

Abstract
We investigate methods for extracting the potential of mean force (PMF) governing ion permeation from molecular dynamics simulations (MD) using gramicidin A as a prototypical narrow ion channel. It is possible to obtain well-converged meaningful PMFs using all-atom MD, which predict experimental observables within order-of-magnitude agreement with experimental results. This was possible by careful attention to issues of statistical convergence of the PMF, finite size effects, and lipid hydrocarbon chain polarizability. When comparing the modern all-atom force fields of CHARMM27 and AMBER94, we found that a fairly consistent picture emerges, and that both AMBER94 and CHARMM27 predict observables that are in semiquantitative agreement with both the experimental conductance and dissociation coefficient. Even small changes in the force field, however, result in significant changes in permeation energetics. Furthermore, the full two-dimensional free-energy surface describing permeation reveals the location and magnitude of the central barrier and the location of two binding sites for K+ ion permeation near the channel entrance—i.e., an inner site on-axis and an outer site off-axis. We conclude that the MD-PMF approach is a powerful tool for understanding and predicting the function of narrow ion channels in a manner that is consistent with the atomic and thermally fluctuating nature of proteins.
INTRODUCTION
As computational methods increasingly are used to interpret or predict biomolecular function (1), it becomes important to critically evaluate their suitability. In principle, the molecular dynamics (MD) potential of mean force (PMF) approach offers the best route from computer simulation to experiment (2) in a manner that is consistent with the atomic and thermally fluctuating nature of proteins (3,4). In practice, computational studies of ion permeation face significant challenges due to the widely varying timescales of protein and membrane thermal fluctuations that become relevant when constructing permeation models. These fluctuations range from rapid bond, angle, and torsion fluctuations to side-chain isomerizations and large-scale protein and lipid conformational changes (5). Because these fluctuations underlie all protein function, they need to be incorporated in computational models that aim to interpret biological structure-function relationships (6). Moreover, because these thermal protein fluctuations are associated with large variations in ion energetics (3,7), it becomes essential to explicitly incorporate their effects in order to obtain appropriate equilibrium averages for the parameters of interest.
A direct connection between structure and function cannot easily be obtained via MD simulation, however, because ionic fluxes correspond to transit times of 10–100 ns—meaning that it becomes difficult to establish contact with experimental results (single-channel conductances and ion binding constants). To circumvent this difficulty, macroscopic (e.g., (8–10) or semimicroscopic (e.g., (7,11–13)) physical models, which treat some or all of the system as uniform dielectric media, have been invoked. In this study, we instead chose to keep the fully microscopic treatment and demonstrate that permeation can be accurately described via an equilibrium free energy surface that incorporates all of the thermal fluctuations of the ions, water, protein, and phospholipids and which is free of parameter fitting. For this purpose, we employ MD to sample a statistical ensemble of configurations for a fully explicit, atomistic system which, with ion mobility calculations, is fed into a phenomenological conduction model consistent with the PMF calculation (14,15). We show that this approach can be used to obtain a rigorously defined, well-converged, and consistent free energy surface and demonstrate that present-day MD force fields—in conjunction with present-day computational methods—are, perhaps surprisingly, accurate in describing ion permeation through a narrow pore.
To test the approach, and its ability to predict experimental observables, we chose the gramicidin A (gA) channel as our test case because this channel, with its single file pore, poses an extreme challenge for computational investigations of molecular function. The gA channel structure is known, being a single-stranded, right-handed β6.3-helical dimer (16), which has been thoroughly characterized structurally (17–20) and functionally (21–25). It is also small enough to allow good sampling with rather modest computational resources (26). The gA channel thus is an excellent system for testing how well MD-PMF simulations can be used to predict complex molecular functions. Since the first MD simulations on this molecule in 1984 (27), several studies have improved our understanding of the microscopic mechanisms of ion permeation (for review, see (28)). The aim of this study was to critically examine, and hopefully validate, this approach by directly finding contact with experimental measurements.
A perennial problem in previous studies has been that MD simulations predicted free energies corresponding to rates of ion movement several orders-of-magnitude smaller than the measured rates (27,29,30). Perhaps the most notable example was the pioneering computations by Mackay et al. (27), in 1984, which revealed a barrier of nearly 40 kcal/mol opposing translocation of a Cs+ along the axis of the gA channel (these results were reported in (31)). In more recent studies (29,30), the MD-PMF profiles for K+ across the gA channel were constructed using no more than 80 ps simulations per umbrella sampling window (30) (see also (32)). These calculations also predicted barriers for ion permeation that were several kcal/mol too high to be compatible with experiment (possibly as much as ∼7 kcal/mol, based on the findings of this study), meaning that the predicted rates of ion movement were approximately five orders-of-magnitude too low. This could suggest that the atomic force fields used in MD simulations are not adequately calibrated and therefore unable to sufficiently stabilize ions within the narrow pore, as compared to bulk water. Methodological limitations, such as the construction of starting configurations, equilibration, and simulation times also might account for the poor prediction of experimental observables. Indeed, a recent study in which the sampling of the equilibrium distribution of ions was greatly extended (15), and in which destabilizing effects of periodicity and hydrocarbon polarizability were accounted for, led to a well-converged PMF that allowed for semiquantitative prediction of experimental observables. Therefore, as these computational studies have become more sophisticated (with improved methodologies and computational sampling), they provide increasing confidence that MD-PMF calculations could become a powerful tool for understanding (and eventually predicting) ion permeation (and, by implication, other molecular functions).
The difficulties encountered in MD simulations of ion permeation can be traced largely to the observation that the measured rates of ion permeation for gA channels are very high (at low permeant ion concentrations comparable to predictions based on a simple waterfilled pore immersed in bulk water (33)). This means that there cannot be a major energy barrier for ion movement, such that the energy profile for permeating ions result from almost complete cancellation of two very large opposing contributions: ion dehydration and protein/pore water solvation. MD simulations going back to 1984 (27) identified a possible major role of the single-file water to overcome the large dehydration barrier, and almost complete cancellation of the barrier by a combination of the single-file water and protein was demonstrated when the overall PMF was decomposed into water, protein, and membrane/bulk water contributions (15). This need to accurately represent ion solvation in both extremes of bulk water and almost complete dehydration in a narrow pore poses significant challenges to MD force fields.
The MD-PMF strategy adopted in this study is based on the assumption that the long-time behavior of an ion permeation event is dominated by some rate-determining step(s) and that its dynamical evolution can be described as a progress along some reaction coordinate (2). The true reaction coordinate for a given system is not known a priori, and the MD-PMF strategy consists in choosing some suitable order parameter (e.g., the position of the translocating ion along the channel axis) as a mathematical surrogate for the true reaction coordinate. The underlying assumption is that all other variables fluctuate rapidly, such that they can be integrated over in order to obtain a PMF for the chosen order parameter(s). Different choices of order parameters may provide adequate descriptions of the rate-determining step(s)—and usually yield slightly different PMFs—but the absolute transition rate is insensitive to such choices as long as dissipative factors are considered properly (34,35). In our case, relatively slowly varying degrees of freedom have been associated with the orientation/reorientation of the single file water column (15), which could be important for describing the microscopic dynamical mechanisms of permeation, in particular the kinetics of ion entry/exit. For now, however, we describe the permeation mechanism in terms of the ionic spatial coordinates alone, as a useful simplification.
Within this framework, we pursued strategies for identifying the most vulnerable aspects of MD-PMF simulations with the goal of improving their ability to predict experimental observables. To this end, we examine the dependence of ion conduction observables on the choice of MD force field and explore the sensitivity of the results to small changes in parameters. We conclude: first, that careful attention to the physical system at hand allows for significantly improved predictions of experimental observables; second, that simple changes in a given force field are unlikely to provide significant improvements; and third, that while semiquantitative agreement with experiment can be achieved with a modern all-atom fixed-charge force field, ultimately an electronically polarizable simulation will be required for further improvement.
METHODS AND RESULTS
Ion channel-membrane simulations
Simulations were done with the program CHARMM (36) using the PARAM27 (37) force field (referred to as CHARMM27 from here on), with standard protein (37) and TIP3P water (38) with ion parameters from Beglov and Roux (39). Simulations were also done with the all-atom AMBER PARAM94 (40), using TIP3P water, with ion Lennard-Jones parameters from Aqvist (41), and also with the united-atom GROMOS87 (42) force field, using the SPC water model (43) and ion parameters from Straatsma and Berendsen (44). We employ the AMBER94 force field previously imported into the CHARMM program (45) and imported GROMOS87 into CHARMM for comparison. In each case, the standard CHARMM27 lipid parameters were used (46) so as to isolate the effects of the protein when comparing PMFs. The use of particle-mesh Ewald (47), SHAKE (48), and constant pressure and temperature algorithms (49), have been described previously (15,20).
Systems that consist of a gA helical dimer (Protein Data Bank (PDB) No. 1JNO (18)) embedded in a DMPC bilayer (Fig. 1), were created using extensions of previous membrane-building techniques (50). The choice of starting gA structure is based on evidence (20) that dynamical trajectories starting with the PDB:1JNO structure reproduce experimental solid-state NMR measurements (51) better than the solid-state NMR PDB:1MAG structure (19). Membrane patches of approximately one and three shells of lipid molecules around the gA protein were used in the simulations. These patches consisted of 20 and 96 lipid molecules, and 1080 and 3996 water molecules, respectively. For the smaller one-shell system, hexagonal periodic boundaries of xy-translation length 32.1 Å, as determined from the area of the protein and lipids, and average height ∼74 Å, were imposed on the protein-membrane system. For the larger three-shell system, the hexagon xy-translation length was 61.9 Å, with average height ∼75 Å. Pressure coupling was employed in the z-direction (parallel to the membrane normal); the x,y dimensions of the hexagonal boundaries remained fixed during simulations. A 1 M KCl ionic solution was to used ensure good sampling of the ionic bath; this corresponds to 19 K+ and Cl− pairs in the smaller system and 74 pairs in the larger system. Fig. 1 A shows the gA ion channel embedded in one shell of lipids. Fig. 1, B and C, shows the small and large systems, respectively, from the top with periodic images.
FIGURE 1.

Gramicidin A in the bilayer: (A) one-shell system: gA dimer (yellow); DMPC bilayer atoms C (gray), O (red), N (blue), and P (green); K+ (green spheres) and Cl− (gray spheres); water O (red) and H (white). Within the channel, seven single-file water molecules are drawn as spheres adjacent to a single K+ ion at the channel entrance. The chosen MD frame has a channel axis with tilt angle ∼9° relative to the membrane normal vector z. Some lipid molecules and electrolyte from neighboring images are visible. (B) One-shell system (CPK color with green lipid C atoms) with hexagonal periodic images (CPK color with gray lipid C atoms) viewed along the membrane normal. Water molecules and ions have been removed for clarity. (C) Three-shell (large) system with hexagonal periodic images.
The selection of starting configurations for umbrella sampling PMF calculation was influenced by our observations from unbiased simulation. First, we need to consider the occurrence of side-chain isomerizations (20). Trp-9 isomerizations occur on the nanosecond timeframe, meaning that sampling long enough to get an equilibrium distribution of rotameric states presents a challenge. Based on comparisons with solid-state NMR observables, we determined that the dominant Trp-9 rotameric state is the one suggested by solution-state NMR (17,18). Fig. 2 shows the distribution of side-chain conformations, with the dominant rotamer highlighted with a solid rectangular box. It is important to ensure that the protein remains near its dominant structure. This side-chain rotamer of Trp-9 was maintained by a flat-bottom harmonic potential with force constant 100 kcal/mol/rad2. To maintain 140 < χ1 < 250°, the harmonic potential was activated for dihedral values χ1 < 150° or χ1 > 240°. Similarly, to maintain 0 < χ2 < 150° the potential was activated for dihedral values χ2 < 10° or χ2 > 140°. Thus, the side chain experiences no force within the indicated box (to allow the usual thermal fluctuations), but provides stiff opposition if the side chains attempt to change rotameric state.
FIGURE 2.

Observed rotameric states of the Trp-9 residues in the gA channel during 47 ns of unbiased simulation (adapted from (20)). The solid box highlights the correct rotameric state and indicates the placement of a two-dimensional flat-bottomed restraint to maintain this rotamer during PMF calculations.
Secondly, we observed occasional interference by phospholipid headgroups near the channel entrances (in particular for the larger three-shell system). Fig. 3 shows two-dimensional lipid distributions around the gA channel for the one- and three-shell systems. The proximity of the lipid headgroup to the channel entrance is evident in this sample of trajectory for the three-shell system. When choosing initial configurations for the PMF calculation, a test was done to see if any lipid atoms were within 4 Å of the channel axis. No constraints were applied to control lipids during the simulations.
FIGURE 3.

Density of lipid heavy atoms around the gA dimer. Density is plotted as a two-dimensional histogram in axial and radial directions with respect to the protein. The one-shell histogram is an average over 10 ns of simulation whereas the three-shell system is an average over only the first 2 ns of a 10-ns simulation to highlight the presence of a lipid density over the channel entrance.
Another observation from unbiased simulation was that occasionally water may protrude deep into the membrane nearby the channel protein. This is consistent with the experimentally observed water mole fractions of the order of 10−4 in bulk C7–C16 hydrocarbons (52), which is likely to be enhanced further by the presence of protein. While this is a natural occurrence, we wished to begin with a similar environment for all window simulations. Thus if water molecules were penetrating very deep into the membrane near the protein (|z| ≤ 6 Å), the initial configuration was discarded. No constraints were applied to prevent water penetration into the bilayer during the production simulations; during the 1–2 ns/window simulation period, the equilibrium sampling therefore did include configurations with water penetration. We did not pursue this question further.
Next, while the water inside the channel maintains a single-file column without any manipulation, occasionally small gaps in that column may occur; especially when an ion is in close proximity to the channel entrance. To test if a system configuration was suitable for a starting point for a window, the maximum space between waters inside the channel was computed. This was done by checking for any gap greater than 1.5 Å in the range −10.5 ≤ z ≤ 10.5 Å. No constraints were used to maintain the pore water structure during simulations. (These breaks in the water column may become important when predicting, or interpreting, the diffusion coefficient of the ion-water column within the pore.)
Constraints were applied to ensure the membrane and protein are kept near the center of the periodic box. A weak harmonic planar constraint, of force constant 5 kcal/mol/Å2 was applied to the z-coordinate of the center of mass of the lipid bilayer to prevent drifting. A weak center-of-mass constraint was also applied on the xy position of the center of mass of the channel by application of a cylindrical harmonic constraint of force constant 5 kcal/mol/Å2. These constraints have no effect on the z position of the channel relative to the membrane, nor the tilting of the channel. They only act to center the membrane and channel independently and have no impact on results.
Reaction coordinate for ion permeation
Following the general statistical mechanical equilibrium theory formulated in Roux (53) and reviewed in Roux et al. (2), the system can be separated into pore and bulk regions, which allows the definition of the free energy surface 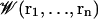 generated for pore occupation by n ions with coordinates ri (i = 1, n). One can form a hierarchy of n-ion PMFs for different occupancy states of the pore (53). For low-to-moderate ionic concentration, the gA channel should be occupied by just one cation (25,54), and a one-ion PMF,
generated for pore occupation by n ions with coordinates ri (i = 1, n). One can form a hierarchy of n-ion PMFs for different occupancy states of the pore (53). For low-to-moderate ionic concentration, the gA channel should be occupied by just one cation (25,54), and a one-ion PMF, 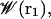 will reveal much about the function of this ion channel in this regime. This one-ion PMF can be written in terms of a configurational integral (see Eq. 13 in (53)). (As noted above, to achieve satisfactory sampling, the simulations were done using a 1 M KCl solution where the channel may be occupied also by two ions, as noted below; this will not affect any of our conclusions, except that they pertain only to the one-ion case.)
will reveal much about the function of this ion channel in this regime. This one-ion PMF can be written in terms of a configurational integral (see Eq. 13 in (53)). (As noted above, to achieve satisfactory sampling, the simulations were done using a 1 M KCl solution where the channel may be occupied also by two ions, as noted below; this will not affect any of our conclusions, except that they pertain only to the one-ion case.)
To calculate a meaningful one-ion PMF,  we must choose a pore region which is almost exclusively occupied by a single ion. Table 1 shows distributions of ion occupancies on either side of the channel (nleft, nright) as a function of the size of an exclusion sphere, centered on the origin, based on analysis of 10.9 ns of unbiased simulation. A suitable choice for this radius appears to be 14 Å, above which it becomes possible to see two cations on one side of the channel within the sphere. Furthermore, with this radius, an anion is found inside the sphere during only 1% of simulation, which is evidence of valence selectivity of the gA channel (this question will be examined further in a separate study). Still, 19% of the time, one cation may be bound at both entrances of the channel. To calculate a one-ion PMF, starting configurations were chosen such that only the one ion was in the 14 Å sphere, and during biased simulation other ions (cations and anions) were excluded with a repulsive flat-bottom spherical harmonic restraint with force constant 5 kcal/mol, applied to other ions only when they enter this exclusion sphere. Thus, our simulation methodology is designed to compute the one-ion PMF from MD simulations that rule out multiple pore occupancy (as would occur with 1.0 M K+ in the aqueous solution (54)).
we must choose a pore region which is almost exclusively occupied by a single ion. Table 1 shows distributions of ion occupancies on either side of the channel (nleft, nright) as a function of the size of an exclusion sphere, centered on the origin, based on analysis of 10.9 ns of unbiased simulation. A suitable choice for this radius appears to be 14 Å, above which it becomes possible to see two cations on one side of the channel within the sphere. Furthermore, with this radius, an anion is found inside the sphere during only 1% of simulation, which is evidence of valence selectivity of the gA channel (this question will be examined further in a separate study). Still, 19% of the time, one cation may be bound at both entrances of the channel. To calculate a one-ion PMF, starting configurations were chosen such that only the one ion was in the 14 Å sphere, and during biased simulation other ions (cations and anions) were excluded with a repulsive flat-bottom spherical harmonic restraint with force constant 5 kcal/mol, applied to other ions only when they enter this exclusion sphere. Thus, our simulation methodology is designed to compute the one-ion PMF from MD simulations that rule out multiple pore occupancy (as would occur with 1.0 M K+ in the aqueous solution (54)).
TABLE 1.
Pore ion occupancy: for spherical radii, ranging from 9 to 20 Å, the distribution of ion occupancies, n (0, 1, or 2 within the sphere on each side of the center), are given as percentage of time
% Cation occupation 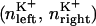
|
% Anion occupation 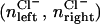
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radius | (0,0) | (1,0) | (1,1) | (2,0) | (2,1) | (2,2) | (0,0) | (1,0) | (1,1) | (2,0) | (2,1) | (2,2) |
| 9 Å | 96 | 4 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| 10 Å | 89 | 11 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| 11 Å | 76 | 24 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| 12 Å | 68 | 32 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| 13 Å | 54 | 38 | 8 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 |
| 14 Å | 48 | 33 | 19 | 0 | 0 | 0 | 99 | 1 | 0 | 0 | 0 | 0 |
| 15 Å | 44 | 31 | 24 | 1 | 0 | 0 | 98 | 2 | 0 | 0 | 0 | 0 |
| 16 Å | 40 | 31 | 28 | 1 | 0 | 0 | 95 | 5 | 0 | 0 | 0 | 0 |
| 17 Å | 34 | 32 | 32 | 1 | 1 | 0 | 91 | 9 | 0 | 0 | 0 | 0 |
| 18 Å | 29 | 32 | 36 | 1 | 2 | 0 | 83 | 15 | 0 | 2 | 0 | 0 |
| 19 Å | 24 | 31 | 38 | 1 | 6 | 0 | 73 | 23 | 1 | 3 | 0 | 0 |
| 20 Å | 19 | 31 | 38 | 1 | 10 | 1 | 61 | 32 | 2 | 5 | 0 | 0 |
The ion occupancy is listed here as (nleft, nright) and the percent of the time in the MD simulation is listed. Occupancies have been symmetrized such that (1, 0) ≡ (1, 0) + (0, 1), for example. The optimal choice of single-ion region is indicated in bold.
The objective of the present calculations is to establish contact with experimental conductance measurements. Net ion movement flow through the channel is driven by a trans-membrane potential difference that arises from a very small ionic charge imbalance between intra- and extracellular spaces, widely distributed across the membrane-water interface (55). This potential is changing in a direction normal to the membrane surface, and the chosen reaction coordinate must include a coordinate, z, parallel to the membrane normal vector to allow for the computation of ionic fluxes due to the potential difference. Therefore, we used the z-component of the distance of the ion to the center-of-mass (CoM) of the gA dimer. In a later section, we discuss the significance of this choice by comparing to the instantaneous channel CoM-axis (monomer CoM to monomer CoM).
Because the ion is confined within a narrow region in the xy-plane within the channel, one may assume that the equilibrium distribution of lateral displacements is obtained quickly and thus may be integrated away, leading to the one-dimensional PMF W(z), or free-energy profile (2,53). However, as reported previously (15), the ion becomes unbounded in the xy-plane at a distance of 14–15 Å from the channel center. Thus, this one-dimensional profile has limited significance outside the channel because integration over all xy extents will cause the PMF to tend toward –∞. To obtain an unambiguous free energy profile one must restrict the lateral displacement of the ion. In the current computations, a flat-bottom cylindrical constraint with radius 8 Å (relative to the center of mass of the dimer) of force constant 10 kcal/mol/Å2 (applied only outside 8 Å) was employed. Without this restraint, the shape of the PMF near the channel entrances is ill-defined and the bulk reference value is meaningless because it is determined by the extent of sampling, as it may have been in previous attempts (30). (The influence of the cylindrical restraint can be, and is, rigorously accounted for in the present analysis.)
The one-dimensional PMF and convergence
Initial configurations for the simulations to calculate the PMF were chosen by searching a 4-ns sample of unbiased MD trajectory for a frame in which the ion exists very close to the reaction coordinate and the center of the window (within 1 Å). When no configuration was found with a K+ ion near the center of a window, as was the case deep within the channel, a nearby water molecule was exchanged with the outermost ion. The trajectory was searched until a water oxygen atom was located close to that point, and that water molecule was exchanged with the K+ ion furthermost from the channel.
We calculated the PMF W(z) using umbrella sampling (56). This requires a set of equally spaced simulations {i}, biased by window functions  that hold the ion near positions along the z-axis. We simulated 101 independent windows, defined by harmonic potential functions, positioned at 0.5 Å increments in z = (−20, +30) Å. Harmonic potentials have a force constant 10 kcal/mol/Å2, chosen to ensure overlap of neighboring windows. For each of the 101 windows, equilibration was performed for 80 ps before 1–2 ns of trajectory generation on separate CPUs. Ionic distributions were unbiased using the weighted histogram analysis method (WHAM) (57), which consists of solving coupled equations for the optimal estimate for the unbiased density 〈ρ(z)〉. Strict attention was paid to the convergence of these WHAM equations. Achieving convergence in the free energy constants (to, say, 0.001 kcal/mol), does not guarantee a comparable convergence in the PMF, as further iterating the WHAM equations can lead to many kcal/mol changes in the PMF. To guard against this, each 100 iterations we check every point in the PMF for convergence to within 0.001 kcal/mol. This is a very strict criterion and usually requires >10,000 WHAM iterations. A total of 2 ns of trajectory was generated for each of the 81 windows between z = −20 to + 20 Å and 1 ns for each of the 20 windows from z = + 20.5 to + 30 Å. The one-dimensional PMF, W(z), shown in Fig. 4 (defined only between the dotted vertical lines) reveals much about the permeation process, including a high central barrier and local free energy minima throughout the channel.
that hold the ion near positions along the z-axis. We simulated 101 independent windows, defined by harmonic potential functions, positioned at 0.5 Å increments in z = (−20, +30) Å. Harmonic potentials have a force constant 10 kcal/mol/Å2, chosen to ensure overlap of neighboring windows. For each of the 101 windows, equilibration was performed for 80 ps before 1–2 ns of trajectory generation on separate CPUs. Ionic distributions were unbiased using the weighted histogram analysis method (WHAM) (57), which consists of solving coupled equations for the optimal estimate for the unbiased density 〈ρ(z)〉. Strict attention was paid to the convergence of these WHAM equations. Achieving convergence in the free energy constants (to, say, 0.001 kcal/mol), does not guarantee a comparable convergence in the PMF, as further iterating the WHAM equations can lead to many kcal/mol changes in the PMF. To guard against this, each 100 iterations we check every point in the PMF for convergence to within 0.001 kcal/mol. This is a very strict criterion and usually requires >10,000 WHAM iterations. A total of 2 ns of trajectory was generated for each of the 81 windows between z = −20 to + 20 Å and 1 ns for each of the 20 windows from z = + 20.5 to + 30 Å. The one-dimensional PMF, W(z), shown in Fig. 4 (defined only between the dotted vertical lines) reveals much about the permeation process, including a high central barrier and local free energy minima throughout the channel.
FIGURE 4.

One-dimensional PMFs from simulations with the CHARMM27 force field (without artifact correction). (A) PMFs for all 40 × 50 ps blocks in the total of 2 ns simulation/window. Panels B and C reveal asymmetry in the PMF for 1 and 2 ns per window, respectively, by plotting with the mirror image about z = 0 (dashed curves). The symmetrized PMFs for 1 and 2 ns are shown in panel D. Broken vertical lines at |z| = 15 Å indicate that the one-dimensional PMF is not defined approximately beyond those points.
A first measure of the error in the convergence of the PMF can be obtained by examining the asymmetry ∼z = 0 (a spatial convergence). If sampling were complete, the PMF should exhibit perfect symmetry. We compared the PMFs obtained based on simulations for different lengths of time (50 ps to 2 ns for each window). Fig. 4 A shows the 40 PMFs calculated with 50 ps per window (from the total of 2 ns). The asymmetry (PMF z = −15 to 15 Å) varied from −8 to 8 kcal/mol (although the asymmetry arises from incomplete sampling all along the reaction coordinate, all the trajectories are defined to be at 0 kcal/mol at z = −15, meaning that the fluctuations become evident as an asymmetry in the resulting PMFs). Similarly, comparison of all calculations with 100, 200, 300, 500, and 1000 ps per window (not shown) revealed asymmetries of up to ∼8, 7, 5, 3, and 2 kcal/mol, respectively—decreasing with sampling time, as expected. Even after 1 ns of simulation, the PMF is somewhat asymmetric at the channel center (Fig. 4 B). After 2 ns of simulation, the asymmetry at the channel center has diminished to <1 kcal/mol within the pore region (Fig. 4 C). This remaining asymmetry corresponds to an average force of 0.03 kcal/mol/Å across the channel, which is much smaller than typical ensemble-averaged atomic forces (of the order of 10 kcal/mol/Å). This asymmetry is associated with noise originating from all degrees of freedom other than the chosen order parameter (z-coordinate of one ion). We do not pursue this asymmetry further, but note that it can be removed from the analysis by symmetrization, which imposes a constraint on the solutions.
We computed symmetrical PMFs by applying the WHAM equations to the biased ion-density distribution after creating duplicate windows on opposite sides of the channel. The second test of convergence is thus the difference in the symmetrized 1- and 2-ns PMFs (a temporal convergence). Fig. 4 D shows the PMFs following symmetrization for both the 1 and 2 ns calculations. The PMF is well converged with a maximum deviation near the center of ∼0.7 kcal/mol (lower in the 2-ns PMF). The average deviation between the two curves is 0.30 kcal/mol, which becomes our estimate for the convergence error in PMF calculation. We conclude that 1 ns per window is sufficient to obtain a well-converged, symmetrized PMF, and we use 1-ns-per-window simulations for all subsequent PMF calculations in this article. The symmetrized CHARMM27 one-dimensional PMF (after 2 ns) reveals a central barrier of ∼11 kcal/mol with respect to the binding site. This barrier height should be compared with previous estimates (29,30,32) of the order of 15 kcal/mol. There is a deep outer binding site at z = 11.3 Å, and a more shallow inner binding site at 9.7 Å.
As is evident from Fig. 4 A, attempts to estimate the height of the central barrier from a short simulation will result in considerable uncertainty. Though, as noted above, the one-dimensional PMF is not rigorously defined outside the channel, the variation in the value at the channel center relative to ±20 Å provides a measure of the PMF convergence. Thus, when comparing all 40 possible 50-ps blocks in the 2-ns total simulation per window (Fig. 4 A), the central barrier varied by 17 kcal/mol—from as little as just 1.2 kcal/mol to as much as 18.2 kcal/mol. The same analysis for the set of 100-ps nonsymmetrized PMFs leads to values ranging from 5.5 to 18.2 kcal/mol. This range drops to 9.6, 5.8, 5.8, and 4.0 for simulations lasting 200, 300, 500, and 1000 ps/window, respectively. The decreasing range (or uncertainty in the height of the central barrier) with increasing simulation is comforting, but it is important to establish an independent test. In the following section, the barrier height from the full two-dimensional PMF (a well-defined quantity) will be computed and compared to an independent free energy perturbation (FEP) calculation.
Two-dimensional free energy landscape
The range of validity of the one-dimensional PMF is within the bounds of the ion channel. Thus, a complete description of permeation requires a three-dimensional PMF, which can be approximated by a two-dimensional PMF, W(z, r), using radially symmetric configurational integration. Other coordinates may describe slowly varying degrees of freedom in the system. For example, the dipole moment, μ, of the single-file water column was shown, via W(z, μ), to provide a barrier to permeation that is not a function of ion position (15). Equilibrium distributions, biased in z, involving a secondary variable r, 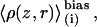 may be readily unbiased to produce a two-dimensional PMF W(z, r). Once the WHAM equations (57) have been iterated to convergence,
may be readily unbiased to produce a two-dimensional PMF W(z, r). Once the WHAM equations (57) have been iterated to convergence, 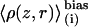 may be unbiased via a straightforward extension of the one-dimensional equation.
may be unbiased via a straightforward extension of the one-dimensional equation.
The resulting two-dimensional PMF (Fig. 5 A), reveals the position of binding sites at the channel entrances and the scale of the free energy barrier experienced by the permeating ion relative to the entrances. It also reveals the extent of lateral ion motion of the ion. Because the two-dimensional PMF is determined in the laboratory frame, lateral movement of the ion relative to the channel, combined with channel tilting, lead to fairly broad free energy wells. This tilt has an impact on the shape of the one-dimensional PMF of Fig. 4 because the ion experiences a greater radial range, with lower free energies, near the channel entrances (raising the barrier with respect to the binding sites compared to the two-dimensional PMF). The action of the cylindrical constraint in the bulk is evident in this graph. It is also evident that the free energy surface is becoming flat away from the channel. Because this two-dimensional PMF is determined only up to a constant, and we need a bulk reference to establish the zero of this free energy surface. To find the correct bulk reference for the two-dimensional PMF, an additional 4-ns simulation (in the absence of a window biasing potential) was used to calculate the bulk ion density and thus the bulk limit, via 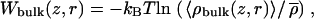 where
where 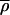 is the K+ density far from the channel (30 < |z| <35 Å). The result is shown in Fig. 5 B. This surface extends out to |z| = 35 Å and reveals a flat PMF away from the channel. The values Wbulk(z, r) and W(z, r) were then matched by linearly interpolating over the range 25 < |z| < 29 Å, for r < 8 Å with the resultant two-dimensional PMF shown in Fig. 5 C. Ion free energies are now known at all positions relative to the bulk. The outer binding site at z = 11.3 Å, can be seen to be −3.2 kcal/mol relative to the bulk. In the narrowest part of the channel, an ion experiences a barrier of 7.2 kcal/mol relative to the bulk.
is the K+ density far from the channel (30 < |z| <35 Å). The result is shown in Fig. 5 B. This surface extends out to |z| = 35 Å and reveals a flat PMF away from the channel. The values Wbulk(z, r) and W(z, r) were then matched by linearly interpolating over the range 25 < |z| < 29 Å, for r < 8 Å with the resultant two-dimensional PMF shown in Fig. 5 C. Ion free energies are now known at all positions relative to the bulk. The outer binding site at z = 11.3 Å, can be seen to be −3.2 kcal/mol relative to the bulk. In the narrowest part of the channel, an ion experiences a barrier of 7.2 kcal/mol relative to the bulk.
FIGURE 5.

Creating a two-dimensional PMF. (A) The PMF obtained from two-dimensional unbiasing of equilibrium distributions from umbrella sampling simulations. (B) The PMF obtained by analysis of bulk ion densities. (C) A blend of panels A and B using linear interpolation, similar to that in Allen et al. (15), but with extended range.
An independent check of the barrier height in W(z, r) was obtained by free energy perturbation calculations, where an ion on-axis at the center of the channel (z = 0) was interchanged alchemically with a water on-axis in the bulk (z = 30 Å) (15) The estimated free energy 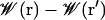 was 8.6 ± 0.4 kcal/mol, consistent with the value obtained from the two-dimensional PMF, and suggests an uncertainty of the order of 1 kcal/mol.
was 8.6 ± 0.4 kcal/mol, consistent with the value obtained from the two-dimensional PMF, and suggests an uncertainty of the order of 1 kcal/mol.
A comparison of reaction coordinates
While a coordinate parallel to the membrane normal is necessary for conduction calculation, the free energy surface governing ion permeation also can be studied along an instantaneous channel CoM-axis to reveal in more detail the ion-protein-water interplay during permeation. Such a PMF is interesting for describing the free energy of the ion relative to the ion channel β-helix. We chose the time-varying vector passing through the center of mass of monomer 1 and monomer 2 of the gA dimer to create a different PMF, based on umbrella sampling along a coordinate that is the ion distance from the center of mass of the channel dimer, projected onto the axis connecting the two centers of mass of the monomers. As before, we apply a lateral constraint in the form of an 8 Å flat-bottom cylinder centered on the instantaneous channel CoM-axis to ensure a well-defined region of sampling outside the channel.
We expect the two PMFs to differ because the channel on average tilts 12° with respect to the bilayer normal (15), which contributes to a radial displacement of ions away from the channel center. Fig. 6 shows the average tilt of the channel as a function of ion position (averaged over 1-ns umbrella sampling for each window). The average tilt decreases steadily as the ion moves from the channel center (where it experiences a maximum of 16°) to the entrances. The decreased tilt near the entrances can be rationalized by considering the interaction between the ion and the bulk electrolyte as a function of depth. The attractive force between an ion in the pore and the bulk solution and membrane interface is stronger when the ion is nearer either interface (58). As a result, the ion-interface interactions will exert a torque on the tilted channel, which will tend to reduce the tilt when the ion is further from the channel center (closer to the interface), as observed in Fig. 6.
FIGURE 6.

Average channel tilt angle (from average cosine of the angle separating the channel axis and membrane normal, z) as a function of ion position z for the small, one-shell, system. Results have been symmetrized by averaging windows on each side of z = 0. Error bars are not shown for clarity. The average standard deviation in ion position is 0.25 Å and that of tilt angle is 3.6°.
The two-dimensional PMF as a function of distance along the instantaneous CoM-axis and radial displacement is shown in Fig. 7 A, with the fixed-frame PMF in Fig. 7 B for comparison. This figure focuses in on the region −20 ≤ z ≤ 20 and r ≤ 8 Å to highlight the effect of choice of coordinate frame. Unlike the two-dimensional PMF of Fig. 5, the PMF has not been merged with a larger spanning bulk region and the zero has been set from the average in the region 18 ≤ z ≤ 20 and r ≤ 8 Å. Based on the shape of the free energy surface in Fig. 5, one may conclude that the consequences of the choice of bulk reference region is <1 kcal/mol, which can be ignored because this PMF is primarily for illustration purposes.
FIGURE 7.

Two-dimensional PMFs in the pore region for two different reaction coordinates: the position along the instantaneous channel axis (monomer center-of-mass to the monomer center-of-mass) (A), and the projection of the distance to the center-of-mass onto the membrane normal, the z-axis (B). Each surface is based on a calculation with a symmetrized biased density. Only the first 1 ns of simulation for each window is included.
The shape of the two-dimensional free energy surface near the channel axis is markedly different as a result of this choice of coordinate frame. The two-dimensional PMF in fixed-membrane normal vector frame (Fig. 5 B) has wide, low free-energy vestibules whereas the two-dimensional PMF in the instantaneous CoM-axis vector frame (Fig. 5 A) does not. Secondly, the size of the barrier in the two-dimensional PMF has dropped from 7.2 (Fig. 5 B) to 5.5 kcal/mol (Fig. 5 A) relative to the bulk. The fixed-frame PMF appears to get narrower at the center, but this simply reflects the widening of the PMF near the entrances due to tilting. Differences in the free energies for lateral displacements away from axis, due to the increased cross section as a result of tilting in the fixed z-PMF, maybe too small to see in this 1 kcal/mol contoured map. Thirdly, the outer binding sites can be seen to be off-axis; a fact that was hidden the PMF of Fig. 5 due to the channel tilting smearing out this feature. Finally, the inner binding sites at 9.7 Å are now clearly visible—and are deeper than the outer sites in the CoM-axis PMF. In this coordinate frame the inner binding site is ∼−3.5 kcal/mol, whereas the outer binding site is ∼−3.0 kcal/mol, relative to the bulk. This is very different from the original fixed-membrane normal vector frame where the inner binding site was ∼−0.5 kcal/mol and the outer binding site ∼−3.2 kcal/mol. The difference arises because the outer binding sites are off-axis, and therefore correspond to a greater spatial volume, such that the integrated one-dimensional PMF has a global minimum there. An off-axis outer binding site implies greater translational freedom of the ion in this region and, perhaps, less distortion of the protein backbone. In any case, the existence of two cation binding sites, with a slight preference for outer site binding in the case of K+, is consistent with the analysis of NMR data (59)—but in conflict with the x-ray scattering results of Olah et al. (60), which shows the major binding site to be at 9.5 Å.
The one-dimensional PMF from this instantaneous CoM-axis umbrella sampling calculation is shown in Fig. 8, together with the 1-ns symmetrized PMF for the original z-coordinate PMF of Fig. 4 C. Not surprisingly, this one-dimensional PMF along a channel-axis vector is not that different to the PMF along the z-vector. The reason for this is that the lateral displacement corresponding to the average tilt is <0.2 Å near the binding sites. One would expect that the depth of the binding sites will change slightly: because of the greater lateral displacements in the z-vector frame (away from z = 0) relative to the instantaneous channel CoM-axis frame, the binding sites should be (a little) deeper (relative to the center of the channel) in the original fixed frame PMF along the fixed z-vector. This is the case in Fig. 8, although the difference is just a fraction of a kcal/mol. There also are small differences in the center of the channel where the original fixed-frame PMF experiences a slightly higher barrier. Given the uncertainties of the PMF and the nontrivial relationship between tilting and the PMF, the origin of the small differences in the PMFs remains unclear.
FIGURE 8.

One-dimensional PMFs from simulations with the CHARMM27 force field (without artifact corrections) using two different reaction coordinates: the position along the instantaneous channel axis (solid) and the projection of the distance from the center of mass onto the membrane normal, z (dashed). Each curve has been symmetrized (via biased density) and only the first 1 ns of simulation for each window is included.
The dependence of the free energy barrier in the one-dimensional PMF on the choice of coordinate used in the umbrella sampling computations could be cause for concern. It therefore is important to recall that the absolute transition rate involves not only the PMF, but also dissipative contributions (e.g., diffusion, friction, memory function, transmission coefficient), which themselves depend on the choice of order parameters (34). Within reasonable restrictions, the resulting absolute transition rate is invariant with respect to the choice of order parameter despite differences in the free energy barrier; see Hinsen and Roux (35), for example. Differences between the CoM-axis and z-axis results do not imply that the MD-PMF computational strategy yields inconsistent results, rather the different reaction coordinates highlight different aspects of the underlying process. The calculations with the instantaneous channel CoM-axis vector choice of reaction coordinate, for example, reveal details about the passage of an ion through the channel, which were hidden in the fixed z-axis PMF calculations. First, the ion in the outer binding site is not entirely within the narrow single-file column, as would be anticipated by knowing it is solvated by three water molecules (15,29,59). Second, a K+ at this site prefers to stay off-axis, which may be a more general result (see (61)). The inner binding site is within the narrow single-file region proper, where the cation is solvated by just two water molecules (15), and stays closer to the channel axis. In the present calculations, a K+ appears to preferentially reside in the outer binding site because the radially integrated (one-dimensional) PMF results in a global minimum at that distance from the channel center, consistent with NMR analysis (59) but contradictory to earlier x-ray scattering results (60). Yet, our original choice of fixed-membrane normal vector z remains the only choice for computing the single-channel conductance with an applied membrane potential difference. Before we can compute the conductance from the PMF, we need to account for simulation artifacts that affect our estimates.
Correcting for simulation artifacts
A spurious destabilization of the ion is caused by the finite size and the periodicity of the system. In addition, in current MD force fields the hydrocarbon chains of the lipid molecules are nonpolarizable, meaning that they have an effective dielectric constant of 1 (62), which is quite different from the value deduced for the bilayer hydrophobic core (63,64) and measured for bulk hydrocarbons (65), ∼2. These artifacts can be approximately corrected using a continuum electrostatic approximation (15,66) utilizing trajectories to average over protein and single-file water configurations. We estimate these corrections for an ion near the channel axis and apply to the one-dimensional PMF calculations (with the two-dimensional PMFs remaining uncorrected). However, as we shall show in the following section, all comparisons with experimental measurements can be formulated using the one-dimensional PMF.
To implement the corrections, we establish a free energy cycle (Fig. 9). The corrected PMF Wcorr(z; εm = 2; L = ∞), with the correct membrane dielectric constant, εm = 2, and for an infinite membrane with no periodic effect, may be obtained from the MD-PMF WMD(z; εm = 1; L = L0), with respect to some reference position far from the channel, z′, where the dielectric constant of the membrane is εm = 1 and the periodic length is L = L0 in the xy-plane. The relation may be written as
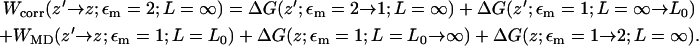 |
(1) |
This may be rewritten as
 |
(2) |
where WMD(z) and Wcorr(z) are defined relative to z′,
 |
(3) |
and
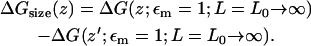 |
(4) |
FIGURE 9.

The free energy cycle illustrates the sequence of correction calculations required for a PMF calculated with finite system size and nonpolarizable membrane. The gA channel is shown as yellow; high dielectric (ε = 80) bulk water as blue; membrane core with ε = 1 as white; membrane core with correct hydrocarbon dielectric constant (ε = 2) as gray; pore water molecules as red (O) and white (H) circles; and K+ as green circles with + sign.
To estimate the various corrections, we embarked on a series of finite-difference Poisson calculations, using the PBEQ module of CHARMM, averaging over several instantaneous configurations (snapshots) extracted from MD trajectories of the protein and its water contents. For each ion position, we extracted 50 snapshots from 500 ps of trajectory from umbrella sampling windows between z = 0 and 20 Å in 2 Å increments. For each ion position, Poisson's equation was solved both with and without the ion present (by setting the K charge to +1 or 0), with membrane dielectric constant εm = 1 or 2, and for systems of different sizes to obtain corrections for our PMFs calculated using small (Fig. 4) and large membranes (reported below). These sizes included the one-shell (small) system, the three-shell (large) system, and a very large system that represents an infinite membrane (similar in size to a five-shell system not reported here) with image translation vector 90 Å. The membrane core was assumed to be 25 Å thick (comparable to the hydrophobic thickness of a DMPC bilayer (67)) and was assigned a dielectric constant εm. Bulk water (and the membrane/solution interface) was assigned a dielectric constant of 80. Because we average over thermal fluctuations of the explicit protein and channel water we assume a dielectric constant of 1 for the protein (εp) and channel water (εc) regions. The protein and any water molecules within −12.5 ≤ z ≤ 12.5 Å and within a cylinder of radius 3 Å (centered on the channel axis) were extracted from each frame of the trajectory. Protein and channel water were oriented with respect to the membrane normal at each frame, and a water-sized reentrant probe was used to assign dielectric constants (3). This set of Poisson solutions are all that is needed to compute the ensemble-averaged corrections in Eqs. 3 and 4.
Periodic boundaries are orthorhombic in the PBEQ algorithm whereas they are hexagonal in the MD-PMF. We chose to match the area of the unit cell so as to equate the number of channels per unit area. To do so, the orthorhombic xy-translation vector for PBEQ images was set to 29.9 Å for the one-shell system, instead of 32.1 Å for the hexagonal boundaries. In the three-shell case the length was set to 57.6 Å instead of 61.9 Å. To test the significance of this approximation, we performed calculations with explicitly included hexagonal and cubic periodic xy images of the MD averaged protein and ion in the PBEQ module of CHARMM. The results for hexagonal and cubic images differed by just 0.19 kcal/mol, suggesting that the corrections are not significantly affected by this choice.
To make these profiles more readily applicable as corrections (requiring interpolation onto a finer grid), we fitted the MD-averaged Poisson solutions with solutions for a single structure using optimized dielectric constants of protein (and channel water). This was done with a single MD-averaged structure of the protein with implicit water inside the pore. The MD-averaged protein structure was obtained by constrained minimization of the PDB:1JNO structure to MD-averaged heavy atom positions. The dielectric constants εp = εc were optimized to obtain the best numerical fit the MD data.
Estimates, obtained from averages over MD trajectories, show that correcting for the spurious destabilization leads to a −1.6 kcal/mol correction (at the channel center, relative to the bulk) for the one-shell system (Fig. 9 A). Fits using values of εm = εp = 1 and 2 envelop the MD solutions, and the optimal choice was found to be εp = εc = 1.25 (Fig. 9 A). For the three-shell system, the correction for periodicity had a maximum amplitude of 0.03 kcal/mol. The spurious destabilization has decayed almost completely when the system size is doubled.
Correcting for the effect of the dielectric constant of the hydrocarbon chains leads to a further −2.1 kcal/mol stabilization of the ion in the center of the channel, as revealed in Fig. 9 B. The MD-averaged data were fitted with a Poisson solution using εp = εc = 1.75. To test the extent of the effect of polarizability of the phospholipid hydrocarbon tails on the stability of the ion, we previously carried out calculations using Drude oscillators (68) to describe the hydrocarbon chains (15). A similar approach using a grid of polarizable point-dipoles was used by Åqvist and Warshel in 1989 (69). In this case, the change in the stabilization of the ion was −3.6 ± 0.3 kcal/mol, which can be compared to our estimate ΔGdiel(0) from the Poisson solutions (−2.1 kcal/mol). The continuum electrostatics approximation captures the effect of the polarizability, but may underestimate its magnitude.
It is also possible to estimate the effect of high electrolyte concentration (70) by approximating the effect of reducing the concentration from 1 M to a level of 0.1 M that better corresponds to the single-ion regime
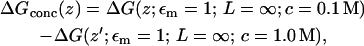 |
(5) |
using the Poisson-Boltzmann equation (see (70)). As expected, the effect of reducing the ionic concentration from 1 M to 0.1 M is a small additional stabilization of −0.2 kcal/mol at the channel center, as seen in Fig. 10 C.
FIGURE 10.

Corrections applied to the one-dimensional PMF to correct for simulation artifacts. (A) Poisson size correction (one shell of lipids → infinite bilayer); (B) Poisson membrane dielectric constant correction (εm = 1 → 2); (C) Poisson-Boltzmann concentration correction (1 M → 0.1 M). Data points represent calculations which are ensemble averages of Poisson solutions using a set of MD protein and channel-water coordinates. Short-dashed curves in panels A and B are corrections that use a single MD-averaged structure with the dielectric constant of the protein and channel water of 1. Long-dashed curves are corrections that assume the dielectric constant of the protein and channel water to be 2. The solid curves in panels A and B employ protein/channel-water dielectric constants of 1.25 and 1.75, respectively, as a fit to the calculated MD averages. In panel C, the dielectric constant was assumed to be 1.5.
Fig. 11 A shows the PMF for the small system after 1 ns of simulation per window with the CHARMM27 force field after corrections for periodicity, membrane dielectric constant, and high concentration (solid curve). The barrier is significantly lower, at ∼8.1 kcal/mol, with respect to the deepest point (the outer binding site). With this estimate of the free energy profile, we are in a position to compare the predictions of our calculations to experimental binding and conductance measurements.
FIGURE 11.

(A) One-dimensional CHARMM27 PMFs (from 1 ns simulation per window) before (dashed) and after (solid) artifact corrections. The dotted curve shows the corrected PMF with scaled hydrocarbon dielectric correction (see text) to be referred to as the “Drude-corrected” CHARMM27 PMF. (B) comparison of PMFs, before and after corrections, between small (20 lipids, red) and large (96 lipid, blue) membranes. The PMFs have been matched at z = 20 Å. PMFs from 2 ns/window simulation (not shown) experience barriers that are 0.7 kcal/mol less than the plotted 1 ns/window PMFs (with or without Drude-oscillator-scaled corrections).
For comparison we also show a corrected PMF with a scaled ΔGdiel correction in Fig. 11 A as a dotted curve. The correction has been scaled by a factor of 3.6/2.1 based on our calculations of the effect of lipid chain polarizability with the Drude oscillator model (and shall be referred to as the “Drude-corrected PMF”). The resulting PMF has a barrier that is reduced by a further 1.5 kcal/mol.
To test the accuracy of our size correction of Fig. 9 A, and to further evaluate the consistency and convergence of the results, we obtained a PMF with the larger three-shell membrane system. Fig. 11 B compares the one- and three-shell PMFs (using 1 ns of simulation in both cases) before corrections as dashed curves. The two PMFs differ by 1.6 kcal/mol at the channel center, a difference that is similar to the size correction for the one-shell system (Fig. 10). The solid curves in Fig. 11 B show our final results with all corrections (size, dielectric of the membrane, and concentration). The two PMFs now look very similar and differ on average by 0.28 kcal/mol. The correction obtained with Poisson solutions therefore accounts for reduced membrane size.
Channel binding and conductance
Experimental observables may be predicted from the calculated MD-PMFs to ascertain their agreement with electrophysiology. Here we outline the strategy by which we use the equilibrium free energy calculations to predict macroscopic observables and apply these methods using our corrected PMF.
Equilibrium dissociation constants
The equilibrium single-ion dissociation constant KD can be expressed either in terms of the three-dimensional one-ion three-dimensional PMF,  or in terms of the one-dimensional PMF, W(z), (see (53)) as long as the sampling of the lateral motion (i.e., with a cylindrical restraint) and corresponding offset constant are incorporated correctly (15). The dissociation constant for the channel (encompassed by −15 < z < 15 Å) from the corrected 1-ns PMF (Fig. 11) is 0.30 M. The 2 ns/window PMF for the CHARMM27 force field from Fig. 4 d, after artifact corrections (not shown) yields a dissociation coefficient of 0.21 M. This estimate is in within the range of experimental values determined from NMR and conductance studies: 0.017 M (71) (measured in dodecylphosphocholine micelles); 0.019–0.73 M (72) (in aqueous lysophosphatidylcholine dispersions); 0.035 M (54) (in glycerylmonoolein bilayers); and a recent estimate of 0.07 M (25) (in diphytanoylphosphatidylcholine/n-decane bilayers). When integrating over the regions defining the individual sites, ions will bind to the outer (10.2 < z < 12.5 Å) and inner (6.9 < z < 10.2 Å) sites with dissociation constants of 0.83 M and 3.6 M, respectively. Previous studies have found mixed results for the most stable position for a Na+ (29,73,74). Experimentally, the major cation binding site for both Na+ and K+ is the inner site (60,71). Significant K+ binding may also occur further from the channel center, as Tian and Cross (59) show that the Na+-induced chemical shift of Leu-10 to Trp-11 linkage exceeds that of Leu-12 to Trp-13, whereas the reverse is the case for K+.
or in terms of the one-dimensional PMF, W(z), (see (53)) as long as the sampling of the lateral motion (i.e., with a cylindrical restraint) and corresponding offset constant are incorporated correctly (15). The dissociation constant for the channel (encompassed by −15 < z < 15 Å) from the corrected 1-ns PMF (Fig. 11) is 0.30 M. The 2 ns/window PMF for the CHARMM27 force field from Fig. 4 d, after artifact corrections (not shown) yields a dissociation coefficient of 0.21 M. This estimate is in within the range of experimental values determined from NMR and conductance studies: 0.017 M (71) (measured in dodecylphosphocholine micelles); 0.019–0.73 M (72) (in aqueous lysophosphatidylcholine dispersions); 0.035 M (54) (in glycerylmonoolein bilayers); and a recent estimate of 0.07 M (25) (in diphytanoylphosphatidylcholine/n-decane bilayers). When integrating over the regions defining the individual sites, ions will bind to the outer (10.2 < z < 12.5 Å) and inner (6.9 < z < 10.2 Å) sites with dissociation constants of 0.83 M and 3.6 M, respectively. Previous studies have found mixed results for the most stable position for a Na+ (29,73,74). Experimentally, the major cation binding site for both Na+ and K+ is the inner site (60,71). Significant K+ binding may also occur further from the channel center, as Tian and Cross (59) show that the Na+-induced chemical shift of Leu-10 to Trp-11 linkage exceeds that of Leu-12 to Trp-13, whereas the reverse is the case for K+.
Channel conductance
To ascertain the magnitude of the current that can pass through the channel, the net stationary flux of ions across the channel can be calculated using a one-dimensional one-ion pore Nernst-Planck theory of Levitt (75) (see also (2) for application to MD-PMF). The ion flow is governed by the total PMF, Wtot, which can be expressed as a sum of the equilibrium PMF W, dominated by local molecular interactions, and the interaction of atomic charges with the transmembrane scalar potential, φmp; see Eqs. 38 and 39 in Roux (53). The one-ion pore NP theory of Levitt (75) provides a convenient route to quickly obtain a rough estimate of the maximum conductance given a one-dimensional PMF. There are alternative approaches that, however, must be considered with caution. For example, utilizing the one-dimensional MD-PMF in any three-dimensional model of permeation as done by Corry and Chung (76) requires additional assumptions and approximations about the width, the length, and the shape of the pore, as well as the absolute value of the free energy in order to construct a complete three-dimensional PMF (the ambiguities in such an operation are well illustrated by considering Fig. 5).
The charge distribution changes considerably as the ion moves, with most of the change being due to the dipole moment of the pore water (15). To gauge the contributions from the coupling to the system dipole moment to the trans-membrane potential, we express the total PMF as a cumulant expansion in the dipole moment of the system (see Eq. 36 in (53),
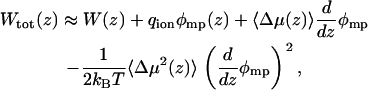 |
(6) |
where Δμ(z) is the deviation from the mean value of the dipole moment of the pore system as a function of the ion position. The value φmp is estimated by solving the modified Poison-Boltzmann equation (2,53) for an MD-averaged gA structure in a 25 Å membrane slab of dielectric constant 2, using 1 M KCl salt in the bulk regions with dielectric constant 80. Poisson-Boltzmann solutions for φmp, together with histograms of the dipole moment of the single-file water column obtained from all umbrella sampling trajectories (with symmetrization), have been used to estimate the perturbations to the equilibrium PMF of Eq. 6. The charge distribution within the pore region is represented by an expansion in the dipole moment, dominated by the single-file water column in Eq. 6, and the dielectric constant is set to 1 inside the protein and channel regions. We also evaluate 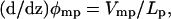 the value where Lp is the length of the pore region. The value Lp is equal to the diameter of the sphere defining the single-ion region (28 Å) and is within the range where the one-dimensional PMF is meaningful (|z| < 15 Å).
the value where Lp is the length of the pore region. The value Lp is equal to the diameter of the sphere defining the single-ion region (28 Å) and is within the range where the one-dimensional PMF is meaningful (|z| < 15 Å).
Conductance calculations are complicated by the coupling of the dipole to the membrane potential, with the linear term in Δμ(z) influencing the flow of ions and the quadratic term creating an additional barrier as a consequence of the bimodal nature of the dipole distribution in the bulk (15). Fig. 12 A shows the membrane potential term qionφmp of Eq. 6, increasing linearly across the channel. Fig. 12 B shows the first correction of Eq. 6 (due to the linear term in the dipole moment), and Fig. 12 C the second correction (due to the quadratic term). The sum of all three terms representing the coupling of the system charge distribution to the applied voltage is shown in Fig. 6 D (with curves from Fig. 6 A superimposed as thin curves). When 100 mV is applied across the membrane (solid curves in Fig. 12), the linear and quadratic dipole corrections have maximum amplitudes of only 0.15 and 0.04 kcal/mol, respectively. Because of the different voltage dependencies, the quadratic term becomes substantial at larger applied potentials. At 500 mV (dash-dot curves), the corrections approach 1 kcal/mol and, as the voltage applied across the membrane is increased, these corrections may have considerable effect on the conductance.
FIGURE 12.

Terms in the cumulant expansion Eq. 6. (A) The membrane potential for 100 mV (solid), 250 mV (dashed), and 500 mV (dash-dot) applied potential difference from solutions to the modified Poisson-Boltzmann equation with MD-averaged structure. The linear dipole (B) and quadratic dipole (C) cumulant corrections are shown in panels B and C, respectively. The sum of all terms for each voltage difference are plotted in panel D as thick curves. The thin curves superimposed in panel D are the qionφmp terms from panel A.
The above discussion is aimed at a correct and accurate computation of the channel conductance at the high potentials that are needed to do a full kinetic analysis based on current-concentration-voltage data (25,77). At lower voltages (comparable to the cell membrane potential), the effects of the coupling of the charge distribution to the membrane potential are small and may be neglected. Presently, we aim to gauge only the order-of-magnitude ability of MD to reproduce experimental observables and neglect these corrections in the following. With symmetric concentrations on both sides of the membrane and low membrane potential, the maximum conductance can be obtained directly from the one-dimensional PMF and a one-dimensional diffusion profile (78), assuming that the pore can be, at most, occupied by a single ion (it is a one-ion Nernst-Planck theory). This description provides only an order-of-magnitude estimate because we ignore multiple-ion occupancy at high concentration (73), as well as the effects of aqueous diffusion limitations (79), which should be valid in the limit of high concentrations.
The diffusion coefficient, D(z), required to calculate the flux may be estimated in different ways, where the general requirement is a method that separates the local dissipative forces from the systematic forces arising from the PMF. We employ a method based on the Laplace transform of the velocity autocorrelation function, using an analysis of the generalized Langevin equation for an harmonic oscillator (80), with the expression for the diffusion constant given in Crouzy et al. (81). To estimate the value of the limit as the Laplace transform variable s → 0, we linearly extrapolate from the range 15 ≤ s ≤ 35. Fig. 13 shows that the (symmetrized) axial ion diffusion coefficient D(z) is approximately two-thirds of bulk diffusion coefficient within the channel.
FIGURE 13.

K+ ion diffusion profile. Calculated values of the axial component of the ion diffusion coefficient, for each window simulation, are drawn with a solid line. All values have been symmetrized (unlike Fig. 6 in (15)) and scaled relative to the calculated bulk value of 0.37 Å2/ps. The fit (dashed line) is a sigmoidal function.
The maximum conductance based on the corrected (solid curve) PMF of Fig. 10 is 0.81 pS, 30-fold less than the experimental value of 21 pS (in DPhPC bilayers with 1 M KCl at 100 mV (82). The 2 ns/window PMF of Fig. 4 d (after artifact corrections, not shown) has a central barrier 0.7 kcal/mol less than the 1-ns/window PMF, and the gmax estimated using this 2 ns PMF is 1.54 pS, approaching order-of-magnitude agreement with experiment. The agreement is encouraging because the remaining discrepancy could be accounted for by only small changes/corrections to the PMF. The first correction to consider is associated with lipid acyl-chain polarizability. Because the Poisson correction for lipid chain dielectric constant may have been underestimated, based on calculations with a polarizable lipid chains, we consider a 1-ns PMF in which the dielectric correction scaled by a factor 3.6/2.1 (dotted curve in Fig. 11 A). This PMF has a barrier that is reduced by a further 1.5 kcal/mol, as compared to the corrected (solid curve) PMF of Fig. 10, and the predicted gmax = 5.1 pS. If we apply the same scaled correction to the 2-ns PMF (not shown), we obtain a maximum conductance of 9.3 pS, which is only twofold less than the experimental value.
The dissociation constant from the 1 ns/window PMF determined with this Drude-scaled correction is 0.21 M, in approximate agreement with experiment. Recalculating KD using the scaled-corrected 2 ns/window PMF yields a value of 0.14 M—approaching agreement with experimental estimates. Overall, these results suggest that, when all simulation artifacts are considered, the PMF may be within a kcal/mol of the true free energy profile for ion permeation through gA channels.
Investigating force fields and ion parameters
All biomolecular force fields are adjusted to reproduce the fundamental microscopic and thermodynamics properties of simple systems as accurately as possible (within the constraints of the mathematical functional form that is used). When using a force field, it is essential to avoid system-specific adjustments of the force-field parameters, as this would compromise attempts to gain mechanistic insights. Equally important, one should strive to identify the elementary (or physical) properties that would be expected to have a significant impact on a system of interest. For example, the energetics of ion permeation through the narrow gramicidin channel would be expected to be directly affected by the magnitude of ion-water and ion-peptide interactions, and indirectly by water-water, water-peptide, and peptide-peptide interactions. To ascertain and illustrate the sensitivity of the results to these different contributions, we repeated our PMF calculations with different commonly used biomolecular force fields. The three force fields we compare are CHARMM27, AMBER94, and the united-atom GROMOS87 force fields which have been used in several studies of ion permeation through the KcsA potassium channel; CHARMM27 (29,83–85), AMBER94 (86,87), and GROMOS87 (88–90). The version of the GROMOS force field implemented (GROMOS87) is that commonly employed in the GROMACS program (force-field ffgmx), which has been widely used in the aforementioned ion-channel simulations, despite the more recent improvements in that force field (e.g., Oostenbrink et al. (91)).
Table 2 includes a comparison of the gas phase interaction energies between a K+ and a single water (first column) or N-methyl-acetamide (NMA, second column) molecule with each force field, as well as experimental and ab initio calculations (variations on the CHARMM27 force field will be described below). All three force fields approximately reproduce the ion-water interaction, but only CHARMM27 and AMBER94 come close to reproducing the ion-NMA interaction energy. The magnitude of the interaction between an ion and a single NMA molecule with the GROMOS87 force field is 8.2 kcal/mol less than the lower limit of the ab initio result in magnitude.
TABLE 2.
Ion gas phase water and NMA interaction energies and liquid NMA free energies of charging (absolute values, referred to as “solvation free energies”) for different force fields
| K-water gas phase
|
K-NMA gas phase
|
K solvation free energy in liquid NMA
|
|
|---|---|---|---|
| Force field | (kcal/mol) | ||
| Experiment | 17.9 | 28.3–32.3 | – |
| Ab initio | 15.9–17.6 | 24.8–31.7 | – |
| CHARMM, σKO = 3.20 Å | 18.9 | 28.0 | 101.3 |
| CHARMM27+, σKO = 3.30 Å | 18.9 | 26.5 | 97.6 |
| CHARMM, σKO = 3.35 Å | 18.9 | 25.8 | 92.6 |
| CHARMM27, σKO = 3.46 Å | 18.9 | 24.2 | 89.2 |
| CHARMM27−, σKO = 3.69 Å | 18.9 | 21.6 | 82.0 |
| AMBER94 | 18.2 | 23.7 | 81.8 |
| GROMOS87 | 17.8 | 16.6 | 71.6 |
Results for CHARMM27, AMBER94, and GROMOS87 force fields, as well as variations on CHARMM27, where the K+-carbonyl O Lennard-Jones σ has been modified, are given. Values for gas phase interactions with water and NMA have been taken from Roux and Bernèche (98).
To further quantify how the force field represents the solvation of an ion by the protein backbone, we carried out FEP calculations on the charging of the ion in liquid NMA. Though the solvation free energy of K+ in bulk-NMA is not known experimentally, it is anticipated to be slightly more negative than the hydration free energy based on results with formamide (92).
For the solvation free energy computation, a sphere of 100 NMA molecules was simulated with the spherical solvent boundary potential method (39); the bulk solvent surrounding the sphere was represented by a uniform dielectric of 178.9 (93). The ion, fixed at the center, has its charge slowly turned off in a series of 11 windows after which the WHAM method is used to extract the free energy of charging. A total simulation time of 200 ps was used for each free energy calculation. A comparison of charging free energies is listed in Table 2. The standard CHARMM27 force field predicts a free energy of 89.2 kcal/mol, in comparison to AMBER94 and GROMOS87, which yield values of 81.8 and 71.6 kcal/mol, respectively.
Within limits it is possible to adjust the 6-12 Lennard-Jones parameter of the ion-carbonyl oxygen, without changing the atomic charge distribution, to reproduce the solvation free energy of K+ in liquid NMA. This method was used by Aqvist for the SPC water model (41), and by Roux and Beglov for the TIP3P water model (39), to develop the LJ parameters of the small ions. Such adjustments are justified because the additive σij = (σii + σjj)/2 or multiplicative 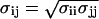 combination rules that are used to generate the atom-atom LJ pair parameters are very limited. In the case of CHARMM27 and AMBER94, such small empirical adjustment of the LJ parameters can be done without affecting the structure of the force fields. In the case of the GROMOS87, the situation is more difficult because the underestimated ion-carbonyl interactions are directly related to the atomic charge distribution of the protein backbone. Indeed, in comparison with CHARMM27 and AMBER94, the backbone partial charges with GROMOS87 are much smaller in magnitude: the C, O, N, and H charges are +0.51, −0.51, −0.47, and +0.31 e for CHARMM27; +0.60, −0.57, −0.42, and +0.27 e for AMBER94; and +0.38, −0.38, −0.28, and +0.28 e for GROMOS87. Therefore, it is not possible to obtain an acceptable solvation free energy of cations in liquid NMA using the GROMOS87 force field without modifying the atomic partial charges assigned to the backbone. Because of the significantly underestimated ion-carbonyl interactions, one would anticipate that the GROMOS87 force field will yield a larger barrier in the PMF.
combination rules that are used to generate the atom-atom LJ pair parameters are very limited. In the case of CHARMM27 and AMBER94, such small empirical adjustment of the LJ parameters can be done without affecting the structure of the force fields. In the case of the GROMOS87, the situation is more difficult because the underestimated ion-carbonyl interactions are directly related to the atomic charge distribution of the protein backbone. Indeed, in comparison with CHARMM27 and AMBER94, the backbone partial charges with GROMOS87 are much smaller in magnitude: the C, O, N, and H charges are +0.51, −0.51, −0.47, and +0.31 e for CHARMM27; +0.60, −0.57, −0.42, and +0.27 e for AMBER94; and +0.38, −0.38, −0.28, and +0.28 e for GROMOS87. Therefore, it is not possible to obtain an acceptable solvation free energy of cations in liquid NMA using the GROMOS87 force field without modifying the atomic partial charges assigned to the backbone. Because of the significantly underestimated ion-carbonyl interactions, one would anticipate that the GROMOS87 force field will yield a larger barrier in the PMF.
We have previously shown, through mean force decomposition, that within the channel the protein is responsible for up to half of the ion's solvation free energy (15). Based on the above liquid NMA calculations, we anticipate that a PMF of ion permeation obtained with the GROMOS87 force field will have a barrier that is up to 5 kcal/mol higher than that obtained with AMBER94.
Fig. 14 compares the PMFs calculated with CHARMM27, AMBER94, and the united-atom GROMOS87 force fields. The results are fairly consistent for the all-atom force fields of AMBER94 and CHARMM27, possessing similar barrier heights, though there are small differences in the PMF shapes (on average 1.0 kcal/mol). In particular the binding sites have been shifted upward with the AMBER94 force field and the inner binding site is less distinct than in the CHARMM27 PMF. Overall, however, the two force fields show a similar stabilization of ions by the channel protein. Table 3 lists dissociation constants and maximum conductances for CHARMM27, AMBER94, and GROMOS87 force fields (as well as PMFs calculated using various modifications to CHARMM27, to be discussed below). The AMBER94 maximum conductance is 6.9 pS, comparable to the experimental value; the dissociation constant, however, is much too high, indicative of the weak binding/solvation predicted with this force field. When comparing the observables predicted using AMBER94 and CHARMM27, AMBER94 predicts gmax and KD to be eightfold higher than the values predicted using CHARMM27. The opposite trends in the predicted conductances and ion affinities demonstrate the delicate counterbalance between conductance and binding when predicting experimental observables.
FIGURE 14.

One-dimensional PMFs using the CHARMM27, AMBER94, and GROMOS87 force fields. All PMFs have been calculated from 1-ns/window simulations and have been corrected for simulation artifacts.
TABLE 3.
Calculated observables for different force fields and parameters
| gmax (pS) | KD (mol/L) | |
|---|---|---|
| Experiment | 21* | 0.017 M,† 0.019−0.73 M‡ |
| 0.035 M,§ 0.07 ± 0.01 M¶ | ||
| 2 ns/window | ||
| CHARMM27 | 1.54 | 0.21 |
| Drude-corrected CHARMM27 | 9.27 | 0.14 |
| 1 ns/window | ||
| CHARMM27 | 0.81 | 0.30 |
| Drude-corrected CHARMM27 | 5.1 | 0.21 |
| CHARMM27− | 0.026 | 5.8 |
| CHARMM27+ | 0.54 | 0.0015 |
| AMBER94 | 6.9 | 2.4 |
| Drude-corrected AMBER94 | 28.1 | 1.6 |
| GROMOS87 | 0.0069 | 2.8 |
| Drude-corrected GROMOS87 | 0.050 | 2.6 |
Maximum conductances and dissociation constants are shown for CHARMM27, AMBER94, and GROMOS87 force fields, as well as variations of CHARMM27 described in Table 2. Because not all PMFs are defined out to z = ±30 Å, the reference for the calculation of KD was chosen such that W(20) = 0, instead of the previous convention: W(30) = 0 (15). Also, previously binding to the channel was measured from −12.5 < z < 12.5 Å, but to allow for some PMFs having very deep and broad binding sites, binding must be calculated in a greater range of −15 < z < 15 Å (for all force fields to allow for comparison). The “Drude-corrected” entries correspond to the corrected PMF with scaled dielectric correction (for CHARMM27 with 1 ns/window, this is the dotted curve of Fig. 11). See footnotes for experimental measurements.
Busath et al. (82).
Jing et al. (71).
Hinton et al. (72).
Thompson et al. (54).
Andersen et al. (25).
The GROMOS87 PMF in Fig. 14 is very different from that obtained with the other force fields with a central barrier that is ∼5 kcal/mol higher. This was to be expected based on the reduced protein polarity, and this force field's inability to produce reasonable liquid NMA solvation free energies. The maximum conductance for this PMF, from Table 3, is almost four orders-of-magnitude too small. Similarly, the ion binding sites are almost nonexistent, with a dissociation constant of 2.8 M. This large deviation from experiment shows that GROMOS87 is unable to describe ion permeation in this channel.
Both AMBER94 and CHARMM27 predict observables that are in semi-quantitative agreement with experimental results. The different shapes of the PMFs, and the opposite trends in the deviations between predicted observables and experimental results show that the results of MD simulations are quite sensitive to the choice of force field. The CHARMM27 (using the Drude-scaled dielectric correction) predicts results to within an order-of-magnitude of experimental conductance and binding data. Similar improvements are also obtained with AMBER94 (as seen in Table 3), but there is a limit to how close to experiment one may come with any traditional fixed-charge force field. One factor determining the shape of the PMF (including the relative depth of the binding sites), is the strength of the ion-protein interaction, where there is particular uncertainty about the ion-protein Lennard-Jones parameter (94). Whereas data for liquid amides suggest that the free energy of solvation of K+ in liquid NMA is similar to the free energy of hydration of K+ in liquid water (92), the force field also should reproduce (approximately) the gas phase interaction of the ion with NMA. These two values are difficult to reproduce simultaneously using a nonpolarizable force field (94,95). Without the nonadditive many-body interactions arising from induced polarization effects, the overall stabilization provided by a coordination shell of carbonyls surrounding a cation will be overestimated by a parameterization that matches gas-phase ion-NMA interaction (73,95). In a nonpolarizable force field, a compromise between local and global energetics is achieved through the parameterization of the ion-oxygen Lennard-Jones 6-12 interaction.
To explore the effects of small changes in the Lennard-Jones parameters on these fixed-charge force field results, we carried out simulations with modified CHARMM27 parameters (Table 2). The CHARMM27 force field gives a K+-liquid NMA free energy ∼7 kcal/mol more negative than the K+-bulk water free energy and gives a gas phase interaction with NMA slightly less than the lower bound of the ab initio results. To explore the sensitivity of permeation energetics to precise ion parameterizations, and thus to investigate the robustness of the force fields, we chose two different modifications of CHARMM27 to compute new PMFs. The first, called CHARMM27−, where the − symbol denotes weaker ion-protein interaction, has an increased K+-O Lennard-Jones radius, σ, leading to a liquid NMA charging free energy similar to that of bulk water. (This is also similar to the modification used in studies of KcsA permeation (83).) The second, called CHARMM27+, where the + symbol denotes a stronger ion-protein interaction, has a reduced K+-O Lennard-Jones radius, to give a gas phase interaction energy within the ab initio range—and nearer the lower bound seen experimentally.
The PMF calculated using the CHARMM27 + force field is shown as a dashed curve in Fig. 15. Because the parameter was chosen to reproduce the K+-NMA interaction energy in gas phase, the central barrier in the PMF is reduced and the depths of the binding sites increased. The central barrier height is only a few kcal/mol above the channel entrances but remains at ∼8 kcal/mol with respect to the binding sites. With this force field there is a broad deep free energy well at the channel entrance and the inner and outer binding sites are of similar depth. In fact, this was predicted based on the increased involvement of the protein carbonyls in solvating the ion at the inner binding site (15). The CHARMM27− force field is shown as a dash-dot curve in Fig. 15. The central barrier has increased dramatically, to almost 11 kcal/mol because σ has been modified to give a K+ charging free energy in liquid NMA similar to that in bulk water. Moreover, the binding sites have almost vanished. We conclude that, calculated, the PMFs are remarkably sensitive to very small changes in the MD force field.
FIGURE 15.

One-dimensional PMFs using the CHARMM27 force field and variations CHARMM27+ and CHARMM27−. The dashed curve uses the CHARMM27+ force field (as defined in Table 2 with modification to K+-O Lennard-Jones interaction). The dash-dot curve uses the CHARMM27− force field, also defined in Table 2. All PMFs have been calculated from 1-ns/window simulations and have been corrected for simulation artifacts.
When computing the maximum conductances and dissociation constants using these two variations of the force field parameters (Table 3), the CHARMM27+ PMF leads to a lower gmax than the unmodified CHARMM27 PMF, and a dissociation constant that is at least an order-of-magnitude too low. Using the CHARMM27− PMF there is almost no binding and the conductance is three orders-of-magnitude too low. The unmodified CHARMM27 (and to a similar extent AMBER94) force fields strike a good compromise between gas and liquid phase properties and allow for the best predictions of gA ion permeation.
DISCUSSION AND CONCLUSION
We have shown that it is possible to use MD-PMF simulations to reliably predict experimental observables with semiquantitative accuracy. We could do so by implementing the procedures required to obtain a well-defined PMF in one and two dimensions. These procedures include: the creation of a realistic ion channel-membrane system; application of force field potentials that accurately describe the interactions between ions, water, protein, and phospholipids; the choice of reaction coordinate that adequately captures the permeation process; and the construction of a conduction model consistent with the definition of the free energy surface. The key step was the construction of a two-dimensional PMF, as a function of both axial and radial coordinates, to characterize the free energy surface governing the permeation of K+ through a gA channel embedded in a phospholipid bilayer—with the correct bulk reference value. This free energy surface reveals the details of the equilibrium distribution of ions inside and around the ion channel, including the height of the central barrier and the depths and positions of the K+ binding sites. Of particular interest is that, when we recalculated the PMF in the instantaneous channel CoM-axis frame, we saw that the ion is binding off-axis when in the outer binding site. The prediction of two cation binding site, at the entrance of the channel is in accord with previous experimental results (59,60), as well as previous MD simulations (29,74). There is divergence among the experimental results, however, with the x-ray scattering results of Olah et al. (62), predicting the predominant K+ binding to occur deeper within the channel (at our inner site), whereas a preferred outer-site binding would be inferred from the solid-state NMR results of Tian and Cross (61). So, even though we find that K+ preferentially binds to the outer site, in agreement with the analysis of solid state NMR data (59), it remains unclear whether this, in fact, is the preferred K+ binding site.
To validate the absolute energy scale deduced from the two-dimensional PMF, we performed free energy perturbation computational alchemy, in which a K+ ion in the center of the channel alchemically was transformed into a water molecule. The two-dimensional PMF could be converted into a one-dimensional PMF by integrating over lateral displacements, with attention to sampling of x,y ionic coordinates in the bulk. We have demonstrated that this one-dimensional PMF allows for the prediction of rates of ion permeation and energetics of ion binding—for comparison with experimental flux and binding measurement.
A perennial concern in MD simulations is whether the configurational sampling is adequate. To assess this issue, we compared the PMFs obtained based on different simulation times (50 ps to 2 ns for each window). This analysis revealed that the PMF was highly asymmetric for short (50–100 ps) simulation times, and that the asymmetry was reduced to <1 kcal/mol after 2 ns/window. This asymmetry corresponds to a very small force across the length of the channel which can be avoided by symmetrization. The symmetrized PMFs over 1-and 2-ns simulation times, were converged (on average) to within 0.3 kcal/mol (Fig. 4 D). Given this convergence, it was possible to investigate the effects of a range of different simulation parameters on the PMF using only 1 ns simulation per window. The large asymmetry and variation in central barrier height obtained with short simulations demonstrate that previous simulations using a set of short (<100 ps) nonsymmetrized PMFs (30,32) may have considerable uncertainty. As a comparison, we investigated PMFs obtained by sequential sampling of windows across the channel, e.g., the dragging procedure used in the aforementioned articles, by calculating PMFs (using CHARMM27) from −10 to +10 Å with 80-ps sampling sequentially in each 0.2 Å window and then from +10 to −10 Å (T.W. Allen, O.S. Andersen, and B. Roux, unpublished results). These forward and reverse PMFs were different in shape and quite asymmetrical. With short simulation times it is no surprise that the two PMFs were different, but in both cases the free energy increased as the PMF progressed from one side of the channel to the other, to yield a hysteresis of ∼8 kcal/mol and a difference in barrier height of ∼7 kcal/mol. The more robust approach is to place the ions along the reaction coordinate and simulate for at least 1 ns per window.
Though encouraging, the PMF from the 2-ns simulations still has a barrier of 7 kcal/mol with respect to the bulk solution (based on the two-dimensional PMF), which is several kcal/mol too high to be compatible with measured rates of ion movement. We identified the physical basis for most of this discrepancy, namely the systematic artifacts that arise in the simulation of finite nonpolarizable membrane systems, and implemented reasonable procedures to correct for these artifacts. We confirmed our continuum-based size correction by recalculating the PMF with a larger (three-shell) membrane system. Before correcting for the simulation artifacts, the two PMFs differed by as much as 1.8 kcal/mol; after correction, the agreement between the small and large systems differed on average by just 0.3 kcal/mol after 1 ns of simulation. We further tested the hydrocarbon dielectric corrections using calculations with a polarizable force field and found that the continuum electrostatic approximation, though capturing the effect of lipid polarizability, may have underestimated the magnitude of the effect.
We conclude that an appropriately calculated and corrected PMF allows for predictions that are in order-of-magnitude agreement with experimental binding and conductance measurements. Using our best estimate, the 2 ns/window Drude-corrected PMF, we obtain agreement with experimental conductance and binding to within a factor of 2. We further conclude that the nonpolarizable CHARMM27 force field, as well as the AMBER94 force field, does a surprisingly good job of modeling ions within narrow pores. But the permeation model used to extrapolate from equilibrium PMF to observable mean conductance is approximate. It uses a one-ion PMF to predict the conductance at saturating concentrations, where the channel would be occupied by more than one ion. Furthermore, whereas the magnitude of the predicted conductance is determined primarily by the free energy barrier in the PMF, dissipative factors such as the ion mobility within the channel also have a direct impact. The calculated diffusion coefficient within the pore is approximately two-thirds of the bulk diffusion coefficient, being four-to-fivefold larger than would be predicted for a single-file column of ion and water moving through the channel as a single cooperative unit (96). The diffusion coefficient would be higher if the cooperative unit was smaller than the complete single file, as suggested by our finding of small gaps in the water column, but the result suggests an uncertainty in our estimate of the diffusion coefficient—and possibly an overestimate of the channel conductance, which still would remain of the same order of magnitude.
When comparing different force fields, AMBER94 yields a PMF that is fairly similar to that obtained using CHARMM27, demonstrating overall consistency among modern all-atom force fields used in MD. There are, nonetheless, differences between the PMFs determined using AMBER94 and CHARMM27, which reflect subtle differences in the protein force fields. Overall, the two force fields are equivalent, insofar as the improved prediction of gmax using AMBER94, as compared to the prediction using CHARMM27, is matched by a worse prediction of KD. Application of the Drude-scaled dielectric correction (see Table 3) does, however, tend to suggest that CHARMM27 provides the most balanced force field for studying permeation through this narrow ion channel. The total barrier for ion movement is expected to be well correlated with the relative solvation in water and liquid NMA. But the fine structure of the free energy profile through gA will depend also on the strength of the β-helix hydrogen-bonds and the backbone φ-ψ dihedral potential terms, which are different between AMBER94 and CHARMM27 force fields; e.g., AMBER94 has a tendency to favor α-helical conformations relative to CHARMM27 (97).
In contrast, the GROMOS87 united-atom force field yields predictions that were in rather severe disagreement with both ion binding and conductance measurements, which can be traced to the low polarity of the protein backbone in that force field. We conclude that widely used GROMOS87 appears to be unsuitable for studies of ion permeation through narrow ion channels One should note that the most recent all-atom GROMOS force field version 53A of Oostenbrink et al. (91) probably does not suffer from these problems.
Though AMBER94 and CHARMM27 can approximately reproduce both experimental binding and maximum conductance measurements, there is a limit to how close to experiment one can come within this framework—as evident also in the differences between the PMFs calculated using AMBER94 and CHARMM27. To analyze the roles of force-field parameters in the PMF calculation, we explored the effect of CHARMM27 force-field modifications that have direct impact on bulk solvation and gas phase interaction energies of a K+ ion with NMA molecules. The PMF and macroscopic observables are quite sensitive to small changes in the K+-O Lennard-Jones σ; a parameter that has been varied in previous studies. Changes on the order of one-tenth of an Å are sufficient to destroy the order-of-magnitude agreement with experiment. It is in this context also important that simple adjustments of the force field provide little, if any, improvement in the PMF—in terms of improved ability to predict experimental observables, whereas significant improvement was obtained by addressing the physical limitations inherent in the standard implementations of MD, the neglected system size, and acyl-chain electronic polarizability (Fig. 11 A, Table 3).
What needs to be done to improve the agreement with experiment beyond what is possible using the unmodified CHARMM27 force fields? Because the best estimate for a system-artifact-corrected PMF predicts ion conductances and binding constants that are within an order of magnitude of experimental values, the PMF is likely to be correct within ∼1 kcal/mol of the true free energy profile for permeation. Protein and water polarizability would be expected to play some role in ion permeation through a narrow pore, and neglect of polarizability is likely to account for most of the remaining discrepancies. It is not sufficient, however, to just introduce polarizability—except in the very special case of the lipid acyl chains—because the force fields have been calibrated to reproduce a wide variety of experimental results, that each in their own way depend on interactions between polarizable atoms. It will be necessary to develop, and calibrate, a new set of force fields de novo. Previously, we have shown, through mean force decomposition, that the single file of water inside the channel plays a surprisingly large role in stabilizing the ion; accounting for almost half of the free energy of solvation of the ion deep within the channel (15). In this region, the channel water is approximately twice as important as the protein in terms of stabilizing the ion, −38 kcal/mol as compared to −19 kcal/mol (15). It thus becomes important to ascertain, what is the role of water polarizability inside the channel? This question was explored previously (15), in calculations of the charging free energy of a K+ in a model single-file column of water surrounded by a cavity reaction field arising from bulk water. FEP calculations were done with the nonpolarizable TIP3P water model (38) as well as the Drude oscillator-based polarizable water model SWM4-DP (68). These calculations showed that the role of water polarizability is of the order of 0.1−0.5 kcal/mol, indicating that the nonpolarizable TIP3P water model adequately describes two extremes of ion solvation: by the bulk water; and by the anisotropic single-file water. But, while the effect of polarizability may be small, the predicted channel properties are extremely sensitive to small changes in the PMF. Reproducing quantitatively the local interaction of a cation with a carbonyl group (related to the gas phase ion-NMA complex) as well as the global magnitude of the free energy barrier through gA (related to bulk ion solvation in liquid NMA) will require taking induced nonadditive many-body polarization effects into account (95). When the next generation of polarizable force field is developed, one will also need to pay careful attention to the solvation free energy of ions in water and also in various organic liquids (amides, alcohols, etc…)—in addition to the gas phase interaction energies. It is in this relevant context that variations in the nonpolarizable CHARMM27 force field (CHARMM27− and CHARMM27+) did not improve the predictions, whereas introducing Drude-oscillator-based lipid acyl-chain polarizability did so.
The moderately optimistic conclusions of this study, pertaining to the use of MD-PMF simulations to reliably predict experimental observables are encouraging, because MD remains the only viable method for calculating the forces acting on an ion in a narrow pore undergoing thermal fluctuations (3). Previous attempts to validate the use of MD-PMF as a tool to predict experimental observables (29,30,32) were less successful than this study. The three key elements underlying the different outcomes and assessments are:
Attention to the convergence of the PMF calculations, which was possible by the availability of more powerful computational resources.
Attention to the proper referencing of the PMFs relative to the bulk solution.
The identification of, and correction for, artifacts that would introduce systematic errors into any MD-PMF-based predictions.
Taken together, these elements ensure that it is possible to compare with experimental data by using the one-ion PMF to predict KD and gmax. The conductances reported here are, however, the extrapolated gmax values in the limit of very high ion concentration (>10 KD); we do not consider the impact of double-ion occupancy, which will be nontrivial (25,73). This becomes important when comparing the predictions of different permeation models. Indeed, any comparison among permeation models can be meaningful only when all efforts are made to ensure that the different approaches are describing the same physical situation. Employing a PMF out of context (with respect to dimensionality, pore occupancy, concentration regime, or timescale) would be expected to lead to questionable predictions.
There is reason to be optimistic about the use of all-atom MD-PMF as a tool to understand protein function, as evidenced by the convergence and consistency of the PMF obtained in this study. Despite the limitations of the conduction model (the neglect of double-ion occupancy and uncertainty about the precise value of the diffusion coefficient), computational studies on gA ion permeation constitutes a suitable system for critically evaluating these approaches to understand ion conduction through narrow pores, which remains one of the most challenging of problems in computational biophysics.
Acknowledgments
Note added in proof: We have performed calculations to demonstrate further the robustness of the PMF calculations reported here since acceptance. Regarding the application of a spherical constraint to both cations and anions to maintain a 1-ion pore, we have repeated the PMF calculation with only a constraint on cations (other than the one in question), leaving anions unbiased, and found a very similar PMF with inner and outer binding sites of unaltered depths (though a small shift toward the channel center of the inner site by 0.3–0.4 Å, remaining in agreement with the position determined experimentally). We note that when the PMF was recalculated without any Umbrella sampling bias, but holding an ion inside the range 7.5–13.5 Å, the inner and outer binding site positions and depths were approximately reproduced. We have also recalculated the PMF without the lateral restraint that is used to provide a well-defined region in the bulk for the ion to occupy, and found no noticeable changes in the PMF within the pore, with the PMF dropping steadily as the ion departs the channel, as expected.
The National Center for Supercomputing Applications Origin supercomputer was used for some reported large-membrane unbiased simulations before the PMF calculations.
This work was supported by the Revson and Keck Foundations (to T.W.A.) and National Institutes of Health grants No. GM21342 (to O.S.A.), No. GM62342 (to B.R.), and No. GM70791 (to O.S.A. and B.R).
Benoit Roux's present address is Institute of Molecular Pediatric Sciences Center for Integrative Science, University of Chicago, Chicago, IL.
References
- 1.Karplus, M. 2002. Molecular dynamics simulations of biomolecules. Acc. Chem. Res. 35:321–323. [DOI] [PubMed] [Google Scholar]
- 2.Roux, B., T. Allen, S. Bernèche, and W. Im. 2004. Theoretical and computational models of biological ion channels. Q. Rev. Biophys. 37:15–103. [DOI] [PubMed] [Google Scholar]
- 3.Allen, T. W., O. S. Andersen, and B. Roux. 2004. On the importance of flexibility in studies of ion permeation. J. Gen. Physiol. 124:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindorff-Larsen, K., R. Best, M. Depristo, C. Dobson, and M. Vendruscolo. 2005. Simultaneous determination of protein structure and dynamics. Nature. 433:128–132. [DOI] [PubMed] [Google Scholar]
- 5.Karplus, M., and J. McCammon. 1981. The internal dynamics of globular proteins. CRC Crit. Rev. Biochem. 9:293–349. [DOI] [PubMed] [Google Scholar]
- 6.Karplus, M., and G. Petsko. 1990. Molecular dynamics simulations in biology. Nature. 347:631–639. [DOI] [PubMed] [Google Scholar]
- 7.Mamonov, A. B., R. D. Coalson, A. Nitzan, and M. G. Kurnikova. 2003. The role of the dielectric barrier in narrow biological channels: a novel composite approach to modeling single-channel currents. Biophys. J. 84:3646–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurnikova, M., R. Coalson, P. Graf, and A. Nitzan. 1999. A lattice relaxation algorithm for three-dimensional Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys. J. 76:642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadler, B., U. Hollerbach, and R. S. Eisenberg. 2003. Dielectric boundary force and its crucial role in gramicidin. Phys. Rev. E. 68:021905. [DOI] [PubMed] [Google Scholar]
- 10.Edwards, S., B. Corry, S. Kuyucak, and S.-H. Chung. 2002. Continuum electrostatics fails to describe ion permeation in the gramicidin channel. Biophys. J. 83:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mashl, R., Y. Tang, J. Schnitzer, and E. Jakobsson. 2001. Hierarchical approach to predicting permeation in ion channels. Biophys. J. 81:2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung, S. H., T. Allen, and S. Kuyucak. 2002. Conducting state properties of the KcsA potassium channel from molecular and Brownian dynamics simulations. Biophys. J. 82:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorman, V. L., and P. C. Jordan. 2004. Ionic permeation free energy in gramicidin: a semi-microscopic perspective. Biophys. J. 86:3529–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernèche, S., and B. Roux. 2003. A microscopic view of ion conduction through the KcsA K+ channel. Proc. Natl. Acad. Sci. USA. 100:8644–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urry, D. W. 1971. The gramicidin A transmembrane channel: a proposed πLD helix. Proc. Natl. Acad. Sci. USA. 68:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arseniev, A. S., A. L. Lomize, I. L. Barsukov, and V. F. Bystrov. 1986. Gramicidin A transmembrane ion-channel three-dimensional structure reconstruction based on NMR spectroscopy and energy refinement. (In Russian.). Biol. Membr. 3:1077–1104. [Google Scholar]
- 18.Townsley, L. E., W. A. Tucker, S. Sham, and J. F. Hinton. 2001. Structures of gramicidins A, B, and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 40:11676–11686. [DOI] [PubMed] [Google Scholar]
- 19.Ketchem, R. R., B. Roux, and T. A. Cross. 1997. High resolution refinement of a solid-state NMR-derived structure of gramicidin A in a lipid bilayer environment. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- 20.Allen, T. W., O. S. Andersen, and B. Roux. 2003. The structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 125:9868–9877. [DOI] [PubMed] [Google Scholar]
- 21.Hladky, S. B., B. Urban, and D. Haydon. 1979. Ion movements in pores formed by gramicidin A. Membr. Transport Processes. 3:89–103. [Google Scholar]
- 22.Eisenman, G., and R. Horn. 1983. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. J. Membr. Biol. 76:197–225. [DOI] [PubMed] [Google Scholar]
- 23.Andersen, O., and R. Koeppe. 1992. Molecular determinants of channel function. Physiol. Rev. 72:S89–S158. [DOI] [PubMed] [Google Scholar]
- 24.Busath, D. 1993. The use of physical methods in determining gramicidin channel structure and function. Annu. Rev. Physiol. 55:473–501. [DOI] [PubMed] [Google Scholar]
- 25.Andersen, O. S., R. E. Koeppe II, and B. Roux. 2005. Gramicidin channels. IEEE Trans. Nanobiosci. 4:10–20. [DOI] [PubMed] [Google Scholar]
- 26.Faraldo-Gomez, J. D., L. R. Forrest, M. Baaden, P. J. Bond, C. Domene, G. Patargias, J. Cuthbertson, and M. S. P. Sansom. 2004. Conformational sampling and dynamics of membrane proteins from 10-nanosecond computer simulations. Proteins. 57:783–791. [DOI] [PubMed] [Google Scholar]
- 27.Mackay, D. H. J., P. H. Berens, K. R. Wilson, and A. T. Hagler. 1984. Structure and dynamics of ion transport through gramicidin A. Biophys. J. 46:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roux, B. 2002. Computational studies of the gramicidin channel. Acc. Chem. Res. 35:366–375. [DOI] [PubMed] [Google Scholar]
- 29.Roux, B., and M. Karplus. 1993. Ion transport in the gramicidin channel: free energy of the solvated right-handed dimer in a model membrane. J. Am. Chem. Soc. 115:3250–3262. [Google Scholar]
- 30.Allen, T. W., T. Bastug, S. Kuyucak, and S. H. Chung. 2003. Gramicidin-A channel as a test ground for molecular dynamics force fields. Biophys. J. 84:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan, P. C. 1987. Microscopic approach to ion transport through transmembrane channels. The model system gramicidin. J. Phys. Chem. 91:6582–6591. [Google Scholar]
- 32.Batug, T., and S. Kuyucak. 2005. Test of molecular dynamics force fields in gramicidin A. Eur. Biophys. J. 34:377–382. [DOI] [PubMed] [Google Scholar]
- 33.Andersen, O. S. 1984. Gramicidin channels. Annu. Rev. Physiol. 46:531–548. [DOI] [PubMed] [Google Scholar]
- 34.Chandler, D. 1978. Statistical mechanics of isomerization dynamics in liquids and the transition state approximation. J. Chem. Phys. 68:2959–2970. [Google Scholar]
- 35.Hinsen, K., and B. Roux. 1997. Potential of mean force and reaction rates for proton transfer in acetylacetone. J. Chem. Phys. 106:3567–3577. [Google Scholar]
- 36.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 37.MacKerell, A. D., Jr., D. Bashford, M. Bellot, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, B. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 39.Beglov, D., and B. Roux. 1994. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 100:9050–9063. [Google Scholar]
- 40.Cornell, W., P. Cieplak, C. Bayly, I. Gould, K. Merz, Jr., D. Ferguson, D. Spellmeyer, T. Fox, J. Caldwell, and P. Kollman. 1995. A second generation force field for the simulation of proteins and nucleic acids. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 41.Åqvist, J. 1990. Ion water interaction potential derived from free energy perturbation simulations. J. Phys. Chem. 94:8021–8024. [Google Scholar]
- 42.van Gunsteren, W., X. Daura, and A. Mark. 1999. GROMOS force field. In Encyclopaedia of Computational Chemistry, Vol. 2. E.-I.-C. P. von Ragu Schelyer, editor. John Wiley & Sons, London. 1211–1216.
- 43.Berendsen, H., J. Postma, W. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to proteins hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands. 331–342.
- 44.Straatsma, T. P., and H. J. C. Berendsen. 1988. Free energy of ionic hydration: analysis of a thermodynamic intergration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys. 89:5876–5886. [Google Scholar]
- 45.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second-generation force field for the simulation of proteins and nucleic acids. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 46.Schlenkrich, M., J. Brickmann, A. J. MacKerell, and M. Karplus. 1996. An empirical potential energy function for phospholipids: criteria for parameters optimization and applications. In Biological Membranes. A Molecular Perspective from Computation and Experiment. K. Merz, and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- 47.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 48.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equation of motions of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Chem. 23:327–341. [Google Scholar]
- 49.Feller, S. E., Y. H. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- 50.Woolf, T., and B. Roux. 1996. Structure, energetics and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins Struct. Funct. Genet. 24:92–114. [DOI] [PubMed] [Google Scholar]
- 51.Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- 52.Schatzberg, P. 1963. Solubilities of water in several normal alkanes from c7 to c16. J. Phys. Chem. 67:776–779. [Google Scholar]
- 53.Roux, B. 1999. Statistical mechanical equilibrium theory of selective ion channels. Biophys. J. 77:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson, N., G. Thompson, C. D. Cole, M. Cotten, T. A. Cross, and D. D. Busath. 2001. Noncontact dipole effects on channel permeation. IV. Kinetic model of 5F-Trp13 gramicidin A currents. Biophys. J. 81:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roux, B. 1997. The influence of the membrane potential on the free energy of an intrinsic protein. Biophys. J. 73:2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torrie, G. M., and J. P. Valleau. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23:187–199. [Google Scholar]
- 57.Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13:1011–1021. [Google Scholar]
- 58.Parsegian, A. 1969. Energy of an ion crossing a low dielectric membrane: solution to four relevant electrostatic problems. Nature. 221:844–846. [DOI] [PubMed] [Google Scholar]
- 59.Tian, F., and T. Cross. 1999. Cation transport: an example of structural based selectivity. J. Mol. Biol. 285:1993–2003. [DOI] [PubMed] [Google Scholar]
- 60.Olah, G. A., H. W. Huang, W. Liu, and Y. Wu. 1991. Location of ion-binding sites in the gramicidin channel by x-ray diffraction. J. Mol. Biol. 218:847–858. [DOI] [PubMed] [Google Scholar]
- 61.Finkelstein, A., and O. S. Andersen. 1981. The gramicidin A channel: a review of its permeability characteristics with special reference to the single-file aspect of transport. J. Membr. Biol. 59:155–171. [DOI] [PubMed] [Google Scholar]
- 62.Stern, H. A., and S. E. Feller. 2003. Calculation of the dielectric permittivity profile for a nonuniform system: application to a lipid bilayer simulation. J. Chem. Phys. 118:3401–3412. [Google Scholar]
- 63.Huang, W., and D. G. Levitt. 1977. Theoretical calculation of the dielectric constant of a bilayer membrane. Biophys. J. 17:111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simon, S. A., and T. J. McIntosh. 1986. Depth of water penetration into bilayers. Methods Enzymol. 127:511–521. [DOI] [PubMed] [Google Scholar]
- 65.Lide, D. R. (Editor-in-Chief). 1992. CRC Handbook of Chemistry and Physics, 72nd Ed. CRC Press, Boston, MA.
- 66.Hunenberger, P. H., and J. A. McCammon. 1999. Ewald artifacts in computer simulations of ionic solvation and ion-ion interaction: a continuum electrostatics study. J. Chem. Phys. 110:1856–1872. [Google Scholar]
- 67.Lewis, B. A., and D. M. Engelman. 1983. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166:211–217. [DOI] [PubMed] [Google Scholar]
- 68.Lamoureux, G., A. D. MacKerell Jr., and B. Roux. 2003. A simple polarizable model of water based on classical drude oscillators. J. Chem. Phys. 119:5185–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Åqvist, J., and A. Warshel. 1989. Energetics of ion permeation through membrane channels. Solvation of Na+ by gramicidin A. Biophys. J. 56:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jordan, P. C., R. J. Bacquet, J. A. McCammon, and P. Tran. 1989. How electrolyte shielding influences the electrical potential in transmembrane ion channels. Biophys. J. 55:1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jing, N., K. U. Prasad, and D. W. Urry. 1995. The determination of binding constants of micellar-packaged gramicidin A by 13C-and 23Na-NMR. Biochim. Biophys. Acta. 1238:1–11. [DOI] [PubMed] [Google Scholar]
- 72.Hinton, J. F., W. L. Whaley, D. C. Shungu, R. E. Koeppe II, and F. S. Millett. 1986. Equilibrium binding constant for the group I metal cations with gramicidin-A determined by competition studies and Tl+ 205 nuclear magnetic resonance spectroscopy. Biophys. J. 50:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roux, B., B. Prod'hom, and M. Karplus. 1995. Ion transport in the gramicidin channel: molecular dynamics study of single and double occupancy. Biophys. J. 68:876–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woolf, T., and B. Roux. 1997. The binding site of sodium in the gramicidin A channel: a comparison of molecular dynamics simulations with solid state NMR data. Biophys. J. 72:1930–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levitt, D. 1986. Interpretation of biological channel flux data—reaction-rate theory versus continuum theory. Annu. Rev. Biophys. Chem. 15:29–57. [DOI] [PubMed] [Google Scholar]
- 76.Corry, B., and S. H. Chung. 2005. Influence of protein flexibility on the electrostatic energy landscape in gramicidin a. Eur. Biophys. J. 34:208–216. [DOI] [PubMed] [Google Scholar]
- 77.Becker, M. D., R. E. Koeppe, and O. S. Andersen. 1992. Amino acid substitutions and ion channel function. Model-dependent conclusions. Biophys. J. 62:25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roux, B., and M. Karplus. 1991. Ion transport in a gramicidin-like channel: dynamics and mobility. J. Phys. Chem. 95:4856–4868. [Google Scholar]
- 79.Andersen, O. S. 1983. Ion movement through gramicidin A channels. Studies on the diffusion-controlled association step. Biophys. J. 41:147–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berne, B. J., M. Borkovec, and J. E. Straub. 1988. Classical and modern methods in reaction rate theory. J. Phys. Chem. 92:3711–3725. [Google Scholar]
- 81.Crouzy, S., T. Woolf, and B. Roux. 1994. A molecular dynamics study of gating in dioxolane-linked gramicidin A channels. Biophys. J. 67:1370–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Busath, D. D., C. D. Thulin, R. W. Hendershot, L. R. Phillips, P. Maughan, C. D. Cole, N. C. Bingham, S. Morrison, L. C. Baird, R. J. Hendershot, M. Cotten, and T. A. Cross. 2003. Noncontact dipole. Biophys. J. 75:2830–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- 84.Allen, T. W., S. Kuyucak, and S. H. Chung. 1999. Molecular dynamics study of the KcsA potassium channel. Biophys. J. 77:2502–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Allen, T., A. Bliznyuk, A. Rendell, S. Kuyucak, and S. Chung. 2000. The potassium channel: structure, selectivity and diffusion. J. Chem. Phys. 112:8191–8204. [Google Scholar]
- 86.Guidoni, L., V. Torre, and P. Carloni. 1999. Potassium and sodium binding to the outer mouth of the K+ channel. Biochemistry. 38:8599–8604. [DOI] [PubMed] [Google Scholar]
- 87.Guidoni, L., V. Torre, and P. Carloni. 2000. Water and potassium dynamics inside the KcsA K+ channel. FEBS Lett. 477:37–42. [DOI] [PubMed] [Google Scholar]
- 88.Shrivastava, I., and M. Sansom. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 78:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biggin, P., G. Smith, I. Shrivastava, S. Choe, and M. Sansom. 2001. Potassium and sodium ions in a potassium channel studied by molecular dynamics simulations. Biochim. Biophys. Acta. 1510:1–9. [DOI] [PubMed] [Google Scholar]
- 90.Åqvist, J., and V. Luzhkov. 2000. Ion permeation mechanism of the potassium channel. Nature. 404:881–884. [DOI] [PubMed] [Google Scholar]
- 91.Oostenbrink, C., A. Villa, A. E. Mark, and W. F. van Gunsteren. 2004. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53a5 and 53a6. J. Comput. Chem. 25:1656–1676. [DOI] [PubMed] [Google Scholar]
- 92.Cox, B. G., G. R. Hedwig, A. J. Parker, and D. W. Watts. 1974. Solvation of ions. XIX. Thermodynamic properties for transfer of single ions between protic and dipolar aprotic solvents. Aust. J. Chem. 27:477–501. [Google Scholar]
- 93.Nathan, W. I., R. M. Meighan, and R. H. Cole. 1964. Dielectric properties of alkyl amides. II. Liquid dielectric constant and loss. J. Phys. Chem. 68:509–515. [Google Scholar]
- 94.Roux, B., and S. Bernèche. 2002. On the potential functions used in molecular dynamics simulations of ion channels. Biophys. J. 82:1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roux, B. 1993. Nonadditivity in cation-peptide interactions: a molecular dynamics and ab initio study of Na+ in the gramicidin channel. Chem. Phys. Lett. 212:231–240. [Google Scholar]
- 96.Finkelstein, A., and P. A. Rosenberg. 1979. Membrane Transport Processes, Vol. 3. C.F. Stevens and R.W. Tsien, editors. Raven Press, New York. 73–88.
- 97.Mackerell, A. D., Jr. 2004. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 25:1584–1604. [DOI] [PubMed] [Google Scholar]
- 98.Roux, B., and S. Bernèche. 2002. On the potential functions used in molecular dynamics simulations of ion channels. Biophys. J. 82:1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]