

Abstract
Conformational changes in the light illuminated intermediate (pB) of photoactive yellow protein (PYP) were studied from a viewpoint of the diffusion coefficient (D) change of several N-truncated PYPs, which lacked the N-terminal 6, 15, or 23 amino acid residues (T6, T15, and T23, respectively). For intact PYP (i-PYP), D of pB (DpB) was ∼11% lower than that (DpG) of the ground state (pG) species. The difference in D (DpG − DpB) decreased upon cleavage of the N-terminal region in the order of i-PYP>T6>T15>T23. This trend clearly showed that conformational change in the N-terminal group is the main reason for the slower diffusion of pB. This slower diffusion was interpreted in terms of the unfolding of the two α-helices in the N-terminal region, increasing the intermolecular interactions due to hydrogen bonding with water molecules. The increase in friction per one residue by the unfolding of the α-helix was estimated to be 0.3 × 10−12 kg/s. The conformational change in the N-terminal group upon photoillumination is discussed.
INTRODUCTION
Elucidation of the molecular mechanism of photoreceptors that control biological processes has been a target of intensive investigations in biophysics and biology as well as in chemistry. Conformational change of proteins plays an essential role in many biological functions. Photoreactive yellow protein (PYP) has been a prototype for such protein researches (1–30). PYP is a relatively small (14 kDa) water-soluble protein, which is considered as a blue light photoreceptor for a negative phototactic response. It possesses a chromophore, p-coumaric acid (4-hydroxycinnamic acid) covalently bound to the side chain of Cys-69 via a thioester linkage (3,4,9). Upon photoexcitation of the chromophore, PYP exhibits a photocyclic reaction. The ground state species (pG) is converted into a spectrally red-shifted intermediate (pR1, also called I1 or PYPL) within <2 ns (6). Subsequently, pR1 is converted to pR2 without any spectral change (7,8). The pR2 species decays on the submillisecond timescale to a blue-shifted intermediate (pB′, I2 or PYPM), which is converted to pB, again without changes in the spectrum (9–11). This pB finally returns to pG on a timescale of seconds (9). The change in the structure of the chromophore should be relayed to the conformational change of the protein to activate a downstream signal transduction. The reaction kinetics as well as the conformational change associated with this reaction have been very interesting and stimulating topics of research (1–30). The conformational change has been studied by a variety of techniques. For example, it has been shown that the conformation of pB is very much different from that of pG by using kinetics experiments by ultraviolet-visible absorption spectroscopy (19,24,25), by use of hydrophobic probe molecules (23), by monitoring heat capacity changes (22), by measurements of hydrogen/deuterium (H/D) exchange rates of hydrophobicity (24), and by diffusion coefficient (D) measurements (7,8).
In particular, the D-value has recently been recognized to be a unique and useful property to probe the conformational change of proteins. It reflects not only molecular size but also the intermolecular interaction between the protein and water (31,32). More interestingly, it has been shown that D not only provides steady-state characteristics but also that one can use this property to characterize the conformation of unstable intermediate species during reaction dynamics in real time by the pulsed laser induced transient grating (TG) technique (33–36). For example, it has been reported that D of cytochrome c gradually increases concomitant with the protein folding process with a time constant of several tens of milliseconds (33–35). Eitoku et al. (36) have studied spectrally silent conformational dynamics of the light-oxygen-voltage sensing (LOV) domain of a phototropin by measuring the time dependence of D and have found that D decreased with a time constant of 2 ms. This change in D was explained in terms of the unfolding of the α-helices in this protein. In the case of the PYP reaction, characteristic features of the TG signal were observed upon photoexcitation of PYP, and it was shown that D of the intermediate species pB is smaller than that of pG (7,8). The smaller D in the pB state implied that significant conformational change took place upon the pB formation so that the friction for the translational diffusion is increased. However, the molecular origin of such changes in D was unknown. If the molecular origin is revealed for this reaction, it would be an important clue to understanding which conformational changes are essential in the PYP photoreaction. Previously, it was found that the mutation of one amino acid residue did not change D of pG or pB, even though the reaction rate changed considerably after this mutation (7). This negligible effect of the single residue mutation was consistent with the expectation that the D-value is governed by the global structure of the protein, not its local structure, as will be discussed later. For further investigation of the origin of this change in D, we here studied D changes upon the transformation from pG to pB for certain N-terminal truncated PYPs, which lacked the N-terminal 6, 15, or 23 amino acid residues (abbreviated as T6, T15, and T23, respectively).
Previously, using the fact that N-terminal truncated PYPs possess much longer pB state lifetimes than intact PYP (i-PYP) (27), several properties of the pB state of the N-truncated PYPs were characterized. Small angle x-ray scattering (SAXS) experiments demonstrated that the radius of gyration (Rg) of pB was 110 pm larger than that of pG for T6 and that this increase in radius was reduced when more N-terminal amino acid residues were cleaved (70 pm for T15). However, further reduction was not observed after more extensive truncation (70 pm for T23) (28). This observation suggested that the positions 6–15 were important for the conformational change. However, the circular dichroism (CD) intensity simultaneously decreased upon formation of pB even for T23, indicating that α-helices besides those in the N-terminal region may be unfolded in pB (29). Changes in the infrared spectrum were observed in the amide I region upon the pB formation, and this band was partially attributed to the antiparallel β-structure (29). These observations suggested that the β-sheet and N-terminal region were involved in the conformational change. Recently, the NMR structure of a PYP lacking 25 N-terminal residues (Δ25) has been reported (30), showing that the central β-sheet and a small part of one α-helix in the helical connector region were still intact but that other portions were highly flexible in the pB state.
In this work, we found a drastic and systematic change in ΔD(= DpG − DpB) depending on the length of the cleaved N-terminal domain. These changes could be explained by the conformational changes (unfolding) of α-helices in the N-terminal group, which one would expect to accompany the conversion of the hydrogen bonding network from intramolecular to intermolecular. Therefore, this finding is clear evidence of a change in the solvation structure around the N-terminal region upon the pB formation. PYP has two short α-helices in the N-terminal domain, two in the Per-Arnt-Sim (PAS) core, and one in the helical connector. The role of these α-helices is discussed below. Furthermore, this was a unique and valuable opportunity to study changes in friction upon the unfolding of an α-helix. We calculated the increase in friction caused by the unfolding of the α-helix, and thus obtained basic data that will be used for examining the influence of α-helices on D in future researches.
EXPERIMENTAL
The experimental setup for the TG experiments was similar to that reported in previous articles (7,8,20). A XeCl excimer laser-pumped dye laser (Lamda Physik, Göttingen, Germany; Compex 102×c, Lumonics Hyper Dye 300; λ = 465 nm) beam was split into two by a beam splitter and crossed inside a quartz sample cell (optical pathlength 2 mm). The laser power of the excitation was <5 μJ/pulse. The resulting refractive index modulation (transient grating) in the sample was probed by a He-Ne laser (633 nm) as a Bragg diffracted signal (TG signal). The TG signal was detected by a photomultiplier tube (Hamamatsu Photonics, Hamamatsu, Japan; R-928), and fed into a digital oscilloscope (Tektronix, Beaverton, OR; 2430A). The TG signal was averaged by a microcomputer to improve the signal/noise (S/N) ratio. The repetition rate of the excitation beam was <0.05 Hz to avoid multiple excitation of the photoproduct of PYP. The transient absorption signals of PYP were measured by a setup described elsewhere (27). Dynamic light scattering measurements on PYP were performed by using an FDLS-3000 system (Otsuka Electronics, Osaka, Japan).
PYP was prepared using a heterologous overexpression system in Escherichia coli as reported previously (16). Truncated PYPs lacking N-terminal 6, 15, and 23 amino acids (T6, T15, and T23) were prepared by partial digestion with chymotrypsin followed by isolation using diethylaminoethyl-Sepharose column chromatography (27). Briefly, PYP (100 mg) was partially digested by chymotrypsin (1 mg) for 16 h at 20°C. The reaction mixture was applied to diethylaminoethyl-Sepharose column and truncated PYPs were eluted by 0–200 mM NaCl. The fractions showing a single band at an apparent molecular weight of 13,500, 14,400, or 17,800 in SDS-PAGE were collected. MALDI TOF mass spectroscopy for these samples showed a single peak at 11,490.3, 12,347.6, and 13,306.8, respectively, which are in good agreement with the calculated values for T23 (11,488.0), T15 (12,347.0), and T6 (13,306.0). PYP was dissolved in 10 mM Tris-HCl (pH 8.0). Absorbance was ∼0.5 with a 2-mm pathlength in each experiment.
RESULTS
Before presenting the results of our studies on N-truncated PYPs, it may be instructive to explain some characteristic features of the TG signal of i-PYP. Figs. 1 a and 2 a depict the TG signals observed after photoexcitation of i-PYP. The TG signal intensity is proportional to the square of the refractive index change (δn) upon photoexcitation as long as the absorption change is negligible at the probe wavelength (37–39), which was satisfied under these experimental conditions. Furthermore, under these conditions, the sources of the TG signal were mostly the temperature change and the presence (or depletion) of chemical species. The former contribution is called the thermal grating signal and the latter, the species grating signal. The temperature changes are induced by energy released upon relaxation of the photoexcited state, and by chemical reaction.
FIGURE 1.

Initial part of the thermal grating signals after photoexcitation of intact PYP (i-PYP) and N-truncated PYPs (T6, T15, and T23) in the buffer solution. The observed signals and the best fitted curves are shown by the dotted lines and the solid lines, respectively.
FIGURE 2.

Observed TG signals of i-PYP and N-truncated PYPs (T6, T15, and T23) in a longer time region. The observed signals and the best fitted curves are shown by the dotted lines and the solid lines, respectively. The signals are roughly normalized at the first peak intensity.
The features of the TG signal are briefly summarized as follows (7,8). The signal rises quickly after photoexcitation with the instrumental response of our system (∼20 ns) and then shows a weak slow-rising component (Fig. 1a). After this, the signal decays to a certain intensity with a rate constant Dthq2, and shows growth-decay curves twice before finally decaying to the baseline (Fig. 2 a).
The slow rising component immediately after photoexcitation has been attributed to a protein conformational change from pR1 to pR2, which does not change the absorption spectrum of PYP. The decays characterized by the rate constant Dthq2 is the thermal grating component. To show these components clearly, the initial rise is depicted in Fig. 1 a on a linear timescale. To determine the lifetime of the slow-rising component with a minimum of ambiguity, the initial part of the signal was fitted by the function:
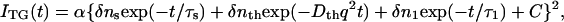 |
(1) |
where α is a constant, τs is the time constant of the slow-rising component, Dth is the thermal diffusivity, q is the grating wave number, τ1 is the time constant of the subsequent pR2→pB kinetics, and C is a constant representing the slower dynamics. To minimize ambiguity in the fitting, we had to reduce the number of adjustable parameters. To do so, we fixed Dthq2 to the decay rate constant of the thermal grating signal of a calorimetric reference sample (bromocresol purple), which rapidly releases the photoexcited state energy as the thermal energy. The τ1-value was fixed at 220 μs, which was the shorter time constant of the pR2→pB kinetics (6). In this way, τs was determined with the least possible ambiguity to be 1.3 ± 0.3 μs. This value was insensitive to the accuracy of the value of τ1 used in the fitting.
The next growth component represents the transformation of pR2 to pB and the last growth-decay curve was attributed to the diffusion of protein pG and pB (Fig. 2 a). This last peak (diffusion peak) appears because D of pB (DpB) is different from that of pG (DpG). Previously, it has been shown that the rates of the rise-and-decay signal were solely determined by the diffusion process, not by the subsequent chemical reaction (7,8). Moreover, the diffusing species was identified based on the sign of the refractive index change as follows: the rate constant of the rising component represents DpGq2 and that of the decaying component corresponds to DpBq2. On the basis of this assignment and the peak profile, one may easily find DpG > DpB (7,8).
The TG signal after the thermal grating (Fig. 2 a) could be expressed well by the following equation (7,8)
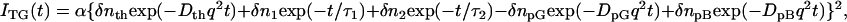 |
(2) |
where the lifetimes τ1 and τ2 represent the pR2→pB kinetics and the preexponential factors represent the refractive index change. The refractive index change due to the thermal grating should possess a negative value at this temperature (δnth < 0), and the other factors were found to be positive. As the difference between DpG and DpB becomes smaller, the two terms of δnpGexp(−DpGq2t) and δnpBexp(−DpBq2t) cancels and the signal intensity becomes weaker. Hence the maximum amplitude of this peak is a good indicator of the difference between DpG and DpB.
The TG signals of N-truncated PYPs are shown in Figs. 1 and 2. The signals up to the diffusion peak were similar to those of i-PYP. As stated above, the initial rise part of the thermal grating signal (Fig. 1, b–d) represents the pR1→pR2 transition, which should be the conformational change occurring distant from the chromophore. Because the N-terminal group is distant from the chromophore, the change in this region could be one of the candidates responsible for this dynamics. If the conformational change of the N-terminal group is really responsible for this change on such a fast timescale, the time constant of the pR1→pR2 transition would be strongly affected by the N-terminal truncation. We fitted the rising part of the thermal grating signal with Eq. 1 using the same method as for i-PYP. It was found that the slow-rising components for all PYPs were very similar (Table 1). (The amplitudes of this component seem to depend on the N-terminal truncation. We consider that this difference among the samples is due to the different amplitude of the thermal grating signal being different from that of the species grating signal. Because the amplitude of the slow-rising component comes mostly from the volume change associated with this transition (7,8), this amplitude has to be compared with the species grating signal intensity. In this respect, if the signal intensity was normalized by the species grating intensity, one found that the amplitude of this slow-rising component was similar for all samples. Hence, we conclude that the effect of the N-terminal truncation is small for the pR1→pR2 transition.) This fact indicates that the conformational change in the N-terminal region does not occur in the initial few microseconds after the photoexcitation.
TABLE 1.
The time constants of the pR1→pR2 transition (τ1/μs and τ2/ms), the diffusion coefficients of pG (DpG/10−10 m2/s) and of pB (DpG/10−10 m2/s), and the change in friction (Δf/10−12 kg/s) upon transformation from pG to pB for intact PYP (i-PYP) and the N-terminal truncated PYPs (T6, T15, and T23
| τs/μs | τ1/μs | τ2/ms | DpG | DpB | Δf | |
|---|---|---|---|---|---|---|
| i-PYP | 1.3 | 250 | 1.5 | 1.33 | 1.18 | 3.9 |
| T6 | 1.4 | 136 | – | 1.33 | 1.21 | 3.1 |
| T15 | 1.4 | 97 | – | 1.27 | 1.15 | 1.5 |
| T23 | 1.7 | 78 | – | 1.13 | 1.11 | 0.7 |
The time τ2 were determined from the rise part of the TG signal (Fig. 1), and those of pR1→pR2 transition were measured by the temporal profiles of the transient absorption signals monitored at 456 nm (Fig. 3). Diffusion coefficients of pG were measured by the dynamic light scattering. D of pB were determined from the temporal profile of the TG signals (Fig. 2). The uncertainties of the values are described in text. The Δf-values were calculated by Eq. 5.
The next rise part represents the pR2→pB kinetics (Fig. 2, b–d). From the signals, it is found that this rate increases slightly in the order i-PYP>T6>T15>T23. For supporting this rate change, the pR2→pB kinetics were monitored by the transient absorption method. The observed signals (ITA) observed over a few milliseconds (Fig. 3) can be reproduced by a single exponential function with a constant term:
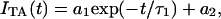 |
(3) |
where the constant term a2 represents the pB contribution, which lasts over a much longer timescale. Indeed, the lifetimes of pB for N-terminal truncated samples have already been reported to be longer when longer N-terminal regions were cleaved; i.e., the time constants were 18 s for T6, 300 s for T15, and 590 s for T23 (27). It is reasonable to assume that in the time range of our observations, this component remained constant. The time constants of the pR2→pB transition are listed in Table 1. The result clearly shows the rate increase, when longer regions were cleaved.
FIGURE 3.

Temporal profiles of transient absorption signal of T6, T15, and T23 probed at 456 nm. The observed signals and the best fitted curves are shown by the dotted lines and the solid lines, respectively. The initial spike-like signals are due to the scattering of the excitation laser light.
The most significant effect of the truncation was observed in the diffusion peak intensity. For example, the peak intensity of T6 (Fig. 2 b) was slightly weaker than that of i-PYP. Using the same consideration as for the i-PYP case, this amplitude reflects the difference between DpG and DpB, that is, the weaker peak amplitude indicates the smaller difference in D (ΔD = DpG − DpB). (In principle, because the final decay rate of the diffusion peak is determined by the protein diffusion and the reverse reaction rate (pB→pG), the lifetime of pB may affect the peak intensity. However, because the pB lifetimes of the N-terminal truncated PYPs (vide supra) are sufficiently longer than the observation time of the TG signal (<100 ms), we can assume that the decay rate of the diffusion peak is solely determined by the protein diffusion process and that its amplitude is solely determined by the difference in D.)
More significantly, the peak amplitude decreases yet more for T15 and T23 (Fig. 2, c and d). In particular, the peak almost disappears for T23. The decreasing trend of the diffusion peak intensity in the order i-PYP>T6>T15>T23 clearly shows that the difference between DpG and DpB decreases upon cleaving the N-terminal group. In other words, it is a clear indication that the change in the N-terminal group is the main factor responsible for the slower diffusion in the pB state.
For quantitative analysis, the signals were fitted by Eq. 2. To reduce the ambiguity of the fitting, D of pG were measured by the light scattering method for the same sample solution. The DpG-values were (1.33 ± 0.05) × 10−10 m2/s for i-PYP and T6, (1.20 ± 0.09) × 10−10 m2/s for T15, and (1.13 ± 0.08) × 10−10 m2/s for T23. To determine ΔD, we fixed the DpG-value at 1.33 × 10−10 m2/s for i-PYP and T6, 1.20 × 10−10 m2/s for T15, and 1.13 × 10−10 m2/s for T23 during the fitting procedure. Furthermore, we used the value of Dthq2 determined from the calorimetric reference sample and τ1, the time constant of pR2→pB kinetics, measured by the transient absorption signal. Therefore, the adjustable parameters in Eq. 2 were DpB and the amplitudes of the exponential terms. Even with these restrictions, the observed TG signals over the wide time range from 10 μs to100 ms could be reproduced well, as shown in Fig. 2. The determined DpB are (1.18 ± 0.03) × 10−10 m2/s for i-PYP, (1.21 ± 0.03) × 10−10 m2/s for T6, (1.15 ± 0.02) × 10−10 m2/s for T15, and (1.11 ± 0.01) × 10−10 m2/s for T23 as listed in Table 1.
DISCUSSION
According to the Stoke-Einstein relationship, D of a spherical molecule with radius r in a continuum medium is given by
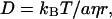 |
(4) |
where kB, T, η, and a are, respectively, the Boltzmann constant, the temperature, the viscosity, and a constant representing the boundary condition between the diffusing molecule and the solvent. This equation assumes that the friction experienced by the diffusing molecule comes solely from the viscosity effect without any specific intermolecular interaction such as hydrogen bonding. For a small molecule, this friction is not large, so that any additional intermolecular interaction could significantly affect D.
On the other hand, because the friction from viscosity would be greater for a large molecule such as a protein, one may expect that changes to friction from intermolecular interactions induced by conformational changes in the protein would be minor compared with those from the viscosity. This is the basis of a commonly used method for determining the molecular size of proteins from their D-values. Contrary to this expectation, D of the intermediate pB of PYP was found, surprisingly, to be smaller than that of pG (7,8). After this finding, it was reported that the increase in friction due to intermolecular hydrogen bonding, induced by the unfolding of α-helices, can actually cause a decrease in D of proteins (33–36) and of a polymer (32); that is, the friction due to the unfolding of an α-helix is not negligible even for macromolecules. In keeping with these results, we suggest that the decrease in D in the pB state of PYP is due to the unfolding of the α-helices.
In this study, we found that ΔD decreases upon cleaving the N-terminal region. This is a clear indication that the additional friction causing ΔD comes from the change in the N-terminal region. The additional friction due to the intermolecular interactions of the N-terminal group may be calculated as follows. The friction coefficient of species i is defined by fi = kBT/Di (species i is pG or pB). The change in friction, Δf, upon the transformation from pG to pB is calculated by
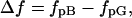 |
(5) |
and is listed in Table 1. The Δf value decreases from 3.9 × 10−12 kg/s for i-PYP to 0.7 × 10−12 kg/s for T23. In particular, the friction decreases significantly upon cleaving the positions 7–15 ((3.1 − 1.5) × 10−12 kg/s = 1.6 × 10−12 kg/s).
PYP has two short α-helices in the N-terminal region (between Ile-11–Leu-15 and Asp-19–Leu-23), two in the PAS core, and one in the helical connector region. Therefore, all α-helices are conserved in T6, but one of the α-helices is cleaved in T15, and two in T23. Using this fact, we may be able to estimate the friction increase per unfolded residue as follows. Taking the difference between fpB and fpG (Δf), the friction change due to the exposure of buried residues by truncation can be eliminated if they do not contribute to the conformational change upon the pB formation. Hence, Δf comes from the friction due to residues that participate in the conformational change. If we assume that the difference in Δf between T6 and T15 (ΔΔf = 1.6 × 10−12 kg/s) is caused by the difference in the length of the α-helix region (five amino acid residues), the friction increase per unfolded residue in an α-helix (Δf0) is roughly estimated to be 0.3 × 10−12 kg/s. This number should be considered as just a rough estimation of the excess friction. However, because there has been no available data on this subject so far, we believe that this value could be a guide for making a connection between D and conformational change for future studies.
If this estimated Δf0 value can be applied to other proteins, the additional friction incurred by unfolding of α-helices may be calculated. For example, the increase in f upon unfolding of an α-helix composed of 100 residues (the size of small globular protein) is calculated to be 3.0 × 10−11 kg/s. If D for such a protein in the native form is assumed to be 1.1 × 10−10 m2/s (cf., D = (0.9–1.1) × 10−10 m2/s for myoglobin (153 residues) (40–41)), D for the unfolded form is estimated at 0.55 × 10−10 m2/s. This estimated value of D is reasonably close to that actually measured for unfolded myoglobin (0.51 × 10−10 m2/s) (40), supporting the values of Δf0 estimated above.
Rg of the pB state have been measured previously for T6, T15, and T23 (28). Although Rg for pB was larger than that of pG for all samples, the radius increased less when the N-terminal amino acid residues were cleaved (110 pm for T6, and 70 pm for T15 and T23). Based on this observation, it was suggested that positions 1–15 are important for the conformational change. This result is consistent with our observation that Δf was reduced by the N-terminal truncation, because the effect of the truncation on Δf is most significant for positions 6–15 (Δf(i-PYP) − Δf(T6) = 0.8 × 10−12 kg/s, Δf(T6) − Δf(T15) = 1.6 × 10−12 kg/s, and Δf(T15) − Δ f(T23) = 0.8 × 10−12 kg/s). However, the fact that cleaving the positions 19–23 further reduces Δf suggests that the α-helix in this region is also unfolded upon pB formation.
The CD measurements on T6, T15, and T23 showed that the CD intensity decreases upon the formation of pB (29). This result suggested that α-helices besides those in the N-terminal region were unfolded in pB, consistent with our observation that D decreases upon the pB formation for T23. This decrease indicates that there is a hydrogen bonding network change in other regions besides the N-terminal region. However, this change is small (Δf(T23) = 0.7 × 10−12 kg/s) compared with the effect of the N-terminal region (Δf(i-PYP) − Δf(T23) = 3.2 × 10−12 kg/s). If we use Δf0 estimated above (0.3 × 10−12 kg/s), the value of Δf(T23) = 0.7 × 10−12 kg/s suggests that two or three residues in the α-helix are unfolded. This minor effect indicates that the hydrogen bonding network may be reorganized in the core region, but that intramolecular hydrogen bonding is still dominant.
From the backbone H/D exchange rate measurement by NMR (24), it was found that the N-terminal domain is of low intrinsic stability. Genetic truncation of the N-terminal domain significantly decreases the extent of cooperativity in the titration curve (30). This N-terminal domain is linked through a hydrogen bonding network with the chromophore-containing domain of the protein. The change in the conformation of the N-terminal region may be caused by this network being linked with the photoisomerization reaction of the chromophore.
It is interesting to note that DpG decreases in the order i-PYP∼T6>T15>T23. Because the radius of gyration decreases upon cleavage of the N-terminal group (28), the above order of DpG is opposite to what is expected from the Stoke-Einstein relationship. The smaller D-value of T23 suggests that the conformation of the chromophore-containing domain is somewhat changed (increasing intermolecular hydrogen bonding) upon removal of the N-terminal group.
Finally, we mention the conformational changes of another photoresponse domain. PYP is a member of the PAS family, and the LOV domain, which is a blue light sensor, is also a member of this family. Previously, TG measurements have shown a significant reduction in D upon photoillumination of a phototropins LOV2 domain with a linker (36). This reduction was explained in terms of the unfolding of the α-helix in the linker moiety, and this unfolding of the α-helix was also reported from NMR measurements (42). The reduction of D was not observed, when the linker region was cleaved. It is particularly interesting to note a similarity with PYP. The LOV2 domain without the linker may correspond to PYP without the N-terminal group. Also, D for the LOV2 domain with the linker could be similar to that of intact PYP, because both of them showed a similar reduction in D, explicable in terms of the α-helices unfolding. For the phototropin case, it was suggested that this α-helix unfolding is important for the biological signal transduction (36,42). Analogously, the same unfolding in the N-terminal region of i-PYP could be responsible for the signal transduction. This result suggests that the D measurement will be an important research tool for detecting protein conformational changes essential for the function of photoresponse proteins.
SUMMARY
Conformational changes in PYP upon photoexcitation were studied from a viewpoint of diffusion coefficient (D) for N-terminal truncated samples. For intact PYP, D of pB (DpB) was ∼11% smaller than that of pG (DpG). This fact indicates that friction for diffusion increases after the conformational changes in the illuminated PYP. Most significantly, the decrease in D after conversion from pG to pB decreases upon cleavage of the N-terminal group in the order of i-PYP>T6>T15>T23. This finding clearly shows that the conformational change at the N-terminal group is the main cause of the change in D and that the solvation structure around this group is significantly altered. This reduction was explained in terms of the increase in the intermolecular interaction by the unfolding of the α-helices in the N-terminal region. Because the pR1→pR2 transition is not affected by the N-terminal truncation, the conformational change in the N-terminal group does not occur in the pR state. The friction increase per residue due to the unfolding of the α-helix is estimated to be 0.3 × 10−12 kg/s. This value may be used for estimating unfolded regions in other protein reactions in future.
Acknowledgments
A part of this study was supported by the Grant-in-Aid (No. 13853002 and No. 15076204) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1.Meyer, T. E. 1985. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim. Biophys. Acta. 806:175–183. [DOI] [PubMed] [Google Scholar]
- 2.Meyer, T. E., G. Tollin, J. H. Hazzard, and M. A. Cusanovich. 1989. Photoactive yellow protein from the purple phototrophic bacterium, Ectothiorhodospira halophila. Quantum yield of photobleaching and effects of temperature, alcohols, glycerol, and sucrose on kinetics of photobleaching and recovery. Biophys. J. 56:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baca, M., G. E. O. Borgstahl, M. Boissinot, P. M. Burke, D. R. Williams, K. A. Slater, and E. D. Getzoff. 1994. Complete chemical structure of photoactive yellow protein: novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry. Biochemistry. 33:14369–14377. [DOI] [PubMed] [Google Scholar]
- 4.Kort, R., H. Vonk, X. Xu, W. D. Hoff, W. Crielaard, and K. J. Hellingwerf. 1996. Evidence for trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 382:73–78. [DOI] [PubMed] [Google Scholar]
- 5.Brudler, R., R. Rammelsberg, T. T. Woo, E. D. Getzoff, and K. Gerwert. 2001. Structure of the I1 early intermediate of photoactive yellow protein by FTIR spectroscopy. Nat. Struct. Biol. 8:265–270. [DOI] [PubMed] [Google Scholar]
- 6.Hoff, W. D., I. H. M. van Stokkum, H. J. van Ramesdonk, M. E. van Brederode, A. M. Brouwer, J. C. Fitch, T. E. Meyer, R. van Grondelle, and K. J. Hellingwerf. 1994. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys. J. 67:1691–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeshita, K., Y. Imamoto, M. Kataoka, K. Mihara, F. Tokunaga, and M. Terazima. 2002. Structural change of site-directed mutants of PYP: new dynamics during pR state. Biophys. J. 83:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeshita, K., Y. Imamoto, M. Kataoka, F. Tokunaga, and M. Terazima. 2002. Themodynamical and transport properties of intermediate states of photo-cyclic reaction of photoactive yellow protein. Biochemistry. 41:3037–3048. [DOI] [PubMed] [Google Scholar]
- 9.Hellingwerf, K. J., J. Hendriks, and T. Gensch. 2003. Photoactive yellow protein, a new type of photoreceptor protein: will this “yellow lab” bring us where we want to go? J. Phys. Chem. A. 107:1082–1094. [Google Scholar]
- 10.Cusanovich, M. A., and T. E. Meyer. 2003. Photoactive yellow protein: a prototypic PAS domain sensory protein and development of a common signaling mechanism. Biochemistry. 42:4759–4770. [DOI] [PubMed] [Google Scholar]
- 11.Losi, A., and S. E. Braslavsky. 2003. The time-resolved thermodynamics of the chromophore-protein interactions in biological photosensors as derived from photothermal measurements. Phys. Chem. Chem. Phys. 5:2739–2750. [Google Scholar]
- 12.Craven, C. J., N. M. Derix, J. Hendriks, R. Boelens, K. J. Hellingwelf, and R. Kaptein. 2000. Probing the nature of the blue-shifted intermediate of photoactive yellow protein in solution by NMR: hydrogen-deuterium exchange data and pH studies. Biochemistry. 39:14392–14399. [DOI] [PubMed] [Google Scholar]
- 13.Genick, U. K., G. E. O. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Šrajer, T. Teng, W. Schildkamp, D. E. McRee, K. Moffat, and E. D. Getzoff. 1997. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science. 275:1471–1475. [DOI] [PubMed] [Google Scholar]
- 14.Genick, U. K., S. Devanathan, T. E. Meyer, I. L. Canestrelli, E. Williams, M. A. Cusanovich, G. Tollin, and E. D. Getzoff. 1997. Active site mutants implicate key residues for control of color and light cycle kinetics of photoactive yellow protein. Biochemistry. 36:8–14. [DOI] [PubMed] [Google Scholar]
- 15.Imamoto, Y., H. Koshimizu, K. Mihara, O. Hisatomi, T. Mizukami, K. Tsujimoto, M. Kataoka, and F. Tokunaga. 2001. Roles of amino acid residues near the chromophore of photoactive yellow protein. Biochemistry. 40:4679–4685. [DOI] [PubMed] [Google Scholar]
- 16.Mihara, K., O. Hisatomi, Y. Imamoto, M. Kataoka, and F. Tokunaga. 1997. Functional expression and site-directed mutagenesis of photoactive yellow protein. J. Biochem. (Tokyo). 121:876–880. [DOI] [PubMed] [Google Scholar]
- 17.Düx, P., G. Rubinstenn, G. W. Vuister, R. Boelens, F. A. A. Mulder, K. Hård, W. D. Hoff, A. R. Kroon, W. Crielaard, K. J. Hellingwerf, and R. Kaptein. 1998. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry. 37:12689–12699. [DOI] [PubMed] [Google Scholar]
- 18.Rubinstenn, G., G. W. Vuister, F. A. A. Mulder, P. Düx, R. Boelens, K. J. Hellingwerf, and R. Kaptein. 1998. Structure and dynamic change of photoactive yellow protein during its photocycle in solution. Nat. Struct. Biol. 5:568–570. [DOI] [PubMed] [Google Scholar]
- 19.Van Brederode, M. E., W. D. Hoff, I. H. M. van Stokkum, M. Groot, and K. J. Hellingwerf. 1996. Protein folding thermodynamics applied to the photocycle of the photoactive yellow protein. Biophys. J. 71:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeshita, K., N. Hirota, Y. Imamoto, M. Kataoka, F. Tokunaga, and M. Terazima. 2000. Temperature-dependent volume change of the initial step of the photoreaction of photoactive yellow protein (PYP) studied by transient grating. J. Am. Chem. Soc. 122:8524–8528. [Google Scholar]
- 21.Lee, B.-C., P. A. Croonquist, T. R. Sosnick, and W. D. Hoff. 2001. 2001. PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J. Biol. Chem. 276:20821–20823. [DOI] [PubMed] [Google Scholar]
- 22.Khan, J. S., Y. Imamoto, M. Kataoka, F. Tokunaga, and M. Terazima. 2005. Time-resolved thermodynamics: heat capacity change of transient species during photo-reaction of PYP. J. Am. Chem. Soc. n press. [DOI] [PubMed]
- 23.Hendriks, J., T. Gensch, L. Hviid, M. A. van der Horst, K. J. Hellingwerf, and J. J. van Thor. 2002. Transient exposure of hydrophobic surface in the photoactive yellow protein monitored with Nile Red. Biophys. J. 82:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoff, W. D., A. Xie, I. H. M. van Stokkum, X. Tang, J. Gural, A. R. Kroon, and K. J. Hellingwerf. 1999. Global conformational changes upon receptor stimulation in photoactive yellow protein. Biochemistry. 38:1009–1017. [DOI] [PubMed] [Google Scholar]
- 25.van der Horst, M. A., I. H. van Stokkum, W. Crielaard, and K. J. Hellingwerf. 2001. The role of the N-terminal domain of photoactive yellow protein in the transient partial unfolding during signalling state formation. FEBS Lett. 497:26–30. [DOI] [PubMed] [Google Scholar]
- 26.Vreede, J., W. Crielaard, K. J. Hellingwerf, and P. G. Bolhuis. 2005. Predicting the signaling state of photoactive yellow protein. Biophys. J. 88:3525–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harigai, M., S. Yasuda, Y. Imamoto, K. Yoshihara, F. Tokunaga, and M. Kataoka. 2001. Amino acids in the N-terminal region regulate the photocycle of photoactive yellow protein. J. Biochem. (Tokyo). 130:51–56. [DOI] [PubMed] [Google Scholar]
- 28.Imamoto, Y., H. Kamikubo, M. Harigai, N. Shimizu, and M. Kataoka. 2002. Light-induced global conformational change of photoactive yellow protein in solution. Biochemistry. 41:13595–13601. [DOI] [PubMed] [Google Scholar]
- 29.Harigai, M., Y. Imamoto, H. Kamikubo, Y. Yamazaki, and M. Kataoka. 2003. Role of an N-terminal loop in the secondary structural change of photoactive yellow protein. Biochemistry. 42:13893–13900. [DOI] [PubMed] [Google Scholar]
- 30.Bernard, C., K. Houben, N. M. Derix, D. Marks, M. A. van der Horst, K. J. Hellingwerf, R. Boelens, R. Kaptein, and N. A. J. van Nuland. 2005. The solution structure of a transient photoreceptor intermediate: Δ25 photoactive yellow protein. Structure. 13:953–962. [DOI] [PubMed] [Google Scholar]
- 31.Cussler, E. L. 1994. Diffusion. Cambridge University Press, Cambridge, UK.
- 32.Inoue, K., N. Baden, and M. Terazima. 2005. Diffusion coefficient and the secondary structure of poly-L-glutamic acid in aqueous solution. J. Phys. Chem. B. n press. [DOI] [PubMed]
- 33.Nada, T., and M. Terazima. 2003. A novel method for study of protein folding kinetics by monitoring diffusion coefficient in time domain. Biophys. J. 85:1876–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishida, S., T. Nada, and M. Terazima. 2004. Kinetics of intermolecular interaction during protein folding of reduced cytochrome c. Biophys. J. 87:2663–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishida, S., T. Nada, and M. Terazima. 2005. Hydrogen bonding dynamics during protein folding of reduced cytochrome c: temperature and denaturant concentration dependence. Biophys. J. 89:2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eitoku, T., Y. Nakasone, D. Matsuoka, S. Tokutomi, and M. Terazima. 2005. Conformational dynamics of phototropin 2 LOV2 domain with the linker upon photoexcitation. J. Am. Chem. Soc. 127:13238–13244. [DOI] [PubMed] [Google Scholar]
- 37.Eichler, H. J., P. Gunter, and D. W. Pohl. 1986. Laser Induced Dynamic Gratings. Springer-Verlag, Berlin, Germany.
- 38.Terazima, M. 1998. Photothermal studies of photophysical and photochemical processes by the transient grating method. Adv. Photochem. 24:255–338. [Google Scholar]
- 39.Terazima, M. 2002. Molecular volume and enthalpy changes associated with irreversible photo-reactions. J. Photochem. Photobiol.C. 24:1–28. [Google Scholar]
- 40.Hilde, K. G., H. Damaschun, R. Misselwitz, M. Muller-Frohne, D. Zirwer, and G. Damaschun. 1994. Compactness of protein molten globules: temperature induced structural changes of the apomyoglobin folding intermediate. Eur. Biophys. J. 23:297–305. [DOI] [PubMed] [Google Scholar]
- 41.Choi, J., and M. Terazima. 2002. Denaturation of a protein monitored by diffusion coefficients: myoglobin. J. Phys. Chem. B. 106:6587–6593. [Google Scholar]
- 42.Harper, S. M., L. C. Neil, and K. H. Gardner. 2003. Structural basis of a phototropin light switch. Science. 301:1541–1544. [DOI] [PubMed] [Google Scholar]