

Abstract
Hydrogen-bearing species in the bone mineral environment were investigated using solid-state NMR spectroscopy of powdered bone, deproteinated bone, and B-type carbonated apatite. Using magic-angle spinning and cross-polarization techniques three types of structurally-bound water were observed in these materials. Two of these water types occupy vacancies within the apatitic mineral crystal in synthetic carbonated apatite and deproteinated bone and serve to stabilize these defect-containing crystals. The third water was observed at the mineral surface in unmodified bone but not in deproteinated bone, suggesting a role for this water in mediating mineral-organic matrix interactions. Direct evidence of monohydrogen phosphate in a 1H NMR spectrum of unmodified bone is presented for the first time. We obtained clear evidence for the presence of hydroxide ion in deproteinated bone by 1H MAS NMR. A 1H-31P heteronuclear correlation experiment provided unambiguous evidence for hydroxide ion in unmodified bone as well. Hydroxide ion in both unmodified and deproteinated bone mineral was found to participate in hydrogen bonding with neighboring water molecules and ions. In unmodified bone mineral hydroxide ion was found, through a 1H-31P heteronuclear correlation experiment, to be confined to a small portion of the mineral crystal, probably the internal portion.
INTRODUCTION
Elucidating the biomineral structure of bone is a significant challenge for traditional structural techniques. Wet chemical methods, x-ray diffraction (XRD), neutron diffraction, IR and Raman spectroscopies, and NMR spectroscopy have all been applied to this highly heterogeneous material and its synthetic calcium phosphate analogs in efforts to characterize its structure (1–9). Despite these efforts it is still not possible to model this material from the molecular level, a vital step in understanding the origins of its mechanical properties. In addition to its biological importance, this avenue of research has relevance in materials research, where biocompatible materials which can be used to replace and even regenerate bone are desired.
Based on the techniques above, the mineral component of bone is generally agreed to be based upon the apatite mineral structure (1,10). Bone mineral contains a substantial amount of carbonate ion (5–8 wt%), believed to substitute mainly in phosphate ion lattice positions (1,9). It is deficient in calcium ion, often containing sodium, potassium, magnesium, and zinc substitutions as well as simple calcium vacancies. The high levels of substitutions and defects in bone mineral crystallites, besides having biological relevance, probably serve to help limit crystal growth. Bone crystallites are very small, plate-shaped crystals ∼5 × 25 × 50 nm in dimension. Mechanical models of bone show the importance of having oriented nanometer-sized mineral crystallites in a collagen matrix to the mechanical properties of bone (11,12). These crystallite dimensions are vital in allowing optimal three-dimensional composite organization of the material. It is somewhat surprising, however, that bone mineral crystallites maintain their integrity, and a carbonated apatite lattice structure, with the high level of crystal defects they contain. It is likely that something stabilizes this lattice structure, either from outside, such as mineral-organic matrix interactions (13), or within the flawed lattice, such as chemical species occupying vacancies in the crystal structure.
In addition to bone mineral and organic matrix, water is an abundant component of bone, accounting for up to 25% by weight (14). Much of this water occurs in pore spaces responsible for nutrient diffusion and contributing to the viscoelastic properties of the material. These pore spaces, with dimensions from millimeters (in trabecular bone) to 0.1 μm, have been extensively investigated (15–17). A much smaller dimension porosity was defined by Neuman and colleagues in the 1950s consisting of the space between mineral crystallites and collagen (18). There have been few attempts to define the role that water in this location plays in the mechanical function of bone (15,17,19). It remains a daunting challenge to distinguish relatively small amounts of structurally bound water molecules amid the bulk water in bone. However, simultaneous derivative thermogravimetric analysis and x-ray diffraction thermal lattice studies found evidence of water that was more tightly bound than surface water in carbonated apatite, a synthetic apatite used as a model of bone mineral that contains carbonate ion and is deficient in hydroxide ion, phosphate ion, and calcium ion (3). Evidence for an architectural and mechanical role for water in bone by solid-state NMR has recently been published as well (19). Further work is needed to characterize this structural water and identify any further structural roles for water in bone.
In this work we have applied a highly selective structural technique, solid-state NMR, to the study of the nanostructure of bone. The magnetic spin properties of nuclei are very sensitive to the surrounding environment. The bonding environment (chemical species), distance and abundance of other magnetic nuclei, and order or disorder of the material all affect the NMR properties in the solid-state. The study of bone by 1H NMR is complicated by the many broad, overlapping signals from both the mineral and the organic matrix. Cross-polarization (CP) NMR techniques are useful for studying heterogeneous materials due to their ability to selectively observe one component of a composite material (8,9,20) and their ability to probe the spatial relationships between molecular species (19,21). Despite these advantages, they remain underutilized in the study of bone. We have applied an advanced CP NMR technique, 1H-31P frequency-switched Lee-Goldburg Heteronuclear Correlation (FSLG HETCOR), to selectively observe only proton signals associated with the mineral component of bone. This technique, based on polarization transfer (via CP) from one nucleus to another, detects protons indirectly through their proximity to phosphorus atoms. Only those protons within <1 nm of phosphorus contribute. The phosphorus NMR signal of bone is dominated by phosphorus in the mineral by a factor of at least 20 over phosphorus associated with the organic matrix, such as phosphorylated proteins (8). Though the use of this two-dimensional technique is still limited by the severe line-broadening in the indirectly detected 1H NMR spectrum due to 1H-1H dipolar coupling during the evolution period, dipolar decoupling techniques can be applied to counter this effect. The very robust line-narrowing technique frequency-switched Lee-Goldburg (FSLG) decoupling (22–25) has been incorporated into the HETCOR pulse sequence (26–28) to obtain a high-resolution 1H spectrum of the bone mineral environment.
We have investigated the 1H environments of three materials to identify the locations and establish the roles of hydrogen-bearing species, and particularly water, in bone mineral and bone architecture. We hypothesize a role for water both in stabilizing the mineral structure and in coupling mineral crystallites to the surrounding bone structures. 1) We have characterized the 1H environment of a synthetic B-type carbonated apatite. In a previous solid-state NMR study comparing calcium phosphate minerals and human bone, Kaflak-Hachulska et al. determined that B-type carbonated apatite was the calcium phosphate mineral most structurally similar to human bone mineral (9). Using this synthetic analog we defined the key structural water and hydroxide species present in a carbonate-substituted apatite lattice. 2) We characterized deproteinated bone, in which at least 90% of the organic matrix component normally found in bone tissue has been removed, specifically the part composed of collagen (29). We compared this biological mineral with the synthetic carbonated apatite to identify similarities and differences in the environments of structural water and hydroxide. Both of these mineral-only materials were compared with 3) 31P-filtered high-resolution 1H NMR spectra of unmodified bone tissue obtained using FSLG HETCOR to a) investigate the proton-bearing species present in the mineral component of bone without any potential compositional changes induced by chemical deproteination and b) characterize proton signals arising from the mineral-organic matrix interface.
EXPERIMENTAL
Sample synthesis and preparation
Synthetic B-type carbonated apatite samples were prepared according to the procedure of Penel et al. (6) and dried overnight at 100°C. Weight percent carbonate was determined by infrared spectroscopy calibrated against carbonated apatite standards analyzed coulometrically (Galbraith Labs, Knoxville, TN) (30). Carbonated apatite samples contained 2.2 wt% carbonate.
Bone samples from two mammalian species, bovine and rat, were employed in this investigation. Though small differences in bone composition exist between species, equally large differences also exist between bones of animals of the same species of different ages and even regions of a single bone remodeled at different times. In this initial study we are interested in bone as a material. In general, we assumed that mammalian bones, and especially mammalian bone mineral, are similar enough to one another to make qualitative comparison useful. This assumption is supported by the work of Johnson et al., which concluded that bovine, corvine, and ovine bone mineral were all similar in composition to one another although being significantly different from the mineral found in ostrich bone (31). In addition, recent NMR and Raman spectroscopic studies have reported strong similarities between mammalian species (8,32). Thus the predominant hydrated chemical species found in the rat bone mineral are likely to correlate well with those found in bovine and human bone mineral as well.
Rat femora from skeletally mature Sprague-Dawley rats six months of age were harvested, stripped of soft tissue and wrapped in gauze, soaked in calcium-buffered saline, and stored at −32°C before handling. All procedures involving animals were in accordance with the relevant state and federal laws and the University of Michigan Committee on the Use and Care of Animals and Occupational Safety and Environmental Health (OSEH) standards. Sections of the rat cortical bone were deproteinated by soaking in a series of hydrazine solutions for a total of 24 h according to a previously published procedure (29). After removal from the hydrazine and thorough washing in a graded series of ethanol/H2O solutions, the brittle bones were ground with mortar and pestle and stored as a powder.
Bovine femora were harvested from freshly slaughtered animals (2–4 years old) at a local slaughterhouse. Animals were roughly age equivalent to rats described above. Femora were stripped of soft tissue, and cortical bone specimens were prepared from central diaphyseal sections. Each diaphysis was sectioned on a band saw into parallelepipeds. Calcium-buffered saline was used during all machining steps to avoid heating the bone and to maintain tissue saturation and ionic balance. Parallelepipeds randomly chosen from an inventory of 10 femora with respect to longitudinal and circumferential location were milled into a powder either cryogenically (∼150 μm particle size) or at room temperature (∼1 mm particle size). Different milling conditions were used to explore the possibility that damage done to bone during milling affected NMR results. However, no spectral changes were noted between samples milled under different conditions. All milling was performed in calcium-buffered saline. Specimens not immediately machined after harvesting or immediately milled after machining were wrapped in calcium-buffered saline saturated gauze and stored at −65°C. Different hydration states were achieved by altering the time samples were exposed to air (10–40% humidity) at room temperature and verified by 1H MAS NMR.
NMR spectroscopy
NMR experiments were performed on a Bruker DSX300 NMR spectrometer (Bruker Instruments, Billerica, MA) using a 4-mm or 7-mm o.d. zirconia sample rotor and a 4-mm or 7-mm double resonance magic-angle probe. Samples were spun at 5–9 kHz. Temperature was controlled to ± 2°C. The temperature for NMR experiments was 25°C unless otherwise stated. In the 1H-31P FSLG HETCOR experiments, the 1H on-resonance power level was 83.7 kHz and the Lee-Goldburg frequency offset was 59.3 kHz. High-power proton decoupling was applied during 31P signal acquisition. Peak-fitting analyses were performed using Peakfit v4.06 (SPSS Inc., Chicago, IL).
To acquire high-resolution 1H spectra of the mineral environment of bone tissue, 1H-31P FSLG HETCOR experiments (26) were performed (Fig. 1) (27,28). In this experiment proton magnetization is indirectly detected through a transfer of magnetization to 31P nuclei in the mineral component of bone. Only those protons close (∼1 nm) to the mineral phosphorus atoms are detected. This is accomplished by preparing 1H magnetization and allowing it to evolve under chemical shift while simultaneously suppressing 1H-1H dipolar coupling that could cause extensive line-broadening with a frequency-switched Lee-Goldburg (FSLG) decoupling pulse sequence (22–25). Suppression of 1H-1H dipolar coupling both improves resolution in the 1H dimension (line narrowing) and prevents loss of signal through fast T2 relaxation. Signal is then transferred to 31P by CP and detected. This two-dimensional experiment is carried out as a function of proton chemical shift evolution time, and the 31P signal is thus encoded with the chemical shifts of the protons close enough to transfer magnetization to phosphorus. A second, F1 dimension, Fourier transform produces an indirectly-detected 1H spectrum. The use of the FSLG decoupling pulse sequence introduces a scaling factor (ideally 0.577) to the 1H chemical shift axis. Experimental parameters can influence the exact scaling obtained. Therefore, the chemical shift axis scaling factor was calibrated to 0.57 using crystalline l-alanine.
FIGURE 1.

Pulse sequence for 1H-31P frequency-switched Lee-Goldburg Heteronuclear Correlation (FSLG HETCOR) experiment. 1H magnetization is prepared and allowed to evolve under chemical shift while 1H-1H dipolar coupling is suppressed, and then cross-polarized to 31P for detection. 31P signal with respect to 1H chemical shift evolution time is Fourier transformed to produce an indirectly detected 1H spectrum.
1H MAS NMR experiments were carried out for all materials at room temperature. Variable-temperature 1H MAS experiments were carried out only for carbonated apatite and powdered deproteinated rat cortical bone at temperatures ranging from 25°C to 225°C. Previously unheated samples were heated in a stepwise manner in the NMR probe using heated ambient air flow and allowed to equilibrate for 10 min before the acquisition of a 1H NMR spectrum. The rotor cap used for variable-temperature experiments included a vent through which vapor created by sample heating could escape. All directly acquired (Bloch decay) 1H NMR spectra were acquired quantitatively, with the delay between scans set to at least 5× the experimentally determined T1 relaxation time. Total integrated areas were determined to quantify total 1H signal lost upon heating of the samples. Integrated areas were corrected for temperature using the standard Boltzmann equation (33). 1H MAS NMR spectra and 1H-31P FSLG HETCOR experiments were performed at room temperature on powdered bovine femoral cortical bone samples at various stages of dehydration in room temperature ambient air. Spectral changes in 1H MAS NMR were used as described previously to monitor the hydration of bone tissue samples (19).
RESULTS
1H MAS NMR of B-type carbonated apatite acquired at temperatures from 25°C to 225°C yielded five distinct proton peaks (Fig. 2, Table 1). The two larger peaks, at 0.1 and 5.4 ppm at room temperature, were assigned as hydroxide ion and water, respectively. This assignment is consistent with previous studies of hydroxyapatite and carbonated apatites (7–9,20). We have assigned the peak at 1.4 ppm to hydroxide ion in a hydrogen-bonding environment. This is consistent with the assignment made for a 1.1 ppm peak observed in a previous 1H NMR study of carbonated apatite, in which the signal was observed to have relaxation properties similar to the 0.1 ppm hydroxide peak and dissimilar to the relaxation of water (9). The peak at 2.2 ppm was assigned to isolated water molecules in the hydroxide ion channel similar to, but slightly downfield of, a water environment found in octacalcium phosphate (7). The water peak at 5.4 ppm was observed to gradually shift upfield as the temperature was increased, until at 225°C two water peaks were observed at 4.3 and 5.5 ppm (Fig. 2). We have assigned these peaks to two distinct water environments in the crystal lattice. Total signal loss for carbonated apatite upon heating to 225°C was 27% after correcting for the different Boltzmann populations (33).
FIGURE 2.

1H MAS NMR spectra of synthetic B-type carbonated apatite mineral at (A) 25°C, (B) 125°C, (C) 175°C, and (D) 225°C. Asterisk indicates spinning sideband. The synthetic mineral contains two hydroxide environments and has two water environments at room temperature, one of which resolves into two distinct environments at higher temperature.
TABLE 1.
Chemical shift assignments for hydrogen-bearing species present in 1H MAS NMR spectra of synthetic B-type carbonated apatite, deproteinated cortical bone, and cortical bone
| Chemical shift (ppm)
|
||||
|---|---|---|---|---|
| Species | Carbonated apatite | Deproteinated cortical bone | Cortical bone | Location/enviornment |
| −OH | 0.1 | 0.4* | 0.4‡ | |
| 0.2† | 0.1§ | Isolated | ||
| −OH | 1.4 | ? | ? | H-bonded to neighboring 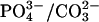
|
| Lipids | – | 1.05, 1.5 | 1.6, 4.0 | Bone lipids |
| H2O | 2.2 | 2.3 | ? | Occupying Isolated −OH vacancies |
| H2O | 5.4* | 5.2* | 5.0‡ | Occupying crystal lattice positions (Ca2+ vacancies?) |
| 4.3, 5.5† | 5.5† | 5.2§ | ||
| H2O/organic matrix | – | – | 6.0–10.0 | Ordered surface layer/possible mineral-organic matrix H-bonds |
 |
– | – | 10.0–14.0 | Substituted for 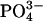
|
Indicates a value from a 1H MAS NMR spectrum acquired at room temperature (25°C).
Indicates a value from a spectrum acquired at 225°C.
Indicates a hydrated sample.
Indicates a dehydrated sample.
1H MAS NMR of deproteinated bone from 25°C to 225°C yielded five distinct proton peaks (Fig. 3, Table 1). Signals analogous to the major hydroxide ion and water signals found in carbonated apatite were seen in these spectra. The hydroxide ion peak was overlapped in the room temperature spectrum, but became more distinct at higher temperatures. A peak fitting analysis indicated that this peak shifted from 0.4 ppm upfield to 0.2 ppm between 75°C to 150°C. The water peak likewise shifted position from 5.2 to 5.5 ppm from room temperature to 225°C (Fig. 3). No second water peak in the 4–5 ppm range was observed at temperatures up to 225°C for deproteinated bone. A single peak in the room temperature spectrum at 1.3 ppm was resolved at higher temperatures into two very sharp peaks at 1.05 and 1.5 ppm (Fig. 3). These peaks were assigned as residual lipids not removed by chemical treatment. 13C CPMAS spectra of deproteinated cortical bone confirmed the presence of lipids at ∼30 ppm (data not shown), consistent with that previously observed in 13C NMR spectra of bone fat and deproteinated bone (31). Finally, a small additional peak was observed at 2.3 ppm (Fig. 3). This peak, similar to that observed in carbonated apatite, was assigned to water molecules in the hydroxide ion channel. Total signal loss for deproteinated rat femoral bone upon heating to 225°C was 63% after correcting for the different Boltzmann populations (33).
FIGURE 3.

1H MAS NMR spectra of deproteinated cortical bone at (A) 25°C, (B) 125°C, (C) 175°C, and (D) 225°C. Deproteinated bone spectra clearly indicate the presence of hydroxide ion and two distinct water environments within the crystallite.
1H MAS NMR spectra were acquired for cortical bone (Fig. 4 A). These spectra were highly overlapped, including signal over a chemical shift range of 0 to ∼15 ppm from hydrogen-bearing species associated with both the mineral and organic components of bone. Sharp peaks at 1.6 and 4.0 ppm were assigned as lipids. The lipid peak at 4.0 ppm is consistent with unsaturated lipid regions. Treatment with hydrazine to deproteinate the bone reduced these regions, shifting this peak upfield to the chemical shift observed for the deproteinated bone. Bulk water was observed in hydrated bone at 5.2 ppm (Table 1). As the cortical bone sample dries in air the sharp bulk water peak is lost from the 1H MAS spectrum, followed by the broadening of the sharp lipid peaks until they cannot be distinguished from other signals (19).
FIGURE 4.

(A) 1H MAS NMR spectrum of partially hydrated cortical bone at room temperature. 1H dimension projection spectra of (B) hydrated, (C) partially hydrated, and (D) dehydrated cortical bone acquired at room temperature using 1H-31P FSLG HETCOR. Bone samples were dehydrated in ambient air (see Experimental). For the purposes of this work we have defined hydrated bone as bone that exhibits a sharp water peak in its 1H NMR spectrum and dehydrated bone as bone that has no sharp peaks in its 1H spectrum and exhibits bulk hydrophobicity. Some water remains in even the dehydrated bone samples; however, enough water has been lost to observe significant changes in the bulk properties of the material. (E) Full 2D 1H-31P FSLG HETCOR spectrum of partially hydrated cortical bone.
1H-31P FSLG HETCOR spectra of cortical bone were acquired (Fig. 4 E) at fully hydrated, partially hydrated, and dehydrated states. A single broad (FWHH = 4 ppm) 31P peak was observed in the directly detected dimension at 3 ± 1 ppm (data not shown). The 1H dimension of these 2-D FSLG HETCOR spectra were analyzed using standard peak-fitting software. All peaks were best described using a Gaussian lineshape, in contrast to the Lorentzian lineshapes that were primarily obtained for the 1H MAS NMR spectra described above. The 1H spectra (Fig. 4, B–D) showed in all cases a peak in the region assigned to hydroxide ion. This peak was observed from 0.4 ppm in the hydrated bone to 0.1 ppm in the dehydrated bone (Table 1). A broad, inhomogeneous water peak was also observed for all cortical bone samples. The maximum of this peak was observed from 5.0 ppm in the hydrated bone to 5.2 ppm in the dehydrated bone (Fig. 4, B–D, Table 1). The hydroxide 1H peak was correlated to a relatively narrow 31P chemical shift range. In contrast, the water 1H signal is spread across the width of the bone 31P signal (Fig. 4 E). The center of the hydroxide 1H peak occurred at a slightly higher chemical shift in the 31P dimension than that of water. FSLG HETCOR spectra of bone also yielded unique signals downfield of the 5 ppm water peak which were not present in the deproteinated bone or synthetic carbonated apatite (Fig. 4, Table 1).
DISCUSSION
Hydration of bone tissue monitored by 1H MAS NMR
We have previously shown that as powdered bone dried in air, the sharp water peak in the 1H MAS NMR spectrum was lost first, followed by the broadening of the lipid peaks until they could not be distinguished (data not shown, see Wilson et al. (19)). When immersed in the pH buffered saline solution bone tissue samples dried to this extent exhibited bulk hydrophobicity, minimizing contact with the solution. This could be due to structural changes induced by drying in the collagen/lipid layer at the particle surface. As described in our previous work, the loss of bulk water was followed by deeper dehydration, with the loss of the ordered hydration shell that has been proposed to mediate the interactions between the mineral and collagen components of bone (19). The terms hydrated, partially hydrated, and dehydrated used in this work are used in accordance with that work and were determined by following changes in the 1H MAS NMR spectra of bone. Hydrated bone is defined as bone that exhibits a sharp water peak in its 1H MAS NMR spectrum and dehydrated bone as bone that has no sharp peaks in its 1H MAS spectrum and exhibits bulk hydrophobicity. Some water remains in even the dehydrated bone samples; however, a significant portion of the water has been lost and changes in the bulk properties of the material are observed.
The bone mineral-H2O-organic matrix interaction
1H MAS NMR spectra of bone were dominated by the organic matrix component. In freshly powdered bone, the 1H spectrum contained a sharp water peak at 5.2 ppm. Two additional sharp peaks at 1.6 and 4 ppm were assigned as lipids. Underneath these sharp peaks the spectrum contained an intense, overlapped feature primarily due to the abundant protons present in collagen. This feature obscures the relatively small 1H signal from structurally important species present in the bone mineral and at the mineral-organic matrix interface, making structural analysis by 1H MAS NMR impossible.
1H-31P FSLG HETCOR spectra of bone were acquired to alleviate this difficulty and provide structural information on the mineral component of bone in the presence of its organic component (Fig. 4, B–E). This technique acts as a filter, allowing observation of only those signals within ∼1 nm of phosphorus nuclei. Phosphorus nuclei in bone tissue primarily exist only in the mineral component; although there are some phosphoproteins, they account for <1 wt% of bone tissue. In contrast, the mineral component of bone accounts for 60–65 wt% of bone, much of which is phosphate ions (14). Cho et al. found that the phosphorus content of bone tissue demineralized with exhaustive treatment with EDTA was negligible compared to that of bone tissue with its mineral component intact (8). Thus HETCOR spectra provide a snapshot of the mineral environment of bone tissue at various stages of drying in air. HETCOR experiments previously performed on bone in the absence of line-narrowing resulted in the almost complete loss of signal downfield of the hydroxide ion peak due to strong dipolar coupling, or a single Gaussian peak due to mobile water with a long enough T2 relaxation time to retain magnetization over the chemical shift evolution period (8,9). Tightly bound, structurally important water molecules are lost from these spectra due to strong dipolar coupling and fast relaxation. Frequency-switched Lee-Goldburg (FSLG) line-narrowing suppressed dipolar coupling during the long (up to 3 ms) chemical shift evolution period of the HETCOR experiment, resulting in greater structural detail in the resulting 1H spectra. Indirectly detected 1H NMR spectra obtained by FSLG HETCOR displayed Gaussian lineshapes, reflecting the inhomogeneous line broadening, or distribution of chemical shift environments within the bone sample. Because the signal intensities in an FSLG HETCOR experiment depend on many different rate constants, including CP transfer rates and T2 relaxation times, signals from different chemical species cannot be quantitatively compared, and overall 1H signal cannot be quantified reliably. Thus the snapshot of the mineral environment obtained is qualitative.
Significant water (5.0–5.2 ppm) and hydroxide ion (0.4–0.1 ppm) peaks were observed in the 1H dimensions of all 1H-31P FSLG HETCOR spectra of bovine cortical bone tissue (Fig. 4, B–E, Table 1). The presence of the hydroxide peak, not visible in the 1H MAS NMR spectrum of bone, is clear evidence that this technique does indeed provide a mineral filter, allowing us to see only those hydrogen-bearing species near mineral phosphorus nuclei.
In addition to the water signal at ∼5.0 ppm, 1H signal was observed in the 1H-31P FSLG HETCOR spectra of cortical bone from 6–10 ppm (Fig. 4, B–E) which was not observed in deproteinated bone (Fig. 3) or synthetic B-type carbonated apatite (Fig. 2). This signal was assigned as water at the surface of bone mineral crystallites in light of this dependence on the presence of the organic matrix and of solid-state NMR dipolar coupling measurements reported in our previous publication (19). Fully hydrated, partially hydrated, and dehydrated bone all showed signal in this region. In the partially hydrated bone, distinct, overlapping features are observed in the region downfield of the 5-ppm water peak (Fig. 4 C). The chemical shift distribution observed reflects the heterogeneity of the mineral and collagen interface and the range of hydration states present within the sample. Although the possibility of some direct collagen amide-mineral contacts has not been ruled out, the chemical shifts observed, downfield of the 5.0–5.5 ppm shifts typically observed for water adsorbed onto an apatite surface (7), suggest water in a highly acidic environment consistent with a hydrogen-bonded network between the mineral phosphate surface and the collagen. Further evidence for the surface water layer is found in the change observed in the 1H projection spectra of bone as a function of the hydration state of the sample. Fully hydrated and dehydrated bone yielded spectra distinctly different from partially hydrated bone. In the 1H projection spectrum of fully hydrated bone, the 6–10 ppm signal appears as a tailing of the 5-ppm water peak rather than as separate distinguishable features (Fig. 4 B). This is consistent with the presence of abundant bulk water in the sample in exchange with the surface water layer. Such exchange on the NMR timescale means that a water molecule would occupy more than a single environment during the course of the experiment, leading to a broadened and upfield-shifted signal. The 1H projection spectrum (Fig. 4 D) of dehydrated bone is very similar in appearance to the 1H spectrum of deproteinated bone (Fig. 3). However, the 5-ppm water peak is not completely homogeneous, as in that material. The extra downfield signal could be residual water at the surface of the mineral crystallites, or possibly protons from collagen or other matrix proteins brought into direct contact with the mineral surface as a result of the loss of the water layer. The evidence presented here and in our previous work suggests water as a third structural component of bone in addition to mineral and collagen.
In addition to the hydration-state dependent spectral features discussed above, there is most likely some unresolved contribution to the bone 1H-31P FSLG HETCOR 1H spectra from the organic matrix itself, since matrix components almost surely exist within 1 nm of the mineral surface. For example, solid-state NMR evidence for a glutamate carboxylate moiety within 5 Å has recently been presented (34). These groups are able to chelate calcium, and they have been proposed as the contact between organic matrix proteins and the mineral. Other recent evidence has shown the presence of an unspecified organic “glue” stretching between mineralized collagen fibers, that might contact the mineral surface at one or both ends (35). This evidence, in combination with that presented above, may allow us to begin to build an overall picture of bone architecture at the molecular level that includes mineral, protein, and structural water. One of the functions of a water layer at the mineral surface may be to mediate the interaction between the mineral surface and the surface of a protein, possibly stabilizing the interaction between a glutamate-containing protein and the mineral. Hansma et al. suggested that the “glue” molecule might be coupled to a mineralized fiber through a water molecule (35). Furthermore, any direct protein-mineral interactions might account for only a small portion of the total mineral surface area available, leaving much of the mineral surface available for hydration.
The 1H signal observed from 10–14 ppm in the 1H-31P FSLG HETCOR spectrum of bone (Fig. 4, B–E, Table 1), and not observed in either of the mineral-only materials (Figs. 2 and 3), indicates protons in a very acidic environment and was assigned as monohydrogen phosphate. This signal is present at all three hydration states. Although it is not completely resolved from the downfield water signal, its chemical shift is too large to be assigned to a neutral water molecule, even one in a strong hydrogen-bonding environment. Monohydrogen phosphate has been observed in calcium phosphate minerals from 10.4 to 16.2 ppm (7). These species have been found to be present in bone using a variety of methods including IR and 31P NMR spectroscopies, but have never before been conclusively identified in 1H NMR of bone minerals. 31P NMR results indicate that the monohydrogen phosphate ions do not exist within a separate mineral phase, but within the apatite phase, taking the place of a small percentage of the orthophosphate groups (36). It is reasonable to assume that, like orthophosphate in bone mineral, they exist in heterogeneous positions with their acidic protons in a range of different hydrogen-bonding environments. It has been suggested that these groups are present primarily near the surface of bone mineral crystallites (21). At the crystallite surface monohydrogen phosphate could be hydrogen bonded to the ordered water layer, or to collagen, or it could be near lipids. These different environments would lead to the observed range of 1H chemical shift values for the acid phosphate ions.
Two types of crystal water observed in bone mineral
The 5.0–5.5-ppm peak in apatites has previously been assigned as surface water, based on the results for synthetic hydroxyapatite (9). However, upon heating surface water was lost in well-crystallized carbonated apatite by 120°C (3). 1H MAS NMR spectra of carbonated apatite and deproteinated bone at temperatures up to 225°C revealed the presence of tightly bound structural waters in these minerals (Figs. 2 and 3, Table 1). Ivanova et al. have also presented evidence for two distinct tightly-bound waters in carbonated apatite not found in stoichiometric hydroxyapatite (3). The chemical shifts of the water observed indicate strong hydrogen-bonding or dipolar interactions with neighboring species. We conclude that these water molecules occupy the vacancies created by substitutions and defects in the crystal lattice, stabilizing the local structure around these vacancies by forming hydrogen-bonding bridges between surrounding ions.
In carbonated apatite two distinct structural water signals were observed with increasing temperature. The single peak at 5.4 ppm gradually shifted upfield to 5.0 ppm as the temperature was increased to 150°C. At 175°C the water peak began to split into two peaks, and by 225°C the spectrum contained two distinct water peaks at 4.3 and 5.5 ppm, respectively (Fig. 2, Table 1). Our interpretation of these changes is that two distinct crystal water environments exist with similar hydrogen-bonding environments at room temperature. As the temperature is increased, one of these peaks is shifted upfield, possibly as a result of increased thermal motion disrupting its hydrogen bonding with surrounding phosphate, calcium, water, and hydroxide ions. Ivanova et al. reported two distinct losses of water, between 250–300°C and 300–350°C, respectively (3). At temperatures of 200–225°C the area of the peak at 5.5 ppm decreased more rapidly than that of the 4.3-ppm peak, indicating that the downfield peak represents the more accessible structural water position.
Deproteinated bone contained structural water up to 225°C as well. However, although bone mineral and synthetic carbonated apatite both contain tightly bound water, it is clear that the bone mineral is more heterogeneous. Both the hydroxide peak and the water peak were broader in the deproteinated bone 1H spectra at every temperature. The water peak in the bone mineral 1H spectra ranged from 2.2–2.9 times the peak width at half-height of the water peak for the synthetic mineral (Figs. 2 and 3). In addition, only a single water peak was observed in this region of the 1H spectra of deproteinated bone up to 225°C instead of the two narrower peaks observed for carbonated apatite. These results are consistent with the poor crystallinity and small size of bone crystallites. A large proportion of each crystallite is at or near its surface, leading to many heterogeneous crystal lattice environments. Bone mineral crystallites may be so disordered that rather than two distinct sites with well-defined chemical environments, the crystallites contain a continuum of defect sites having different crystal surroundings. Also different between these two materials was the total amount of water lost (assuming constant hydroxide content) upon heating. The synthetic carbonated apatite lost 27% of its total 1H signal upon heating to 225°C, whereas the deproteinated bone mineral lost 63%. A significant loss of water took place in the deproteinated bone at a temperature approaching 175°C (Fig. 3 C), a higher temperature than expected for loss of surface-adsorbed water (3). There was no analogous water loss observed in the synthetic carbonated apatite 1H spectrum at that temperature (Fig. 2 C). This water loss is also consistent with small, flawed crystals having a very high surface area. Crystal waters in the biological mineral are close to the surface of the mineral, and lost more easily. Thus, although bone mineral is similar to carbonated apatite, the degree of long-range crystallinity appears much lower for the biological mineral, creating a more heterogeneous and possibly porous mineral structure.
1H-31P FSLG HETCOR spectra of nondeproteinated bone also contained a peak in this 5.0–5.2 ppm region (Fig. 4, B–D, Table 1). This peak was observed to shift slightly downfield as the bone was allowed to dry in air. We have interpreted this as a loss of bulk water while water molecules more tightly bound to the mineral remain.
We assigned the 2.2–2.3 ppm signal in the 1H NMR spectra of carbonated apatite and deproteinated bone as water occupying hydroxide ion positions within the hydroxide channel of the apatite mineral (Figs. 2 and 3). Bone mineral is known to have a greatly reduced hydroxide ion content, compared to stoichiometric hydroxyapatite (5,8). Water molecules occupying only crystal hydroxide positions are relatively isolated (3.4 Å apart), producing the observed upfield chemical shift. Water has previously been observed as upfield as 1.5 ppm in another calcium phosphate mineral, octacalcium phosphate (7). In the 1H-31P FSLG HETCOR spectrum of bone no 1H signal was observed at 2.3 ppm (Fig. 4, B–E). This may indicate that water fills the channel completely and therefore appears downfield of the isolated water chemical shift, unresolved from other crystal-bound water in the bone. Such water could stabilize calcium ions surrounding the hydroxide-deficient hydroxide channel through dipolar interactions. Water in exchange with hydroxide ion may also explain why no hydroxide peak is observed in IR studies of bone (5). Alternatively, there may be no water present in the hydroxide channels of bone; however, we think this unlikely in this highly hydrated tissue. Deproteination of bone may therefore remove collagen protecting the hydroxide channels from dehydration, causing loosely bound waters to be lost and leaving only tightly bound, isolated water molecules and hydroxide ions.
Hydroxide ion in bone mineral
We observed hydroxide ion in synthetic carbonated apatite, deproteinated and nondeproteinated cortical bone (Figs. 2–4, B–E). The chemical shift of hydroxide ion in carbonated apatite was 0.1 ppm, consistent with that previously observed (7–9,20). However, the chemical shift in deproteinated bone at 75°C and in hydrated bovine cortical bone was 0.4 ppm (Table 1). A chemical shift of 0.5 ppm was recently reported for human bone mineral (9). The hydroxide ion peak in the 1H spectra of deproteinated bone shifted upfield to 0.2 ppm as the temperature approached 150°C and remained there up to 225°C (Fig. 3). The hydroxide ion chemical shift in the indirectly detected 1H spectra of bovine cortical bone tissue shifted upfield to 0.1 ppm upon drying in air (Fig. 4, B–D). The 0.4–0.5 ppm chemical shift is consistent with hydroxide ions hydrogen bonded or in chemical exchange with water at the surface of these very small crystallites. The upfield shift upon drying for both bone mineral materials is consistent with loss of that water, and therefore loss of those interactions.
The 2-D 1H-31P FSLG HETCOR spectrum of bone provides information on the correlation between 1H signals and the 31P species to which they are close enough to transfer magnetization through cross-polarization (Fig. 4 E). Although the 31P spectrum of bone tissue contained only a single broad peak, this correlation can still provide information about the crystal environment. It has long been noted that the 31P peak of bone crystallites is broader than that of synthetic apatite minerals (36). This is expected for the high-surface area, poorly crystalline mineral of bone, in which many heterogeneous phosphorus environments exist. In the 1H-31P FSLG HETCOR spectra of bone the hydroxide ion 1H signal occurred over a much narrower 31P chemical shift range than did the water 1H signal (Fig. 4 E), and the center-of-mass of the hydroxide 1H signal was slightly downfield of the center-of-mass of the water signal in the 31P dimension. This result implies that hydroxide ion is present in a limited portion of the crystal, most likely the internal region of the bone mineral crystallites and not near the disordered surface regions. In contrast, water is present in abundance throughout the crystal. This provides another insight into the reduced hydroxide ion content of bone mineral.
A second peak was observed in carbonated apatite that we have assigned to hydroxide ion (Fig. 2, Table 1). This peak occurs at 1.4 ppm, similar to hydroxide ion peaks found in mixed fluorohydroxyapatites (7). In the fluorohydroxyapatites this peak was ascribed to hydroxide ions hydrogen-bonded to neighboring fluoride ions. A previous study of carbonated apatite also assigned a peak near 1 ppm as hydrogen-bonded hydroxide based on its relaxation properties, which were similar to those of the hydroxide ion peak and dissimilar to those of water (9). Thus we concluded that a small amount of hydroxide ion exists in carbonated apatite mineral in a unique, hydrogen-bonded state. We recently reported the presence of hydrogen bonds between hydroxide ions and neighboring phosphate groups in carbonated apatite (19). This was expected to occur near crystal defects, such as calcium defects, and may be one structural consequence of carbonate substitution in the apatite crystal. This signal was not resolved in either of the bone mineral materials, although in the case of the deproteinated bone this region was obscured by signal from residual lipids. The lack of this signal in bone tissue 1H spectra may be a result of insufficient resolution of broad, heterogeneous peaks to observe this peak distinctly. It may also reflect a less ordered hydroxide environment in which the remaining hydroxide ions are not held in rigid interactions with surrounding ions in the crystallite. For example, they may interact with water molecules in the hydroxide channel.
CONCLUSIONS
We have investigated and compared the 1H environments of three different substituted apatite minerals using solid-state NMR to more fully describe the structure in and surrounding the mineral crystallites of bone tissue. We have observed three different structural roles for water in bone. Water was observed in two crystal lattice environments. Crystal-bound structural water occupies vacancies in the imperfect carbonated apatite crystal lattice, providing structural stability by forming hydrogen-bonding bridges between neighboring ions. The presence of this water may explain why these crystallites do not collapse or spontaneously rearrange into more perfect, and less biologically appropriate, mineral crystals. Finally, the water layer observed at the surface of bone mineral crystallites may mechanically couple the mineral and collagen in bone. This water may serve as a cushion against mechanical stress. Two possible mechanisms for this are that 1), water movement allows bone to withstand stress with less deformation and 2), water acts as a sacrificial layer, protecting collagen from shear under uniaxial stress. We have added water to the list of fundamental building blocks that define the molecular-level structural of bone. With a complete model of how these building blocks fit together, it will be possible to accurately model the material and understand the molecular origins of its unique mechanical properties.
Acknowledgments
The authors thank Joey Wallace for contributions to bone sample preparation.
This study was supported by National Institutes of Health R01 AR048969, National Institutes of Health P30 AR46024, National Institutes of Health T90 DK070071, Department of Defense/US Army DAMD17-03-1-0556, Department of Defense/US Army DAMD17-01-1-080, and the Department of Chemistry at the University of Michigan.
References
- 1.Elliott, J. C. 2002. Calcium phosphate biominerals. In Reviews in Mineralogy & Geochemistry, Vol. 48. M. L. Kohn, J. Rakovan, and T. M. Hughes, editors. Minerology Society of America, Washington, DC. 427–453.
- 2.Sudarsanan, K., and R. A. Young. 1969. Significant precision in crystal structural details: Holly Springs hydroxyapatite. Acta Crystallogr. B. 25:1534–1543. [Google Scholar]
- 3.Ivanova, T. I., O. V. Frank-Kamenetskaya, A. B. Kol'tsov, and V. L. Ugolkov. 2001. Crystal structure of calcium-deficient carbonated hydroxyapatite. Thermal decomposition. J. Solid State Chem. 160:340–349. [Google Scholar]
- 4.Loong, C.-K., C. Rey, L. T. Kuhn, C. Combes, Y. Wu, S.-H. Chen, and M. J. Glimcher. 2000. Evidence of hydroxyl-ion deficiency in bone apatites: an inelastic neutron-scattering study. Bone. 26:599–602. [DOI] [PubMed] [Google Scholar]
- 5.Rey, C., J. L. Miquel, L. Facchini, A. P. Legrand, and M. J. Glimcher. 1995. Hydroxyl groups in bone mineral. Bone. 16:583–586. [DOI] [PubMed] [Google Scholar]
- 6.Penel, G., G. Leroy, C. Rey, and E. Bres. 1998. MicroRaman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif. Tissue Int. 63:475–481. [DOI] [PubMed] [Google Scholar]
- 7.Yesinowski, J. P., and H. Eckert. 1987. Hydrogen environments in calcium phosphates: 1H MAS NMR at high spinning speeds. J. Am. Chem. Soc. 109:6274–6282. [Google Scholar]
- 8.Cho, G., Y. Wu, and J. L. Ackerman. 2003. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science. 300:1123–1127. [DOI] [PubMed] [Google Scholar]
- 9.Kaflak-Hachulska, A., A. Samoson, and W. Kolodziejski. 2003. 1H MAS and 1H→ 31P CP/MAS NMR study of human bone mineral. Calcif. Tissue Int. 73:476–486. [DOI] [PubMed] [Google Scholar]
- 10.Dejong, W. F. 1926. La substance minerale dans les os. Recl. Trav. Chim. Pays-Bas. Belg. 45:445–448. [Google Scholar]
- 11.Rho, J.-Y., L. Kuhn-Spearing, and P. Zioupos. 1998. Mechanical properties and the hierarchical structure of bone. Med. Eng. Phys. 20:92–102. [DOI] [PubMed] [Google Scholar]
- 12.Gao, H., B. Ji, I. L. Jager, E. Arzt, and P. Fratzl. 2003. Materials become insensitive to flaws at nanoscale: Lessons from nature.: Section Title: General Biochemistry. Proc. Natl. Acad. Sci. USA. 100:5597–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKee, M. D., and M. T. Kaartinen. 2002. Regulation of biomineralization by bone proteins and their assembly into extracellular matrices: Implications for implant osseointegration. In Aging, Osteoporosis, and Dental Implants. G. Zarb, U. Lekholm, T. Albrektsson, and H. Tenenbaum, editors. Quintessence Publishing, Carol Stream, IL. 191–205.
- 14.American Society for Bone and Mineral Research ASBMR Bone Curriculum. 2004. http://depts.washington.edu/bonebio/ASBMRed/ASBMRed.html. [Online].
- 15.Fernandez-Seara, M. A., S. L. Wehrli, and F. W. Wehrli. 2002. Diffusion of exchangeable water in cortical bone studied by nuclear magnetic resonance. Biophys. J. 82:522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wehrli, F. W., and M. A. Fernandez-Seara. 2005. Nuclear magnetic resonance studies of bone water. Ann. Biomed. Eng. 33:79–86. [DOI] [PubMed] [Google Scholar]
- 17.Cowin, S. C. 1999. Bone poroelasticity. J. Biomech. 32:217–238. [DOI] [PubMed] [Google Scholar]
- 18.Neuman, W. F., T. Y. Toribara, and B. J. Mulryan. 1953. The surface chemistry of bone. VII. The hydration shell. J. Am. Chem. Soc. 75:4239–4242. [Google Scholar]
- 19.Wilson, E., A. Awonusi, M. D. Morris, D. H. Kohn, M. M. J. Tecklenburg, and L. W. Beck. 2005. Highly ordered interstitial water observed in bone by nuclear magnetic resonance. J. Bone Miner. Res. 20:625–634. [DOI] [PubMed] [Google Scholar]
- 20.Santos, R. A., R. A. Wind, and C. E. Bronnimann. 1994. 1H CRAMPS and 1H-31P HetCor experiments on bone, bone mineral, and model calcium phosphate phases. J. Magn. Reson. B. 105:183–187. [DOI] [PubMed] [Google Scholar]
- 21.Wu, Y., J. L. Ackerman, H.-M. Kim, C. Rey, A. Barroug, and M. J. Glimcher. 2002. Nuclear magnetic resonance spin-spin relaxation of the crystals of bone, dental enamel, and synthetic hydroxyapatites. J. Bone Miner. Res. 17:472–480. [DOI] [PubMed] [Google Scholar]
- 22.Haeberlen, U., and J. S. Waugh. 1968. Coherent averaging effects in magnetic resonance. Phys. Rev. 175:453–467. [Google Scholar]
- 23.Mehring, M., and J. S. Waugh. 1972. Magic-angle NMR experiments in solids. Phys. Rev. B. 5:3459–3471. [Google Scholar]
- 24.Bielecki, A., A. C. Kolbert, and M. H. Levitt. 1989. Frequency-switched pulse sequences: homonuclear decoupling and dilute spin NMR in solids. Chem. Phys. Lett. 155:341–346. [Google Scholar]
- 25.Levitt, M. H., A. C. Kolbert, A. Bielecki, and D. J. Ruben. 1993. High-resolution 1H NMR in solids with frequency-switched multiple-pulse sequences. Solid State Nucl. Magn. Reson. 2:151–163. [DOI] [PubMed] [Google Scholar]
- 26.Massiot, D., B. Alonso, F. Fayon, F. Fredoueil, and B. Bujoli. 2001. New NMR developments for structural investigation of proton-bearing materials at different length scales. Solid State Sci. 3:11–16. [Google Scholar]
- 27.van Rossum, B.-J., H. Förster, and H. J. M. de Groot. 1997. High-field and high-speed CP-MAS 13C NMR heteronuclear dipolar-correlation spectroscopy of solids with frequency-switched Lee-Goldburg homonuclear decoupling. J. Magn. Reson. 124:516–519. [Google Scholar]
- 28.van Rossum, B.-J., C. P. de Groot, V. Ladizhansky, S. Vega, and H. J. M. de Groot. 2000. A method for measuring heteronuclear (1H-13C) distances in high speed MAS NMR. J. Am. Chem. Soc. 122:3465–3472. [Google Scholar]
- 29.Termine, J. D., E. D. Eanes, D. J. Greenfield, M. U. Nylen, and R. A. Harper. 1973. Hydrazine-deproteinated bone mineral. Physical and chemical properties. Calcif. Tissue Res. 12:73–82. [DOI] [PubMed] [Google Scholar]
- 30.Featherstone, J. D. B., S. Pearson, and R. Z. LeGeros. 1984. An infrared method for quantification of carbonate in carbonated apatites. Caries Res. 18:63–66. [DOI] [PubMed] [Google Scholar]
- 31.Johnson, G. S., M. R. Mucalo, and M. A. Lorier. 2000. The processing and characterization of animal-derived bone to yield materials with biomedical applications. Part 1: Modifiable porous implants from bovine condyle cancellous bone and characterization of bone materials as a function of processing. J. Mater. Sci. Mater. Med. 11:427–441. [DOI] [PubMed] [Google Scholar]
- 32.de Carmejane, O., M. D. Morris, M. K. Davis, L. Stixrude, M. Tecklenburg, R. M. Rajachar, and D. H. Kohn. 2005. Bone chemical structure response to mechanical stress studied by high pressure Raman spectroscopy. Calcif. Tissue Int. 76:207–213. [DOI] [PubMed] [Google Scholar]
- 33.Harris, R. K. 1986. Nuclear Magnetic Resonance Spectroscopy: A Physicochemical View. Addison Wesley Longman, Essex, UK.
- 34.Jaeger, C., N. S. Groom, E. A. Bowe, A. Horner, M. E. Davies, R. C. Murray, and M. J. Duer. 2005. Investigation of the nature of the protein-mineral interface in bone by solid-state NMR. Chem. Mater. 17:3059–3061. [Google Scholar]
- 35.Fantner, G. E., T. Hassenkam, J. H. Kindt, J. C. Weaver, H. Birkedal, L. Pechenik, J. A. Cutroni, G. A. G. Cidade, G. D. Stucky, D. E. Morse, and P. K. Hansma. 2005. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat. Mater. 4:612–616. [DOI] [PubMed] [Google Scholar]
- 36.Wu, Y., M. J. Glimcher, C. Rey, and J. L. Ackerman. 1994. A unique protonated phosphate group in bone mineral not present in synthetic calcium phosphates: Identification by phosphorus-31 solid state NMR spectroscopy. J. Mol. Biol. 244:423–435. [DOI] [PubMed] [Google Scholar]