in a single barrel, i.e., without translocation. Our simulations reproduce this observation. What we observe is that translocation is most likely for proteins that are far from the native state. Although the available experiments demonstrate that intracavity folding may take place, they do not rule out translocation as a possible mechanism to boost the folding efficiency. One of the possible advantages of the translocation mechanism is that it reduces the probability that incorrectly folded proteins escape into the cytosol. At first sight, it seems that the available experiments indicate that incompletely folded proteins can escape quite easily from the chaperonin complex. In particular, Weissman et al. (25) studied the folding efficiency of a wild-type GroEL-GroES system in the presence of a mutated GroEL molecule that binds nonnative proteins irreversibly. The results indicated a decay in the percentage of folded substrate upon addition of the trapping mutant. In other words, incompletely folded proteins can easily escape from the chaperonin complex and are then removed from the pool of refoldable intermediates. These results, and similar ones reported in Burston et al. (26), would seem to contradict our hypothesis. However, subsequent experiments (27) on the same system as studied in Burston et al. (26) showed that in the presence of crowding agents that mimic the conditions of the cytosol, nonnative proteins did not leave the GroEL complex until they were folded. Hence, it seems that, in the cytosol, incompletely folded proteins do not escape easily from the chaperonin complex. This is in agreement with the scenario that we propose. The scenario suggested by our simulations could be experimentally tested. First of all, it should be possible to verify that proteins can move through the equatorial plane of the GroEL barrel. Moreover, we should expect that the translocation process would be very sensitive to the nature of the disordered protein segments near the flexible region in the equatorial plane. Any chemical modification that would effectively block the hole should decrease the chaperonin activity of the GroEL-GroES complex.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We acknowledge discussions with R. A. Heeren on experimental studies of the GroEL-GroES complex.
This work is part of the research program of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), which is financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). An Nationale Computerfaciliteiten grant for computer time is gratefully acknowledged.
APPENDIX
Design of the folding and of the cavity coating
A given lattice polymer can form a large number of compact conformations. Obviously, every conformation is characterized by a different contact map. Hence, the energy of the polymer depends on its conformation. The mean-field approximation for its entropy is (28)
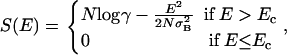 |
(3) |
where N is the number of elements in the chain, σB is the standard deviation of the interaction matrix, and γ is the coordination number for fully compact structures on the lattice. Ec is the (lower) crossing point of the parabola with the abscissa, 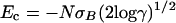 . When the sequence of a heteropolymer is designed in a target configuration, a low-energy state is generated. If the energy EN of this state is lower than Ec, then the system can fold in the target configuration. In the following, we refer to this lowest-energy state as the native state of the heteropolymer. Of course, the native state of a protein should not only have a low energy, it should also be nondegenerate.
. When the sequence of a heteropolymer is designed in a target configuration, a low-energy state is generated. If the energy EN of this state is lower than Ec, then the system can fold in the target configuration. In the following, we refer to this lowest-energy state as the native state of the heteropolymer. Of course, the native state of a protein should not only have a low energy, it should also be nondegenerate.
To design a lattice protein that will fold into a specific conformation, we use the approach described in Shakhnovich and Gutin (17) and Coluzza et al. (18). In this approach, we sample sequences for a given conformation, rather than conformations for a given sequence. The basic trial moves are single-point mutations. As in the conventional Metropolis scheme, the acceptance of trial moves depends on the ratio of the Boltzmann weights of the new and old states. However, if this were the only criterion, there would be a tendency to generate homopolymers that have a highly degenerate ground state, rather than a chain that folds selectively into a desired target structure. To ensure the necessary heterogeneity, we impose the following additional acceptance criterion to the conventional Metropolis scheme:
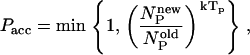 |
where Tp is an arbitrary parameter that plays the role of a temperature, and NP is the number of permutations that are possible for a given set of amino acids. NP is given by the multi-nomial expression
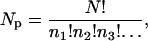 |
(4) |
where N is the total number of monomers and n1, n2, etc. are the number of amino acids of type 1, 2,…. While sampling the sequence space with a Monte Carlo scheme, we keep the temperature (TP) associated with this quantity high. In doing so, we generate a heterogeneous composition of amino acids. The importance of sequence heterogeneity for the design of specific structures is confirmed in our simulation, as it allows us to design heteropolymer sequences that have a nondegenerate native state. There is another, subtler meaning of the “temperature” associated with the structural heterogeneity: it also represents the “frustration” imposed on protein design by the fact that a protein lives in the presence of many other molecules to which it should not bind unspecifically. By increasing this “frustration” temperature, we make it less likely that the protein will form an undesired, specific bond to any of the other proteins in the system. During a Monte Carlo run of several million cycles, a large number of distinct sequences is generated. The sequence S* with the lowest energy is assumed to be the best candidate to fold into the native state. The energy of a given lattice polymer depends on its conformation.
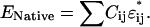 |
(5) |
In this work, we used this scheme to design a lattice heteropolymer to fold to a given target structure. During the design process we do not take in to account any interaction with the cage. In other words, we consider only the intramolecular interaction term in Eq. 1.
To design the interior of a chaperonin, we have to mimic the hydrophilic or hydrophobic nature of the cage while excluding any sequence selectivity. To this end, we employ the approach used in Takagi et al. (5) and Jewett et al. (8) to make a totally structureless cage wall. The interaction matrix that we use is such that strongly hydrophobic amino acids have on average attractive interactions with the other residues; on the other hand, the hydrophilic ones are on average repulsive. As a consequence, a surface covered of amino acids with large positive values of the average pair interaction (API) will be strongly hydrophilic, whereas one with large negative values of API will be mostly hydrophobic. That is, we selected from the interaction matrix the amino acids with the strongest average repulsive interaction APIMax and with the strongest attractive one APIMin:
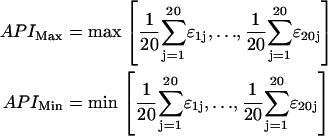 |
(6) |
For the matrix that we used, the amino acid with the largest average attractive interaction is Phe, with a value of APIMax = −0.23 kT, whereas the most repulsive is Arg, APIMin = 0.38 kT. We also considered a cage with a milder attractive interaction, because, as will be discussed below, a cage made with Phe is so attractive that any protein is irreversibly absorbed on the inner surface of the chaperonin. To model a more moderate hydrophobic surface, we used Tyr, for which API = −0.16 kT. We stress that, although the parameters of. Betancourt and Thirumalai (15) and Miyazawa and Jernigan (16) are derived from experimental data, the interaction strengths in our model reproduce the interactions between the amino acids of real proteins only qualitatively. This is no problem for the generic model that we consider, but it would be inadequate for a quantitative description.
Folding
To explore the possible conformations of the lattice polymer, we use four basic Monte Carlo moves: corner-flip, crankshaft, branch rotation, and translation. The corner-flip involves a rotation of 180° of a given particle about the line joining its neighbors along the chain. The crankshaft move is a rotation by 90° of two consecutive particles. A branch rotation is a turn around a randomly chosen pivot particle of the whole section, starting from the pivot particle and going to the end of the chain. The translation is simply a displacement of the center-of-mass of the protein of one lattice unit in a random direction.
We explore the equilibrium properties of the system by sampling the free energy as a function of two order parameters. The first is the number of native intramolecular contacts of the protein in a given conformation
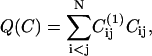 |
(7) |
where 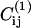 is the contact map of the reference structure, and Cij is the contact map of the instantaneous configuration. Only those contacts that belong to the reference structure contribute a value + 1 to the order parameter. A second order parameter, Qs, allows us to quantify the progress of the extrusion process. It is defined as the total number of residues that are still in the cavity.
is the contact map of the reference structure, and Cij is the contact map of the instantaneous configuration. Only those contacts that belong to the reference structure contribute a value + 1 to the order parameter. A second order parameter, Qs, allows us to quantify the progress of the extrusion process. It is defined as the total number of residues that are still in the cavity.
The quantity that we aim to compute is the free energy F as a function of the two order parameters. To compute F(Q), we used the following relation:
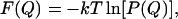 |
(8) |
where F(Q) is the free energy of the state with order parameter Q and P(Q) is the histogram that measures the frequency of occurrence of conformations with order parameter Q. In practice, a direct (brute-force) calculation of this histogram is not efficient, as the system tends to be trapped in local minima, especially at low temperatures. To solve this problem, we incorporated the Monte Carlo sampling approach of Frenkel (29) in the parallel-tempering algorithm of Coluzza et al. (18) and Coluzza and Frenkel (19). This scheme is very efficient in sampling both high and low free-energy states. A more detailed description of the algorithm can be found in Coluzza and Frenkel (20).
References
- 1.Fenton, W. A., and A. L. Horwich. 2003. Chaperonin-mediated protein folding: fate of substrate polypeptide. Q. Rev. Biophys. 36:229–256. [DOI] [PubMed] [Google Scholar]
- 2.Thirumalai, D., D. Klimov, and G. Lorimer. 2003. Caging helps proteins fold. Proc. Natl. Acad. Sci. USA. 100:11195–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young, J. C., V. R. Agashe, K. Siegers, and F. U. Hartl. 2004. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 5:781–791. [DOI] [PubMed] [Google Scholar]
- 4.Sigler, P. B., Z. Xu, H. S. Rye, S. G. Burston, W. A. Fenton, and A. L. Horwich. 1998. Structure and function in GroEL-mediated protein folding. Annu. Rev. Biochem. 67:581–608. [DOI] [PubMed] [Google Scholar]
- 5.Takagi, F., N. Koga, and S. Takada. 2003. How protein thermodynamics and folding mechanisms are altered by the chaperonin cage: molecular simulations. Proc. Natl. Acad. Sci. USA. 100:11367–11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stan, G., B. R. Brooks, and D. Thirumalai. 2005. Probing the “annealing” mechanism of GroEL minichaperone using molecular dynamics simulations. J. Mol. Biol. 350:817–829. [DOI] [PubMed] [Google Scholar]
- 7.van der Vaart, A., J. Ma, and M. Karplus. 2004. The unfolding action of GroEL on a protein substrate. Biophys. J. 87:562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jewett, A. I., A. Baumketner, and J.-E. Shea. 2004. Accelerated folding in the weak hydrophobic environment of a chaperonin cavity: creation of an alternate fast folding pathway. Proc. Natl. Acad. Sci. USA. 101:13192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiyagarajan, P., S. Henderson, and A. Joachimiak. 1996. Solution structures of GroEL and its complex with rhodanese from small-angle neutron scattering. Structure. 4:79–88. [DOI] [PubMed] [Google Scholar]
- 10.Saibil, H. R., D. Zheng, A. M. Roseman, A. S. Hunter, G. M. F. Watson, S. Chen, A. A. Dermauer, B. P. Ohara, S. P. Wood, N. H. Mann, L. K. Barnett, and R. J. Ellis. 1993. ATP induces large quaternary rearrangements in a cage-like chaperonin structure. Curr. Biol. 3:265–273. [DOI] [PubMed] [Google Scholar]
- 11.Saibil, H., and N. A. Ranson. 2002. The chaperonin folding machine. Trends Biochem. Sci. 27:627–632. [DOI] [PubMed] [Google Scholar]
- 12.Rabut, G., and J. Ellenberg. 2001. Nucleocytoplasmic transport: diffusion channel or phase transition? Curr. Biol. 11:R551–R554. [DOI] [PubMed] [Google Scholar]
- 13.Kim, H., and K. J. Shin. 1999. Exact solution of the reversible diffusion-influenced reaction for an isolated pair in three dimensions. Phys. Rev. Lett. 82:1578–1581. [Google Scholar]
- 14.Morrissey, M. P., Z. Ahmed, and E. I. Shakhnovich. 2004. The role of cotranslation in protein folding: a lattice model study. Polymer. 45:557–571. [Google Scholar]
- 15.Betancourt, M., and D. Thirumalai. 1999. Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci. 8:361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazawa, S., and R. Jernigan. 1985. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 18:534–552. [Google Scholar]
- 17.Shakhnovich, E., and A. Gutin. 1993. Engineering of stable and fast-folding sequences of model proteins. Proc. Natl. Acad. Sci. USA. 90:7195–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coluzza, I., H. G. Muller, and D. Frenkel. 2003. Designing refoldable model molecules. Phys. Rev. E. 68:046703. [DOI] [PubMed] [Google Scholar]
- 19.Coluzza, I., and D. Frenkel. 2004. Designing specificity of protein-substrate interactions. Phys. Rev. E. 70:051917. [DOI] [PubMed] [Google Scholar]
- 20.Coluzza, I., and D. Frenkel. 2005. Virtual-move parallel tempering. Chemphyschem. 6:1779–1783. [DOI] [PubMed] [Google Scholar]
- 21.Weissman, J. S., C. M. Hohl, O. Kovalenko, Y. Kashi, S. Chen, K. Braig, H. R. Saibil, W. A. Fenton, and A. L. Norwich. 1995. Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell. 83:577–587. [DOI] [PubMed] [Google Scholar]
- 22.Weissman, J. S., H. S. Rye, W. A. Fenton, J. M. Beechem, and A. L. Horwich. 1996. Characterization of the active intermediate of a GroEL-GroES-mediated protein folding reaction. Cell. 84:481–490. [DOI] [PubMed] [Google Scholar]
- 23.Hayer-Hartl, M., F. Weber, and F. Hartl. 1996. Mechanism of chaperonin action: GroES binding and release can drive GroEL-mediated protein folding in the absence of ATP hydrolysis. EMBO J. 15:6111–6121. [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhry, C., G. W. Farr, M. J. Todd, H. S. Rye, A. T. Brunger, P. D. Adams, A. L. Horwich, and P. B. Sigler. 2003. Role of the gamma-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure and energetics. EMBO J. 22:4877–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weissman, J. S., Y. Kashi, W. A. Fenton, and A. L. Horwich. 1994. GroEL-mediated protein-folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 78:693–702. [DOI] [PubMed] [Google Scholar]
- 26.Burston, S. G., J. S. Weissman, G. W. Farr, W. A. Fenton, and A. L. Norwich. 1996. Release of both native and non-native proteins from a cis-only GroEL ternary complex. Nature. 383:96–99. [DOI] [PubMed] [Google Scholar]
- 27.Martin, J., and F. U. Hartl. 1997. The effect of macromolecular crowding on chaperonin-mediated protein folding. Proc. Natl. Acad. Sci. USA. 94:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derrida, B. 1981. Random-energy model: an exactly solvable model of disordered systems. Phys. Rev. B. 24:2613–2626. [Google Scholar]
- 29.Frenkel, D. 2004. Speed-up of Monte Carlo simulations by sampling of rejected states. Proc. Natl. Acad. Sci. USA. 101:17571–17575. [DOI] [PMC free article] [PubMed] [Google Scholar]