

Abstract
We report on the electrical manipulation of single- and double-stranded oligodeoxynucleotides that are end tethered to gold surfaces in electrolyte solution. The response to alternating repulsive and attractive electric surface fields is studied by time-resolved fluorescence measurements, revealing markedly distinct dynamics for the flexible single-stranded and stiff double-stranded DNA, respectively. Hydrodynamic simulations rationalize this finding and disclose two different kinetic mechanisms: stiff polymers undergo rotation around the anchoring pivot point; flexible polymers, on the other hand, are pulled onto the attracting surface segment by segment.
INTRODUCTION
The behavior of short DNA strands at the solid/liquid interface is of interdisciplinary significance and actively investigated within various realms, including, e.g., DNA-chip technology (1) and DNA-based sensing (2,3), DNA-templated nanostructures (4), molecular mechanics (5), DNA computing (6), the structure of self-assembled monolayers (7), as well as complementary theoretical studies (8).
When grafted to conducting substrates, the conformation of nucleic acid layers can efficiently be controlled by electric fields (9,10). A fundamental understanding of the underlying mechanisms that determine the motion of DNA on biased surfaces is essential, not only for the reason that it might stimulate the development of novel, actively controlled biosensors, but also because the behavior of this charged macromolecule is representative of the wide-ranging class of linear polyelectrolytes.
In the presented experiments, we take advantage of the circumstance that DNA can straightforwardly be examined in its single- (ssDNA) as well as double-stranded (dsDNA) form to study the influence of polymer stiffness on the kinetic properties of these molecules under the action of attractive and repulsive surface forces. The length of the used oligonucleotides (48-mer ∼16 nm for dsDNA) lies in between the mechanical persistence lengths of ssDNA and dsDNA, which are ∼2–3 (11) and 50 (12) nm, respectively, corresponding to the limiting cases of ‘flexible’ and ‘rigid’ charged polymers.
MATERIALS AND METHODS
Experimental
Self-assembled layers of single-stranded oligonucleotides of a mixed, nonself-complementary sequence were prepared on gold surfaces by adsorption from aqueous solution. Details of the sample preparation as well as technical aspects of the measurement are described in Rant et al. (10) and Tinland et al. (13). Gold electrodes of 2.0 mm diameter were prepared on 3-inch single crystalline sapphire wafers by subsequently depositing thin layers of Ti (10 nm), Pt (40 nm), and Au (200 nm) using standard optical lithography and metallization techniques. The Ti layer promotes adhesion between the Au film and the substrate, whereas the Pt interlayer constitutes a precautionary measure to prevent potential diffusion between Ti and Au when cleaning the electrodes in hot solutions. Before DNA adsorption, the electrodes were cleaned in Piranha solution (H2SO4:H2O2 (30%) = 7:3) and exposed to HNO3 (60%) for 15 min each, followed by a final rinse with deionized water.
The sequence of the 48-mer oligonucleotides (obtained from IBA, Goettingen, Germany) was: 5′ HS-(CH2)6 – TAG TCG TAA GCT GAT ATG GCT GAT TAG TCG GAA GCA TCG AAC GCT GAT – Cy3 3′. The 5′ end was modified with a thiol linker to graft the strands chemically to the surface (S-Au bond). Single-stranded oligonucleotides were adsorbed onto the surface by exposing the Au electrodes to Tris-buffered electrolyte solution of low salinity ([Tris] = 10 mM, pH = 7.3) containing 1 μM DNA for 5 min. Afterward, the samples were thoroughly rinsed with buffer solution ([Tris] = 10 mM, [NaCl] = 50 mM, pH = 7.3). Subsequently, a second adsorption step was carried out, during which a monolayer of short spacer molecules, mercaptohexanol (MCH) (obtained from Sigma-Aldrich, St. Louis, MO), was coadsorbed on the DNA-modified surface ([MCH] = 1 mM, [Tris] = 10 mM, [NaCl] = 50 mM, pH = 7.3, incubation time ∼1 h). By forming a dense sublayer, MCH aids to prevent nonspecific interactions between DNA and the gold surface (14). After rinsing the samples again they were finally kept in Tris buffer ([Tris] = 10 mM, pH = 7.3), which was also used during the measurements.
The adsorption process had previously been optimized to yield DNA layers of low surface coverage (13) to prevent steric interactions between neighboring strands (10), which would significantly affect the efficiency of the electrical manipulation. The surface coverage was determined using electrochemical methods (15), yielding 1.7 × 1011 molecules/cm2 (corresponding to a typical interstrand distance of ∼26 nm, larger than the strand contour length).
The upper (3′) ends of the oligonucleotides were dye labeled with a fluorescence marker (Cy3) for optical detection. The emitted fluorescence light was collected by an optical fiber, passed through a 0.5 m monochromator (Jobin Yvon, Longjumeau, France), set to the Cy3 peak emission wavelength (565 nm), and detected by a cooled photomultiplier (Hamamatsu, Hamamatsu City, Japan) operated in single-photon-counting mode.
Electrical potentials were applied to the Au substrates versus an Ag/AgCl reference electrode using a commercially available potentiostat system (Autolab, Ecochemie, Utrecht, The Netherlands) and a Pt counterelectrode. All measurements were conducted in an aqueous, monovalent buffer solution of low salinity (10 mM Tris, pH = 7.3).
After having performed measurements in its single-stranded conformation, the layer was hybridized with nucleic acids of complementary sequence ([cDNA] = 1 μM) in 200 mM NaCl solution. The measurements were then repeated with the same layer in its double-stranded conformation. The hybridization efficiency for low-density ssDNA layers as used here is expected to be nearly 100%, as indicated by, for instance, Herne and Tarlov (14) and our own experience from measuring the molecule density on the surface before and after hybridization by electrochemical means.
Simulations
We performed Brownian dynamics simulations, where the DNA is modeled as a chain of N = 24 elastically connected charged spherical beads with radius a characterized by the stretching and bending moduli including full hydrodynamic interactions and the nonslip surface condition. Similar simulation techniques have been recently used to study the sedimentation of semiflexible polymers (16) and electroosmosis at rough surfaces (17). One simulation bead comprises two nucleic acid monomers and thus a = 0.34 nm. The effective linear charge density of a charged polymer is reduced by counterion condensation (8), leading to a renormalized charge of 1 e per Bjerrum length; this phenomenon is called Manning condensation. Since the Bjerrum length roughly corresponds to the bead diameter 2a in our simulations, we take Manning condensation into account by assuming each bead to bear a charge of 1 e, both for ss- and dsDNA. Electrostatic repulsion between monomers as well as interactions with the surface are included and approximated by an exponentially decaying Debye-Hückel potential, where the characteristic decay length 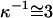 nm is set by the experimental salinity. The persistence length of dsDNA is taken as
nm is set by the experimental salinity. The persistence length of dsDNA is taken as 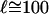 nm and includes electrostatic contributions which are not accounted for by the effective renormalized charge values (8). For ssDNA we set the persistence length to zero and model the polymer as a freely jointed chain with bond length 2a = 0.68 nm. Together with the excluded volume effects between monomers, this generates an effective persistence length of the order of a nanometer. We note that microscopic details of the ssDNA model are not critically important, as the effective elasticity in this case is dominated by electrostatic interactions.
nm and includes electrostatic contributions which are not accounted for by the effective renormalized charge values (8). For ssDNA we set the persistence length to zero and model the polymer as a freely jointed chain with bond length 2a = 0.68 nm. Together with the excluded volume effects between monomers, this generates an effective persistence length of the order of a nanometer. We note that microscopic details of the ssDNA model are not critically important, as the effective elasticity in this case is dominated by electrostatic interactions.
The surface potential is assumed to switch instantaneously from negative to positive bias and vice versa, neglecting more complicated effects due to finite double-layer charging times. One end of the chain is grafted on the surface, and we record the dynamical evolution of the distal end under external potential bias of ±0.125 V at the surface.
RESULTS AND DISCUSSION
The basic principle of manipulating DNA orientations with high persistency by applying alternating bias potentials to the supporting Au electrodes is shown in Fig. 1 (see also Rant et al. (10)). The intrinsically negatively charged DNA is either repelled from or attracted to the surface upon inducing a charge reversal on the electrode. Information about the position of the DNA's top end with respect to the surface is obtained by optical means. Cy3 dye labels attached to the upper (3′) ends of the oligonucleotides are continuously excited by green light from an Argon ion laser (515 nm) while the fluorescence is measured with a photomultiplier. Owing to nonradiative energy transfer to the metal substrate, the fluorescence intensity (F) emitted from the fluorophores depends on their distance (d) to the surface. Theoretical considerations (18) treating the (quenched) emission of an oscillating dipole above a metallic surface yield F ∝ d3 in the range of d-values attainable in the presented experiments.
FIGURE 1.

Electrical manipulation of the conformation of a double-stranded 48-mer DNA layer, demonstrated at low frequency (0.2 Hz) using different waveforms. (Left) The upper panel depicts the bias potential applied to the supporting gold substrate, and the lower panel shows the simultaneously measured fluorescence emission from the dye-labeled layer. (Right) Schematic illustration of the optical probing method. Owing to the distance-dependent, nonradiative energy transfer to the metallic substrate, information about the orientation of the oligonucleotides with respect to the surface can be inferred from the fluorescence intensity emitted by the attached dye labels.
Time-resolved measurements
To elucidate the dynamics involved in the molecular switching process, we present the time-resolved response of a 48-mer DNA layer to a square wave bias potential, which is applied to the supporting electrodes while the fluorescence is detected using boxcar averaging techniques.
A potentiostat, driven by a frequency generator at 300 Hz, is used to control and measure the potential of the gold electrode (topmost panel in Fig. 2). The applied bias steps induce charge reversals on the electrode as revealed by peaks in the electrochemical current (middle panel in Fig. 2). Low direct current components (<0.1 μA/cm2) demonstrate the nearly ideal polarizability of the interface. Integrating the current over time yields the difference in charge which has been accumulating at the electrode surface; its evolution can be perfectly fitted by single exponential functions, signifying the resistive charging of a capacitance. We assign the transient currents to the transport of ions from the bulk solution to the electrode interface, that is, the build-up of the electrochemical ‘double-layer’ (19) which constitutes the polarization of the interface on the solution side.
FIGURE 2.

Time-resolved measurements of the electrically induced, mechanical switching of a 48-mer oligonucleotide layer in electrolyte solution. The upper panels show the electrochemical response of the electrode measured for single-stranded samples (data are representative for double-stranded layers as well). Solid black lines are single exponential fits to the accumulated charge, yielding characteristic time constants of 194 ± 2 μs for both polarities. Relative information of the distance of the DNA's top end to the surface (bottom panel) is inferred from the measured fluorescence intensity. (Hybridizing Cy3-labeled ssDNA strands with strands of complementary sequence results in an enhancement of the dye emission by a factor 1.7. The fluorescence intensity emitted from dsDNA layers has been corrected by this factor before deconvolution to relative height.) Solid lines are single exponential fits to the data. The extracted time constants are listed in Table 1.
The orientation of the DNA layer is inferred from the fluorescence emission of one and the same DNA layer in its single-stranded and double-stranded conformation (time resolution = 20 μs). Due to the macroscopic detection spot (Ø ≈ 0.5 mm) the observed fluorescence resembles an average over an ensemble of a large number of strands (∼3 × 108). However, we point out that as a consequence of the low surface coverage, the motions of individual DNA strands are expected to be largely uncorrelated to those of their neighbors (10). After deconvolution of the fluorescence intensity, we obtain relative information about the distance of the DNA's top end to the surface (d ∝ F1/3).
First, we identify general features of the observed DNA dynamics. When switching to repulsive (negative) bias at t ∼ 1100 μs the DNA layers hardly show a reaction until the interface is counterpolarized, that is, the double-layer is nearly completely charged. (Surprisingly, the delayed onset for the upward motion exhibits a very pronounced characteristic. Presumably, it originates from unspecific interactions of the ‘lying’ DNA with the substrate that impede the ‘stand-up’ process. This would be in agreement with measurements that we performed with DNA layers of higher surface coverage, for which steric interactions prevent the strands from lying down on the surface completely. These experiments did not show as distinct delays in the onset of the repulsion.) Then, the average layer heights increase quickly and eventually saturate at their upper levels; to analyze characteristic time constants from this phase, single exponential functions have been used to phenomenologically fit the data. When applying the positive voltage step (at t ∼ 2750 μs) the response of the DNA layers is more abrupt; however, as long as the charging of the ionic double-layer is still rapid, the DNA layers' downward motion gradually accelerates. Afterward, the layer dynamics again can satisfyingly be approximated by single-exponential fits.
DNA motion and interface polarization
A principal result of Fig. 2 is the observation that the DNA motion is correlated to the double-layer formation. Indeed, changing the double-layer charging time directly affects the dynamics of the DNA switching, as could be verified in experiments with buffer solutions of higher salinity. For instance, when 50 mM NaCl was added to the Tris buffer, the double-layer charging time reduced to 38 μs (due to a higher solution conductance), whereas the upward transition time for dsDNA was found to be as short as 45 μs (see also Rant et al. (10)).
These findings demonstrate that orienting a DNA layer on a surface using moderate (i.e., ‘electrochemically harmless’) bias potentials requires the enhanced electric field within the ionic double layer, since the electrostatic torque acting on an oligonucleotide within a field of typically 1 V/cm (without double layer) is negligible compared to the thermal energy kT. Yet, as the strong gradient in ion concentration (the diffuse double layer) is forming at the interface, the DNA becomes exposed to high field strengths (up to 107 V/cm), so that electrostatic interactions can prevail over entropic effects and a preferential alignment of the DNA strands versus Brownian motion becomes feasible.
Differences in the motion of ssDNA and dsDNA
The observed differences in the traces of ss- and dsDNA are particularly intriguing. For attractive (positive) substrate potentials the height of the single-stranded and double-stranded layer are nearly the same, whereas for repulsive (negative) potentials the height of the single-stranded layer reaches only 80% of the value of the double-stranded layer. This can be attributed to the high flexibility of the single strands, in conjunction with the quickly decaying electric field in solution (cf. also Fig. 4). The upper part of the dsDNA is rigidly connected to its lower segments, which are located within the range of strong electrostatic repulsion (given by the screening length, κ−1 ≈ 3 nm). In contrast, the upper segments of the flexible ssDNA can dangle above its lower part, leading to significant coiling of the molecule. As a result, the average distance of the ssDNA's top end to the surface is reduced compared to dsDNA. This is particularly noteworthy since the contour length of (long) ssDNA is by a factor of ∼1.7 larger when compared to dsDNA of the same base number length (20).
FIGURE 4.

Representative snapshots of the simulation data presented in Fig. 3. The numbers denote timing information in units of 103 simulation steps.
Although the DNA motion is largely governed by the double-layer charging process, Fig. 2 exhibits a remarkable asymmetry in the dynamic transitions. When switching to repulsive electrode potential, the dsDNA layer transforms faster to an upright orientation than the ssDNA, whereas the behavior reverses when switching to attractive potential; now, the single-stranded layer lies down much more quickly than the dsDNA (cf. Table 1). Differences in the upward and downward transition time of the individual layers are especially pronounced for ssDNA, which exhibits a very fast downward motion.
TABLE 1.
Transition time constants τ determined from single exponential fits to experimental and simulated data
| Experiment
|
Simulation τup/τdown
|
|||
|---|---|---|---|---|
| τup (μs) | τdown (μs) | τup/τdown | ||
| ssDNA | 240 | 115 | 2.1 | 2.0 |
| dsDNA | 220 | 235 | 0.9 | 0.7 |
Comparison to simulation
To understand the observed difference in the motion of ss- and dsDNA, we performed Brownian dynamic simulations treating the DNA as a chain of elastically connected beads with full hydrodynamic interactions between monomers. In our simulations, the surface potential is assumed to switch instantaneously from negative to positive bias and vice versa, neglecting more complicated effects due to finite double-layer charging times; consequently, it is not viable to relate absolute timescales to experimental data.
Fig. 3 shows traces of the DNA's top end from 50 simulation runs. Averaged traces (red points) have been computed from the simulation data to permit a comparison between theory and experiment, since an ensemble average is measured in the latter.
FIGURE 3.

Results from simulations. The distance of the DNA's top end (z) as plotted for 50 simulation runs is normalized by the molecular contour length (L0); thin black lines represent single molecule traces; red points denote averaged data. For dsDNA, the blue horizontal lines denote the ‘point of capture’ at which the electrostatic torque equals the thermal energy kT.
We find good qualitative agreement between experiment and simulation: the ensemble layer height for repulsive potential is reduced for ssDNA compared to dsDNA, and the shape of the averaged transitions resembles the experimentally observed features. Table 1 compares the ratios of the evaluated ‘up’ and ‘down’ time constants of simulation (obtained by single exponential fits of the averaged traces, not shown) to experimental data, which show good quantitative agreement.
The upward motion is similar for ss- and dsDNA. First, the lying strands experience a strong repulsion within the electric field that is localized within the first few nanometers close to the surface. Subsequently, their motion is increasingly governed by diffusion, as the upper segments have left the short-ranged interaction region. In contrast to the flexible single strands, the rigid double strands are still ‘pushed’ to some degree by the electric torque exerted on their lower segments during this phase.
The downward motion of ss- and dsDNA differs substantially. When switching to attractive surface potentials, the ensemble height of the single-stranded layer shows a distinct onset and decreases quickly. The narrow distribution of the single molecule traces (cf. Fig. 3) characterizes a collective transition. Representative snapshots depicted in Fig. 4 allow identifying the mechanism that results in the fastest time constant observed: the flexible chain is pulled downward segment by segment by virtue of the high electric field in the proximity of the surface. Note also that the DNA's top end does not have to be displaced considerably in the horizontal plane, which favors the swiftness of the process due to little hydrodynamic drag.
In contrast to the collective transition of the ssDNA, the decrease of the double-stranded layer height is of stochastic nature. As long as the DNA remains in a relatively upright orientation, the electric attraction does not prevail over the influence of thermal motions. However, once it tilts below the ‘point of capture’, which is defined by the critical angle for which the electric torque exerted by the double-layer potential exceeds kT (note that the unit of torque is force times distance and therefore corresponds to energy), the surface field rapidly pulls the strand to the surface by a rotation around its tether point. The random onset of this transition gives rise to a broad distribution of single molecule traces, which in turn results in a slow ensemble decay time.
CONCLUSION
Our experimental studies and simulations of the dynamic behavior of surface-tethered single- and double-stranded oligonucleotides at the polarized solid/liquid interface reveal the following results:
The electrical manipulation of the oligonucleotides' conformation relies on the enhanced field within the ionic polarization layer (double-layer) in solution; thus, the electrically induced motion of DNA at the interface is correlated to the characteristic charging time of the electrochemical double layer.
The equilibrium conformations for repulsive and attractive surface fields as well as the dynamic transitions between them are predominantly governed by interactions of the short-ranged electric field with merely a few DNA segments which are closest to the surface.
For that reason, the stiffness of the polyelectrolyte largely determines its behavior on the surface; for instance, rigid dsDNA can be aligned more efficiently by repulsive electrode potentials than flexible ssDNA.
The dissimilar dynamic behavior which is observed for ss- and dsDNA can be related to the distinct flexibilities of the molecules. When switching from repulsive to attractive surface potentials the molecular kinetics are particularly interesting; whereas the orientation of the rigid dsDNA is governed by stochastic motions before it ‘tips over’ and falls onto the surface, the flexible ssDNA shows a sudden onset and swift transition from an extended to a condensed state on the surface. It is facilitated by an efficient ‘pulling’ of the lowest nucleotides by virtue of the short-ranged field.
Finally, we note that the different kinetic behavior observed here for flexible and stiff polymers should be universally valid for short-ranged surface potentials. The difference between flexible and stiff polymer adsorption/desorption kinetics is expected to grow with increasing polymer length as long as the persistence length of the stiffer polymer is larger than the polymer length.
Acknowledgments
We gratefully acknowledge the help of Y. Yamaguchi with the substrate preparation.
This work was financially supported by the Fujitsu Laboratories of Europe and in part by the Deutsche Forschungsgemeinschaft via SFB 563.
References
- 1.Heller, M. J. 2002. DNA microarray technology: devices, systems, and applications. Annu. Rev. Biomed. Eng. 4:129–153. [DOI] [PubMed] [Google Scholar]
- 2.Tarlov, M. J., and A. B. Steel. 2003. DNA-based sensors. In Biomolecular Films. J. F. Rusling, editor. Marcel Dekker, New York. 545–608.
- 3.Boon, E. M., J. E. Salas, and J. K. Barton. 2002. An electrical probe of protein-DNA interactions. Nat. Biotechnol. 20:282–286. [DOI] [PubMed] [Google Scholar]
- 4.Seeman, N. C. 2003. DNA in a material world. Nature. 421:427–431. [DOI] [PubMed] [Google Scholar]
- 5.Bustamante, C., Z. Bryant, and S. B. Smith. 2003. Ten years of tension: single-molecule DNA mechanics. Nature. 421:423–427. [DOI] [PubMed] [Google Scholar]
- 6.Liu, Q. H., L. M. Wang, A. G. Frutos, A. E. Condon, R. M. Corn, and L. M. Smith. 2000. DNA computing on surfaces. Nature. 403:175–179. [DOI] [PubMed] [Google Scholar]
- 7.Levicky, R., T. M. Herne, M. J. Tarlov, and S. K. Satija. 1998. Using self-assembly to control the structure of DNA monolayers on gold: a neutron reflectivity study. J. Am. Chem. Soc. 120:9787–9792. [Google Scholar]
- 8.Netz, R. R., and D. Andelman. 2003. Neutral and charged polymers at interfaces. Phys. Rep. 380:1–95 and references therein. [Google Scholar]
- 9.Kelley, S. O., J. K. Barton, N. M. Jackson, L. D. McPherson, A. B. Potter, E. M. Spain, M. J. Allen, and M. G. Hill. 1998. Orienting DNA helices on gold using applied electric fields. Langmuir. 14:6781–6784. [Google Scholar]
- 10.Rant, U., K. Arinaga, S. Fujita, N. Yokoyama, G. Abstreiter, and M. Tornow. 2004. Dynamic electrical switching of DNA layers on a metal surface. Nano Lett. 4:2441–2445. [Google Scholar]
- 11.Tinland, B., A. Pluen, J. Sturm, and G. Weill. 1997. Persistence length of single-stranded DNA. Macromolecules. 30:5763–5765. [Google Scholar]
- 12.Smith, S. B., L. Finzi, and C. Bustamante. 1992. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science. 258:1122–1126. [DOI] [PubMed] [Google Scholar]
- 13.Rant, U., K. Arinaga, S. Fujita, N. Yokoyama, G. Abstreiter, and M. Tornow. 2004. Structural properties of oligonucleotide monolayers on gold surfaces probed by fluorescence investigations. Langmuir. 20:10086–10092. [DOI] [PubMed] [Google Scholar]
- 14.Herne, T. M., and M. J. Tarlov. 1997. Characterization of DNA probes immobilized on gold surfaces. J. Am. Chem. Soc. 119:8916–8920. [Google Scholar]
- 15.Steel, A. B., T. M. Herne, and M. J. Tarlov. 1998. Electrochemical quantitation of DNA immobilized on gold. Anal. Chem. 70:4670–4677. [DOI] [PubMed] [Google Scholar]
- 16.Schlagberger, X., and R. R. Netz. 2005. Orientation of elastic rods in homogeneous Stokes flow. Europhys. Lett. 70:129–135. [Google Scholar]
- 17.Kim, Y. W., and R. R. Netz. 2005. Electroosmosis at homogeneous charged surfaces. Europhys. Lett. 72:837–843. [Google Scholar]
- 18.Chance, R. R., A. Prock, and R. Silbey. 1978. Molecular fluorescence and energy transfer near interfaces. Adv. Chem. Phys. 37:1–65. [Google Scholar]
- 19.Bard, A. J., and L. R. Faulkner. 2000. Electrochemical Methods. Wiley & Sons, New York.
- 20.Smith, S. B., Y. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]