

Abstract
The histone acetyl transferase Tip60 (HTATIP) belongs to a multimolecular complex involved in the cellular response to DNA damage. Tip60 participates in cell cycle arrest following DNA damage by allowing p53 to activate p21CIP (p21) expression. We show here that Tip60 and the E1A-associated p400 protein (EP400), which belongs to the Tip60 complex, are also required for DNA damage-induced apoptosis. Tip60 favours the expression of some proapoptotic p53 target genes most likely through the stimulation of p53 DNA binding activity. In contrast, p400 represses p21 expression in unstressed cells, thereby allowing cell cycle progression and DNA damage-induced apoptosis. Tip60 and p400 have thus opposite effects on p21 expression in the absence of DNA damage. We further found that this antagonism relies on the inhibition of Tip60 function by p400, a property that is abolished following DNA damage. Therefore, taken together, our results indicate that Tip60 and p400 play distinct roles in DNA damage-induced apoptosis and underline the importance of the Tip60 complex and its regulation in the proper control of cell fate.
Keywords: apoptosis, cell cycle, DNA damage, E1A-associated p400, Tip60 complex
Introduction
In higher eukaryotes, DNA damages such as double-strand breaks are recognised and repaired by specialised machineries. The signal given by the DNA damage leads to the activation of specific checkpoints (such as the G1/S or the G2/M checkpoints) resulting in the appropriate cell response (Sancar et al, 2004). Progression into the cell cycle is stopped until damages are repaired, or, if the damages are too important, cells can enter the process of apoptosis. Defects in this DNA damage response can result in the accumulation of mutations, which in turn can lead to cell transformation and cancer.
The ATM/ATR pathway plays a critical role in the G1/S and G2/M checkpoints induced in response to DNA damage. The G2/M checkpoint is activated through the phosphorylation of the cdc25 phosphatase resulting in its binding to 14-3-3 proteins and its subsequent nuclear export (Sancar et al, 2004). Consequently, cyclin-dependent kinases remain phosphorylated and inactive, and cells cannot proceed to mitosis. Activation of the G1/S checkpoint by the ATM/ATR pathway is largely dependent on the p53 tumour suppressor (Sancar et al, 2004). P53 is involved in both cell cycle arrest and apoptosis following DNA damage. P53 plays its role, at least in part, through the activation of specific genes. It binds to specific target DNA sequences, which are located in the promoters of genes involved in cell cycle arrest in G1, such as the gene encoding the cyclin/cdk inhibitor p21 (el-Deiry et al, 1993), or genes involved in apoptosis, such as genes encoding the Fas death receptor or the mitochondrial protein Bax (Miyashita et al, 1994; Muller et al, 1998). In the absence of DNA damage, p53 activity is repressed in part by the oncoprotein Mdm2, which binds directly to the p53 activation domain (Momand et al, 1992). Mdm2 is a ring-finger dependent E3 ubiquitin ligase, which ubiquitinates p53 and thereby targets it to proteosomal degradation (Honda et al, 1997; Kubbutat et al, 1997). In addition, Mdm2 suppresses the transcriptional activity of p53 (Momand et al, 1992). Moreover, recent results indicate that Mdm2 can be recruited to p53 binding sites to mediate transcriptional repression through histone ubiquitination (Minsky and Oren, 2004). Following DNA damage, activation of the ATM/ATR pathway results in p53 phosphorylation on multiple sites and in the inhibition of Mdm2 binding (Meek, 2004). P53 is then stabilised and its transcriptional activity is induced. What mediates the decision between a p21-dependent G1 arrest or apoptosis is still not completely clear for the moment (Meek, 2004).
The histone acetyl transferase (HAT) Tip60 also plays a major role in the DNA damage response. Indeed, we and others found that it is required for G1 arrest following actinomycin D treatment or DNA double-strand breaks and for apoptosis following double-strand breaks (Ikura et al, 2000; Berns et al, 2004; Legube et al, 2004). Tip60 has been cloned as a protein of 60 kDa, which binds to the HIV Tat protein (Kamine et al, 1996). Sequence analysis indicates that it belongs to the MYST (from MOZ-Ybf2/Sas3-Sas2-Tip60) family of HATs, which is conserved from yeast to human (Utley and Cote, 2003). The so-called Tip60 complex, purified using an exogenous tagged Tip60, contains various enzymatic activities, including two helicases of opposite polarity, Tip49a and Tip49b, and a chromatin remodelling enzyme from the SWI/SNF family of ATPases, the E1A-associated p400 protein (Ikura et al, 2000; Fuchs et al, 2001). That this complex is relevant to Tip60 function is indicated by the finding that the complex is much more efficient in acetylating histones than purified Tip60 by itself (Ikura et al, 2000). Moreover, the Tip60 complex is structurally and functionally conserved from yeast to human (Ceol and Horvitz, 2004; Doyon et al, 2004). Because of the importance of histone modifications in transcription, many studies have investigated the role of Tip60 and the Tip60 complex in transcriptional control. Tip60 has been found to function as a co-activator for many transcription factors (Brady et al, 1999; Hlubek et al, 2001; Baek et al, 2002), and binding of many members of the Tip60 complex (including Tip60 itself) to some E2F or c-myc regulated-promoters correlates with their transcriptional activation (Frank et al, 2003; Taubert et al, 2004).
In addition to its role in transcription, recent results indicate that Tip60 and the Tip60 complex play a direct role in the cell response to DNA damage, in particular following DNA double-strand breaks. First, in yeast, the NuA4 complex, which is the orthologue of the Tip60 complex, is recruited to sites of DNA breaks through binding to phosphorylated H2A (Downs et al, 2004). Second, in Drosophila, the Tip60 complex is important for the loss of phosphorylated H2Av (a Drosophila histone H2A variant homologous to H2AX) foci following double-strand break repair (Kusch et al, 2004). Drosophila Tip60 (dTip60) acetylates phosphorylated H2Av, thereby favouring its exchange with unphosphorylated H2Av mediated by the Drosophila homologue of p400, Domino. Thirdly, Tip60 directly acetylates ATM and may therefore participate in the signalling pathways controlling the cell response to DNA double-strand breaks (Sun et al, 2005). Finally, Tip60 is a major component of the p53 pathway: We previously found that it is coregulated with p53, since it is also a target of the E3 ubiquitin ligase activity of Mdm2 and since its expression increases following UV irradiation (Legube et al, 2002). Moreover, its expression is required for both the p53-dependent G1 arrest and the expression of p53 dependent genes (Berns et al, 2004; Doyon et al, 2004; Legube et al, 2004).
Here, we observed that the expression of both Tip60 and another component of the Tip60 complex, p400, are required for apoptosis induced by UV irradiation. However, whereas Tip60 favours the expression of p53-dependent proapoptotic genes in agreement with its described role as a p53 cofactor, p400 had an opposite effect and functioned as a repressor of p21 and fas transcription in unirradiated cells.
This surprising result led us to investigate the molecular mechanisms involved. We found that Tip60 was required for efficient binding of p53 to its target promoters. Finally, we demonstrate that the role of p400 in apoptosis is due to its ability to repress p21 expression and therefore to allow cell cycle progression. By manipulating p400 levels, we were able to change cell fate from apoptosis to cell cycle arrest, pointing out the importance of the Tip60 complex and its regulation in the choice between cell cycle arrest or apoptosis following DNA damages.
Results
Tip60 expression is required for UV-C-induced apoptosis
To test whether Tip60 is important for apoptosis controlled by the p53 pathway, we knocked-down Tip60 expression in p53-positive U2OS osteosarcoma cells using specific siRNAs that we previously characterised (Legube et al, 2004). We reproducibly achieved more than 90% of transfection efficiency using siRNAs (data not shown). We then irradiated cells with UV-C in order to induce apoptosis. Cells transfected with control siRNAs underwent massive apoptosis when irradiated, as indicated by extensive chromatin condensation, nucleus size reduction (Figure 1A) and caspase-mediated PARP cleavage (Figure 1B). Knockdown of p53 reduced apoptosis induction (data not shown), indicating that UV-induced apoptosis is, at least in part, p53-dependent. In cells in which Tip60 was knocked-down, virtually no apoptosis could be observed either by direct dapi staining (Figure 1A) or by the analysis of PARP cleavage (Figure 1B), even at a high UV-C dose (80 J/m2). Moreover, this result was obtained using two siRNAs targeting two different regions of the Tip60 mRNA (Tip1 and Tip2, Figure 1B), indicating that it is unlikely to be due to ‘off target' effects of Tip60 siRNAs. These data thus indicate that Tip60 expression is required for apoptosis following UV-irradiation. Importantly, Tip60 is also required for apoptosis induced by other types of DNA damage, since Tip60 knockdown also inhibited apoptosis induced in U2OS cells by doxorubicin (a topo II inhibitor) and by cisplatin (Supplementary data 1).
Figure 1.
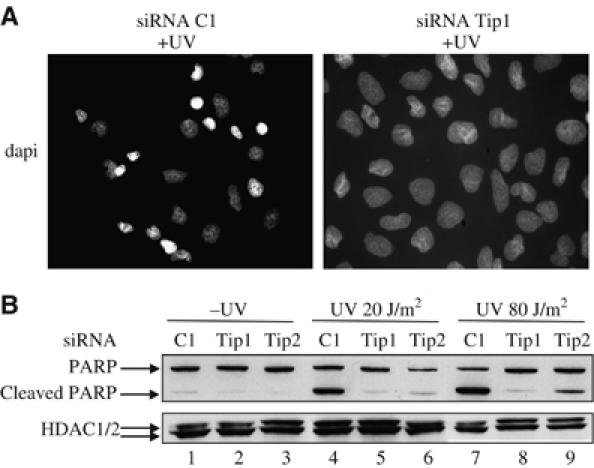
Tip60 is required for p53-dependent apoptosis. (A) U2OS cells were transfected by the indicated siRNAs using oligofectamine. After 48 h, they were UV-irradiated (80 J/m2), fixed with paraformaldehyde after 6 h and stained with dapi. (B) U2OS cells were transfected by the indicated siRNAs with oligofectamine. After 48 h, these cells were UV-irradiated (20 or 80 J/m2) or not, as indicated. After 4 h, Laemmli sample buffer was added to the transfected cells to ensure extraction of total proteins. Total cell extracts were then analysed for the presence of PARP (full-length or cleaved) and HDAC1/2 (as a loading control) by Western blot.
p400 and Tip49b are also required for UV-induced apoptosis
In mammals, Tip60 belongs to a multimolecular complex, the so-called ‘Tip60 complex', which is generally believed to be the active form of the enzyme (Ikura et al, 2000). To test whether Tip60 controls UV-induced apoptosis within the Tip60 complex, we investigated the role of other subunits of the Tip60 complex. We transfected U2OS cells with siRNA targeting the E1A-associated p400 ATPase. This siRNA induced the silencing of p400 expression at the mRNA level (Figure 2A) as well as at the protein level, as shown by the disappearance of a faint 400 kDa band recognised by the anti-p400 antibody and of coprecipitating Tip49b (lower panel) in p400 immunoprecipitates (Figure 2B). We then irradiated cells and assayed caspase-mediated cleavage of PARP. We found that knockdown of p400 resulted in a significant decrease of PARP cleavage even at high UV dose (80 J/m2) (Figure 2C), indicating that p400 expression is required for UV-induced apoptosis. We also found that knockdown of Tip49b had a similar effect (Supplementary data 2). Taken together, these data demonstrate that at least three subunits of the Tip60 complex, Tip60 itself and the ATPases Tip49b and p400 are required for DNA damage-induced apoptosis.
Figure 2.
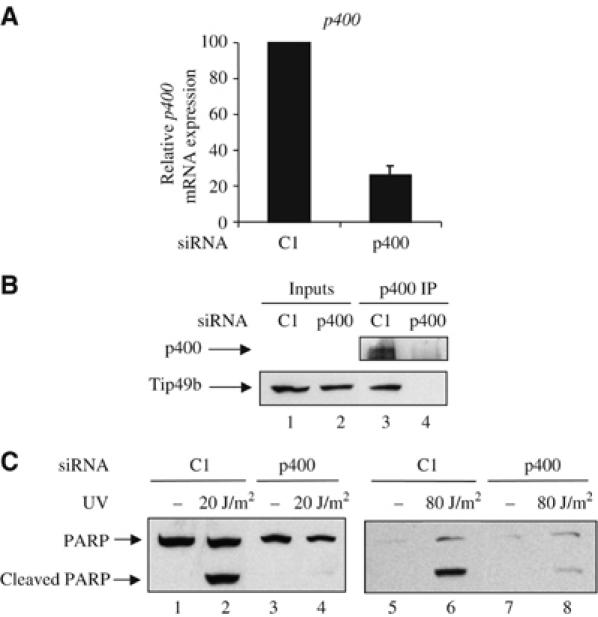
p400 and Tip49b are also required for UV-induced apoptosis. (A) U2OS cells were transfected with the indicated siRNAs using oligofectamine. After 48 h, they were harvested by scraping. Total RNAs were extracted and reverse transcribed. The amount of p400 cDNAs was measured by real-time PCR, divided by the amount of P0 cDNA and calculated relative to 100% for cells transfected with the control siRNAs. (B) Same as in (A), except that whole-cell extracts were prepared, immunoprecipitated with the anti-p400 antibody and blotted using anti-p400 or anti-Tip49b antibody. Also shown is a Western blot monitoring Tip49b levels in input material (p400 Western blots did not work properly on whole cell extracts). (C) U2OS cells were transfected with the indicated siRNAs using oligofectamine. After 48 h, the transfected cells were UV-irradiated (20 or 80 J/m2) or not. After 4 h, Laemmli sample buffer was added to the transfected cells to ensure extraction of total proteins. Cell extracts were then analysed for the presence of PARP by Western blot.
Tip60 and p400 play opposite roles in the expression of p53-dependent genes
UV-induced apoptosis is largely controlled through the p53 transcription factor, which induces the transcriptional activation of proapoptotic target genes, such as genes encoding Fas or Bax (Miyashita et al, 1994; Muller et al, 1998). As Tip60 has already been shown to be involved in transcriptional activation of p53-responsive genes, we tested the effect of the knockdown of Tip60 and p400 on the expression of proapoptotic p53-responsive genes. We irradiated cells with 10 J/m2 of UV and harvested them 16 h later to achieve maximal induction of p53-dependent genes (data not shown). Under these conditions, apoptosis induction was still observed (data not shown). We analysed by reverse transcription followed by quantitative PCR (Q-PCR) the expression of some p53 target genes, including the gene encoding p21, which is already known to be dependent on Tip60 expression (Berns et al, 2004; Doyon et al, 2004; Legube et al, 2004). Induction upon UV irradiation was observed for four p53 target genes (p21, fas, hdm2 and bax) (Figure 3), whereas three p53 target gene (PTEN, PIG3 and Apaf1) were not activated (Supplementary data 3), the reasons for these differences being unclear. Knockdown of Tip60 resulted in a decreased expression of these genes, both in unirradiated cells and following irradiation (Figure 3A and Supplementary data 3), indicating that Tip60 is required for their expression. Surprisingly, knockdown of p400 had a completely different effect: whereas bax, PTEN, PIG3, Apaf1 were unaffected (data not shown), p21, fas and hdm2 were activated to some extent upon p400 knockdown in unirradiated cells (Figure 3B). This activation is unlikely to be due to ‘off-target' effects of the siRNAs, since a similar activation of p21 expression following p400 knockdown was recently described in IMR 90 primary human diploid fibroblasts (Chan et al, 2005). Thus, in unstressed cells, p400 represses the expression of at least three p53 target genes. P400 knockdown did not significantly induce the expression of these genes following UV irradiation (Figure 3B) (or after doxorubicine or cisplatine treatment (Supplementary data 1)), suggesting that the repressive effects of p400 were abolished following DNA damage. Importantly, similar results were obtained in MCF7 cells upon UV irradiation, at least for p21 expression (Supplementary data 4). Taken together, these results indicate that although the expression of Tip60 and p400 are required for UV-induced apoptosis, they play different roles in the expression of some p53-dependent genes. In addition, they suggest that the effects of Tip60 on apoptosis could be dependent, at least in part, on its ability to favour the expression of some proapoptotic p53 target genes, such as fas or bax.
Figure 3.
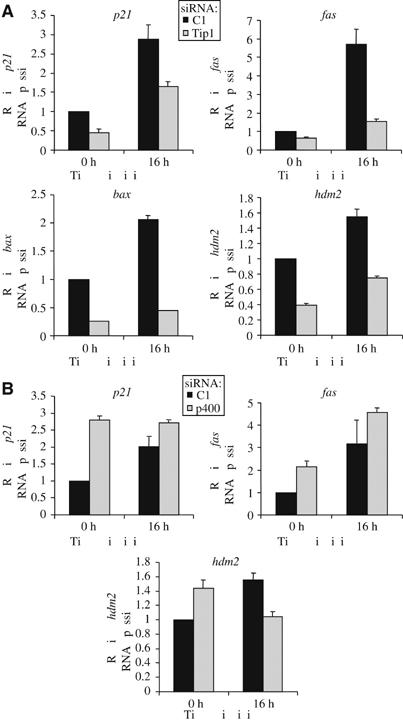
Tip60 and p400 have opposite effects on the expression of p53-dependent genes. (A) U2OS cells were transfected by the C1 siRNAs (black bars) or the Tip siRNAs (grey bars) by electroporation. After 48 h, they were UV-irradiated or not (10 J/m2), as indicated, and harvested 16 h later by scraping. Total RNAs were extracted and reverse transcribed. The amounts of p21, fas, bax and hdm2 cDNA were measured by Q-PCR, divided by the amount of ribosomal phosphoprotein P0 cDNA and calculated relative to 1 for cells transfected by the control siRNAs and not irradiated. A representative experiment is shown. (B) U2OS cells were transfected by the C1 siRNAs (black bars) or the p400 siRNAs (grey bars) by electroporation and treated as in (A). The amounts of p21, fas and hdm2 cDNA were measured by real time PCR and analysed as in (A). A representative experiment is shown.
Tip60 knockdown does not affect p53 activation by UV
We thus investigated further the mechanism by which Tip60 favours the expression of some p53-dependent genes. We first tested whether Tip60 knockdown affects p53 accumulation or activation following UV irradiation. Indeed, Tip60 knockdown is known to result in a decrease in p53 expression in uninduced U2OS cells (Legube et al, 2004). Furthermore, Tip60 expression has recently been shown to be required for efficient ATM activation following DNA double-strand breaks (Sun et al, 2005). Analysis of p53 expression in transfected cells indicates that, as already shown (Legube et al, 2004), knocking down Tip60 slightly decreased p53 levels in uninduced cells (Figure 4A). However, following UV irradiation conditions similar to Figure 3, p53 accumulates to nearly equivalent levels in cells transfected by the control or by the Tip60 siRNAs. Accurate quantification of this experiment indicated that the decrease in p53 levels was lower than 1.2-fold in cells transfected by the Tip60 siRNAs following UV irradiation, indicating that Tip60 knockdown did not significantly affect p53 accumulation under our experimental conditions. We also analysed p53 activation by performing Western blot against various phoshorylated forms of p53. We found that p53 was phosphorylated at S15, S37 and S392 to normal levels in UV-irradiated cells transfected by the Tip60 siRNAs (Figure 4B), indicating that Tip60 knockdown does not inhibit all the signal transduction pathways leading to p53 activation following UV-C irradiation. Since phosphorylation of p53 on serine 15 in response to UV-C irradiation is mediated through the ATR pathway (Tibbetts et al, 1999), our data thus suggest that Tip60 expression, although involved in ATM activation (Sun et al, 2005), is not required for ATR activation. Altogether, although we cannot rule out the possibility that Tip60 is required for a signalling pathway leading to p53 activation though a post-translational modification we have not tested, these results thus suggest that Tip60 plays a role in the p53 pathway downstream of p53 activation, in agreement with our previous finding that Tip60 is required for exogenous p53 to activate efficiently the p21 encoding gene in the absence of DNA damage signalling (Legube et al, 2004).
Figure 4.

Tip60 knockdown does not affect p53 activation by UV-C. (A) U2OS cells were transfected by the indicated siRNAs and UV-irradiated (10 J/m2) 48 h later, where indicated. After 16 h, total cell extracts were prepared and analysed by Western blot for the presence of p53 (upper panel). After stripping, the membrane was blotted using an antibody recognising HDAC1/2 (lower panel). (B) Same as in (A), except that total cell extracts were analysed by Western blot for the presence of S15-, S37- and S392-phosphorylated p53. The stars indicate nonspecific bands.
Physical interaction between Tip60 and p53
Since Tip60 is an HAT, which is involved in transcriptional activation, we investigated whether it could function as a co-activator for p53, which means that it would be recruited by p53 on promoters to mediate transcriptional activation. We first tested whether Tip60 physically interacts with p53. GST pull down experiments using bacterially produced GST-fusion proteins and transfected cell extracts indicated that p53 and Tip60 can interact, at least in vitro (data not shown). Since Tip60 is expressed at low levels in cells and since Tip60 and p53 migrate in the vicinity of the immunoglobulin heavy chain, co-immunoprecipitation of endogenous proteins was not conclusive (data not shown). Therefore, to test whether p53 and Tip60 interaction can be detected in living cells, we raised a cell line derived from U2OS cells and expressing exogenous HA-tagged Tip60. Reverse transcription followed by quantitative RT–PCR indicated that these cells expressed about 30-fold more Tip60 mRNA than the original U2OS cells. We then irradiated or not these cells. UV irradiation led to an increase in the expression of both endogenous p53 and HA-tagged Tip60 (Figure 5A and B), which was expected since both p53 and Tip60 are known to be upregulated in UV-treated cells (Legube et al, 2002; Meek, 2004). Strikingly, immunoprecipitation of endogenous p53 from UV-irradiated cells led to the co-immunoprecipitation of HA-tagged Tip60, indicating that both proteins interact in irradiated cells (Figure 5A). Moreover, less Tip60 was co-immunoprecipitated with p53 in un-irradiated cells, indicating that irradiation of cells induced the accumulation of the p53/Tip60 complex. Similar results were obtained when the immunoprecipitation was performed in the opposite way (Figure 5B). Note, however, that since the expression of the two proteins increased following irradiation, we cannot conclude from these experiments that their ability to interact is induced by irradiation. Nevertheless, these data indicate that Tip60 interacts with p53, in agreement with the hypothesis that it functions as a co-activator for p53.
Figure 5.
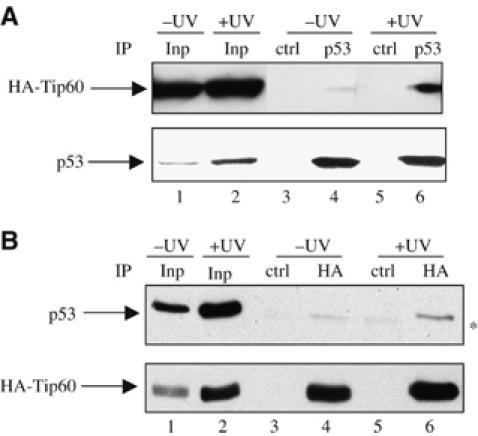
Physical interaction between Tip60 and p53. (A) U2OS cells stably expressing HA-tagged Tip60 were UV-irradiated (10 J/m2) or not. After 16 h, total cell extracts were prepared and subjected to immunoprecipitation using an anti-p53 or an irrelevant antibody (ctrl) as indicated. Immunoprecipitated proteins were then analysed for the presence of p53 and Tip60-HA by Western blot. In total, 10% of inputs (Inp) were also loaded on the gel (lanes 1 and 2). (B) Same as in (A), except that cell extracts were immunoprecipitated by an anti-HA or a control antibody. The star indicates the migration of immunoglobulin heavy chains of the immunoprecipitating antibody, which is slightly detected by the secondary antibody (see e.g. in lane 5).
Tip60 is required for p53 binding on p53 target promoters
If Tip60 functions as a co-activator for p53, then it should not only interact with p53 but also be recruited to p53 target promoters through this interaction. However, our efforts to observe directly Tip60 recruitment to p53-dependent promoters by chromatin immunoprecipitation (ChIP) were unsuccessful, even in the HA-TIP60 overexpressing cell line in which we could readily detect the p53/Tip60 interaction (data not shown). Since transcriptional activation by p53 is known to be regulated at the level of p53 DNA binding, we also tested the effect of Tip60 knockdown on p53 binding to its target promoters by ChIP. We could readily detect p53 binding on the p21 promoter and on the fas promoter, since these sequences were highly enriched in the p53 ChIPs compared to control (Figure 6). No p53 binding was observed on a sequence derived from the β-actin gene, which is not targeted by p53. P53 occupancy was lower on the fas promoter than on the p21 promoter in uninduced cells (Figure 6), certainly reflecting a difference in p53 affinity for the two promoters. As expected, p53 binding to both promoters increased following UV irradiation. Tip60 knockdown resulted in the decrease of p53 presence on the two promoters, both in unirradiated cells and following irradiation (Figure 6). Since Tip60 knockdown did not significantly affect p53 expression following UV irradiation (Figure 4A), this result indicates that knockdown of Tip60 indeed compromises the ability of p53 to bind to its target sequences on the fas and p21 promoters, at least following UV irradiation. Therefore, all together, these experiments indicate that the role of Tip60 in apoptosis is mediated, at least in part, by its ability to favour p53 binding to its target promoters.
Figure 6.
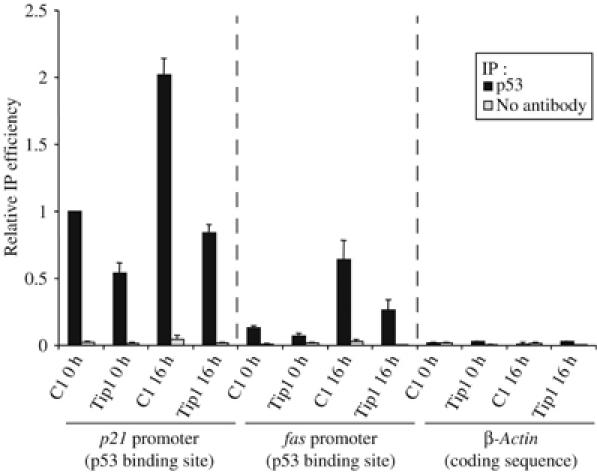
Tip60 is required for p53 binding on p53 target promoters. U2OS cells were transfected by the indicated siRNAs (C1 or Tip1) by electroporation and 48 h later, they were UV-irradiated or not (10 J/m2). After 16 h irradiation, ChIP was performed on these cells using an anti-p53 antibody (black bars) or no antibody as a control (grey bars). The presence of p21 promoter, fas promoter (containing the p53 response element) and β-actin coding sequence in the immunoprecipitates and in the inputs were analysed by real-time PCR. After dividing by the input DNA, the amount of immunoprecipitated sequence was calculated relative to 1 for the amount of p21 promoter immunoprecipitated by the anti-p53 antibody in cells transfected by the control siRNA and not irradiated. The mean and standard deviation from three entirely independent experiments are shown.
p400 knockdown induces a p21-dependent cell cycle arrest
Whereas the effect of Tip60 knockdown on some p53-dependent proapoptotic genes could explain its role in DNA damage-induced apoptosis, the molecular mechanism by which p400 knockdown blocked apoptosis is clearly different, since p400 was not required for the expression of the proapoptotic p53 target genes we tested (see Figure 3). We thus intended to investigate the molecular basis of p400 action. We found that p400 knockdown led to an increased expression of p21 mRNA (see above, Figure 3) and protein (Figure 7A). Since p21 functions as an inhibitor of cyclin/cdks, we also investigated the phosphorylation status of a known important target of cyclin/cdks, the retinoblastoma protein, which is a critical regulator of G1/S progression. We found that p400 knockdown resulted in the appearance of a faster migrating form of the retinoblastoma protein, thereby reflecting its dephosphorylation (Figure 7B). We finally analysed the cell cycle distribution of the transfected cells and we found that p400 knockdown in unstressed cells resulted in a G1 and a G2 cell cycle arrest (Figure 7C). Importantly, this cell cycle arrest is dependent of p21 activation, since it is abolished by the cotransfection of p21 siRNA (Supplementary data 5). Taken together, these data indicate that p400, through the inhibition of p21 expression, allows appropriate Rb phosphorylation and proper cell cycle progression. Similar results were obtained for Tip49b (Supplementary data 2).
Figure 7.
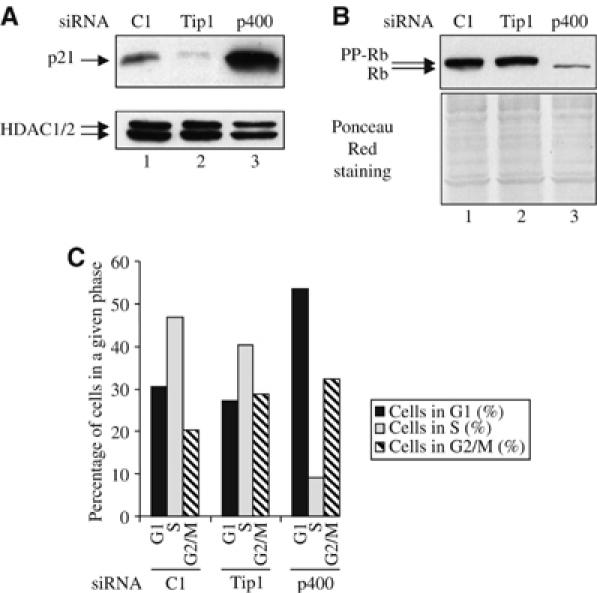
p400 knockdown induces a cell cycle arrest. (A) U2OS cells were transfected with the indicated siRNAs by electroporation. After 48 h, total cell extracts were prepared and analysed for the presence of p21 or HDAC (as a loading control) by Western blot. (B) Same as in (A), except that cell extracts were analysed for the presence of Rb by Western blot and for equal loading by ponceau red staining. (C) U2OS cells were transfected with the indicated siRNAs by electroporation. After 48 h later, the cell cycle distribution of transfected cells was analysed by flow cytometry.
p400 knockdown blocks apoptosis through cell cycle arrest
Since p21 and hypophosphorylated Rb are potent inhibitors of apoptosis (Harbour and Dean, 2000; Seoane et al, 2002), we reasoned that the effect of p400 knockdown on apoptosis (Figure 2) could be related to its ability to induce p21 expression and accumulation of hypophosphorylated Rb. To test this possibility, we prevented p21 accumulation following p400 knockdown by co-transfecting p21 siRNAs. We first checked knockdown efficiency by reverse transcription followed by Q-PCR. We found that p400 and Tip60 knockdown efficiencies were not affected by the presence of p21 siRNAs (Figure 8A). The analysis of p21 mRNA levels showed that, as already observed, p400 and Tip60 knockdown had opposite effects on p21 expression (upregulation and downregulation, respectively), whereas transfection of p21 siRNAs prevented p21 accumulation following p400 knockdown (Figure 8B). Importantly, similar results were observed when p21 levels were analysed by Western blot (Figure 8C). We then irradiated transfected cells and we analysed PARP cleavage, reflecting apoptosis (Figure 8D). We found that, as already observed, knockdown of either Tip60 or p400 resulted in inhibition of UV-induced PARP cleavage. Strikingly, inhibition of p21 expression restored PARP cleavage in cells transfected by p400 siRNAs, but not in cells transfected by Tip60 siRNAs, indicating that inhibition of apoptosis by p400 knockdown is largely dependent on the induction of p21 expression. Moreover, this result also demonstrates that Tip60 and p400 are involved in DNA damage-induced apoptosis through separate mechanisms.
Figure 8.

p400 knockdown blocks apoptosis through p21 induction. (A, B) U2OS cells were transfected with the indicated siRNAs (10 μl of each siRNAs (100 μM)) by electroporation. The amount of siRNAs was kept constant using the control siRNA (C1). After 64 h, cells were harvested. Total RNAs were extracted and reverse transcribed. The amounts of p400, Tip60 (A) and p21 (B) cDNA were measured by real-time PCR and divided by the amount of P0 cDNA and standardised to 100% relative to the amount of cDNA in cells transfected by the C1 siRNA alone. A representative experiment is shown. (C) U2OS transfected as in (A) and (B) were harvested in Laemmli sample buffer and cell extracts were analysed for p21 protein expression by Western blot. (D) U2OS cells analysed in (A) and (B) were UV-irradiated or not (20 J/m2) and 4 h later, cell extracts were prepared and analysed for the presence of PARP by Western blot.
p400 role in cell cycle control is Tip60-dependent
Our results from Figure 3 indicate that p400 and Tip60 play antagonistic roles in p21 gene expression. Since p400 and Tip60 belong to the same complex, we reasoned that one of these proteins could regulate the other one. To test this possibility, we transfected U2OS cells with the two siRNAs together, and we analysed p21 gene expression. We found that both at the mRNA (Figure 9A) and the protein level (Figure 9B), p21 expression in cells transfected by the two siRNA were down to the levels observed in cells transfected by the Tip60 siRNA alone: importantly, since no effect of p400 knockdown could be observed in Tip60 knockdown cells (although the efficiency of p400 knockdown was not affected by the Tip60 siRNAs (data not shown)), the effects of p400 are entirely Tip60-dependent. This led us to investigate whether the effects of p400 knockdown on cell cycle progression were also Tip60-dependent: we found by analysing Rb phosphorylation (Figure 9C) or cell cycle distribution (Figure 9D) that, whereas Tip60 knockdown had no major effect by itself, it was able to reverse completely the effects of p400 knockdown. Thus, the effects of p400 knockdown on p21 gene activation and cell cycle progression are Tip60-dependent, indicating that p400 functions as a negative regulator of Tip60 in unstressed cells.
Figure 9.

p400 functions as an inhibitor of Tip60 in unstressed cells. (A) U2OS cells were transfected with the indicated siRNAs (10 μl of each siRNAs (100 μM)) by electroporation. The amount of siRNAs was kept constant using the control siRNA (C1). After 64 h, cells were harvested. Total RNAs were extracted and reverse transcribed. The amount of p21 cDNA was measured by real-time PCR and divided by the amount of P0 cDNA and standardised to 100% relative to the amount of cDNA in cells transfected by the C1 siRNA alone. A representative experiment is shown. (B) U2OS cells transfected as in (A) were harvested in Laemmli sample buffer and cell extracts were analysed for Rb (top panel), p21 (middle panel) and HDAC (loading control, bottom panel) expression by Western blot. (C) U2OS cells were transfected with the indicated siRNAs as in (A). After 64 h, the cell cycle distribution of transfected cells was analysed by flow cytometry.
Discussion
Tip60 and the p53 pathway
In this manuscript, we analysed the role of two members of the so-called Tip60 complex in the cellular response to DNA damage. Our results, together with previous findings (Berns et al, 2004; Legube et al, 2004) indicate that Tip60 is required both for p53 dependent cell cycle arrest and apoptosis following DNA damage. Importantly, our data (Figure 3 and Supplementary data 3) as well as data obtained by others demonstrate that Tip60 is important for the expression of many p53 target genes (p21, fas, bax, hdm2, Apaf1, PTEN, PIG3) (Berns et al, 2004; Doyon et al, 2004; Legube et al, 2004), indicating that it mediates its effects as a cofactor of p53. How can Tip60 affect p53 function? One possibility could be that Tip60 expression is required for a signalling pathway leading to p53 activation. Although we cannot definitively rule out this possibility, we think it is unlikely, since (i) we found that Tip60 expression is not required for some UV-inducible phosphorylation events on p53 (Figure 4); (ii) we and others previously found that Tip60 knockdown specifically affects the p53-dependent G1 arrest but not the p53-independent G2/M arrest following DNA damage (Berns et al, 2004; Legube et al, 2004), indicating that the pathway leading to the G2/M arrest is intact in Tip60 knocked-down cells; (iii) Tip60 is required for activation of the p21 promoter by overexpressed p53 in the absence of stress signalling (Legube et al, 2004). Thus, Tip60 likely functions downstream of p53 activation. Another important and still unresolved issue is whether the effect of Tip60 on p53-dependent transcription is direct or not. We show that Tip60 and p53 interact in a UV-inducible manner and it has previously been found that TRRAP, a component of the Tip60 complex but present in other complexes as well, binds to p53 and mediates transcriptional activation by p53 (Barlev et al, 2001; Ard et al, 2002), in agreement with the possibility that TRRAP/Tip60 directly regulates p53 activity. Since Tip60 is an HAT, which has been shown to function as a co-activator for many transcription factors, a tempting hypothesis is that Tip60 is recruited to its target promoters by p53 where it would mediate transcriptional activation by p53 through histone acetylation. Our results anyway indicate that Tip60 knockdown resulted in a decreased binding of p53 to its target sequences (Figure 6). The effect of Tip60 on p53-dependent genes expression can be explained by this finding, since transcription of these genes is dependent on p53 binding to their promoters.
What is the mechanism by which Tip60 allows p53 binding to the promoters? Interestingly, the DNA binding activity of p53 is known to be regulated by acetylation (Gu and Roeder, 1997; Luo et al, 2004). It is thus tempting to speculate that Tip60 directly acetylates p53, thereby inducing its ability to bind DNA. It is important to note that, in our hands, bacterially produced GST-Tip60 or Tip60 immunoprecipitated from cells were not able to acetylate recombinant bacterially expressed p53 in vitro (G Legube and D Trouche, unpublished results). The possibility still remains that specific post-translational modifications of p53 or Tip60 or the presence of additional co-factors are required for this acetylation to occur. Alternatively, Tip60 can affect the function of other HATs, such as CBP/p300, which are known to acetylate p53 (Gu and Roeder, 1997). Consistent with this possibility, we recently showed that Tip60 and p300 physically interact and that p300 regulates Tip60 expression (Col et al, 2005). Whether p300 properties are also modified through Tip60 binding remains to be investigated.
p400: a negative regulator of Tip60
Our data also show that p400 knockdown leads to cell cycle arrest, indicating that p400 expression is required for cell cycle progression. The role of p400 in cell cycle progression is due to its ability to repress the expression of p21, a major regulator of cyclin/cdk activity. What is the molecular mechanism involved? We found that p400 on the one hand and Tip60 and p53 on the other have antagonistic roles on p21, fas and hdm2 mRNA expression, since Tip60 or p53 knockdown decrease their expression whereas p400 knockdown stimulates it. Moreover, we observed that Tip60 knockdown results in lower binding of p53 to fas and p21 promoters. Strikingly, p400 knockdown has been shown to favour p53 binding to the p21 promoter (Chan et al, 2005), a finding that we confirmed in U2OS cells (Supplementary data 6). Thus, p400 and Tip60 have also antagonistic roles on p53 binding to the p21 or the fas promoter.
Importantly, we found that the effects of p400 knockdown on p21 and fas expression are entirely Tip60-dependent, indicating that p400 is a negative regulator of Tip60. Altogether, our data indicate that Tip60 favours p53 binding to the p21 or the fas promoter by an unknown mechanism (see above), and that the major role of p400 is to repress this function of Tip60. Consistent with such a mechanism, the effects of p400 are p53-dependent, since the phenotype induced by the p400 siRNA can be reversed, at least partially, by co-transfection of p53 siRNA (Supplementary data 7).
Since Tip60 and p400 are both present in the Tip60 complex, p400 could repress Tip60 function in the context of the Tip60 complex. Strikingly, the Tip60 complex is separated in two different complexes in yeast, the NuA4 complex containing the HAT ESA1, the orthologue of Tip60, and the SWR1 complex, containing the orthologue of p400 (Doyon et al, 2004). It is thus tempting to speculate that the orthologue of the SWR1 complex (i.e. a subcomplex including p400 and Tip49b) could function as a regulatory module within the Tip60 complex, controlling the function of a subcomplex orthologue to the NuA4 complex. Note, however, that we cannot rule out for the moment the possibility that the effect of p400 on Tip60 function is indirect.
Whatever the mechanism, we did not observe any stimulating effects of p400 knockdown on p21, hdm2 or fas expression in UV-irradiated (Figure 3) or doxorubicine- or cisplatine-treated cells (Supplementary data 1), although their expression is still dependent on Tip60. This result suggests that the repressive effects of p400 on Tip60 function are abolished upon DNA damage and that p400 is directly or indirectly a target of DNA damage-induced signal transduction pathways. Further works are required to clarify this point.
Role of the Tip60 complex in the response to DNA damage
Our data together with published data demonstrate that Tip60 expression is required for p53-dependent cell cycle arrest and apoptosis following DNA damage. Here, we observed that two other members of the Tip60 complex, p400 and Tip49b, are also required for apoptosis. Whereas Tip60 controls apoptosis, at least in part, as a co-factor for p53, the primary role of p400 in apoptosis is mediated through the repression of p21 expression in unstressed cells. Indeed, p21 levels are a major determinant in the cell decision to stop proliferating or to enter apoptosis following DNA damage (Seoane et al, 2002). In our study, we were able to change the fate of irradiated cells from apoptosis to cell cycle arrest by manipulating p400 levels. It would be thus interesting to find signals that would lead to such an effect. Indeed, according to our findings, one could predict that cells with lower p400 expression would be less sensitive to DNA-damage induced apoptosis.
Our findings anyway indicate that members of the Tip60 complex play distinct roles in the control of DNA-damage-induced apoptosis, suggesting the existence of subcomplexes with different functions. In agreement with this possibility, a complex similar to the Tip60 complex but devoid of Tip60 has been described (Fuchs et al, 2001), suggesting that the presence of Tip60 within its complex is a regulated event. The relative levels of these subcomplexes are likely to play major roles in the control of cell fate following DNA damage. Clearly, a major challenge in the future is to characterise potential DNA damage-induced modifications of Tip60 subcomplexes formation, and the molecular mechanisms involved. Finally, the finding that members of the Tip60 complex are required for apoptosis following DNA damage indicates that loss of their activity can lead to the accumulation of heritable mutations. The possibility of pathological modifications of expression levels or activity of these proteins in cancer thus deserves investigation.
Materials and methods
Antibodies, siRNAs and plasmids
The anti-PARP, anti-S37P p53 and anti-S392P p53 antibodies were purchased from Cell Signaling Technology, the anti-p53 antibody (DO-1) from Santa Cruz, the anti-S15P p53 from Calbiochem, the anti-HDAC3 antibody (which recognises also HDAC1 and HDAC2) from Transduction Laboratories, the anti-HA antibody from Covance, the anti-Rb (G3-245) and anti-p21 antibodies from Pharmingen (Becton-Dickinson) and the anti-p400 antibody from Abcam. All secondary antibodies were purchased from Amersham. All siRNAs were purchased from Eurogentec and are described in Supplementary data 8. C1 does not recognise any human mRNA. Vectors for standard curves used in Q-PCR were constructed by inserting the various amplification products from cDNA preparations or ChIP inputs into pGEMT-easy vector (Promega), according to the manufacturer's instructions. All PCR inserts were entirely sequenced.
Cell culture, transfections and UV-irradiation
U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with antibiotics and 10% fetal calf serum. A cell line overexpressing HA-Tip60 was constructed following transfection of pcDNA3 HA-Tip60 in U2OS and a 3 weeks selection in G418 (400 μg/ml). Ten resistant clones were picked and one of them was used for the experiments following analysis of recombinant Tip60 expression by Western blot and RT-Q-PCR analysis. For oligofectamine transfection, 150 000 cells were transfected with 4 μl of oligofectamine (Invitrogen), and 10 μl of the double-stranded siRNAs (20 μM) according to the manufacturer's instructions. For electroporation, cells were transfected in 200 μl of serum-free medium with 20 μl of siRNAs (20 or 100 μM) on a Biorad electroporation device set to 250 V, 950 μF. Concerning UV-irradiation, cells were washed with TBS (Tris-buffered saline, 50 mM Tris pH 8.0, 150 mM NaCl) and irradiated as a monolayer with UV-C rays on a UV crosslinker (Hoefer). Culture medium was added immediately after irradiation.
Cell extracts, immunoprecipitation
For Western blotting analysis, cells extracts were prepared by lysing cells directly in Laemmli sample buffer. Immunoprecipitations were performed as previously described (Nicolas et al, 2000).
RNA extraction and reverse transcription
Total RNA was extracted using an RNeasy mini kit (QIAGEN) and eluted with 30 μl of RNase-free water. Two to five microlitres of each purified RNA preparation was reversed-transcribed for 50 min at 42°C in a 20 μL-reaction containing 0.5 μg of oligo-dT or Random Primers (Promega), 1 × AMV RT buffer, 10 U of AMV Reverse Transcriptase (Promega), 40 U of rRNasin (Promega), 10 mM DTT and 0.5 mM deoxynucleotides (Promega). The reaction was stopped by incubating the samples for 15 min at 70°C, and cDNAs were analysed by Q-PCR.
Chromatin immunoprecipitation assay
Formaldehyde was added to the culture medium to a final concentration of 1% and crosslinking was allowed to proceed for 10 min at room temperature. In order to stop the reaction, glycin was added to a final concentration of 0.125 M. After 5 min, cells were washed twice with cold PBS and harvested by scraping. Pelleted cells were first subjected to lysis in the following buffer: Pipes 5 mM pH 8, KCl 85 mM, NP-40 0.5%. This lysis was followed by homogenisation with a Dounce homogeniser and nuclei were harvested by centrifugation. Nuclei were then incubated in nuclear lysis buffer: 50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS and sonicated five times for 10 s at a power setting of 5 and 50% duty cycle (Branson Sonifier 250), so as to obtain DNA fragments of about 500–1000 bp. Samples were diluted 10 times in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.1, 167 mM NaCl) and subjected to a 45 min preclearing with 140 μl of previously blocked protein-A and protein-G beads (Sigma). Blocking was achieved by incubating the agarose beads with 500 μg of BSA and 200 μg of herring sperm DNA for 3 h at 4°C. Precleared samples were incubated overnight at 4°C with antibodies specific for p53 (10 μl) or without antibody as negative control. Immune complexes were then recovered by incubating the samples with 140 μl of blocked protein A/protein G beads for 2 h at 4°C on a rotating wheel. Beads were washed once in dialysis buffer (2 mM EDTA, 50 mM Tris pH 8, 0.2% Sarkosyl) and four times in wash buffer (100 mM Tris pH 8.8, 500 mM LiCl, 1% NP-40, 1% NaDoc). Elution from the beads was achieved by incubation in elution buffer (1% SDS, 100 mM NaHCO3) for 15 min. Crosslink was reversed by adding NaCl and RNase A to the samples and incubating overnight at 62°C. After a 2 h-proteinase K treatment, DNA was purified with the GFX PCR kit (Amersham), and analysed by Q-PCR.
Quantitative PCR analysis
Q-PCR analysis was performed on a i-Cycler device (Biorad) using the platinium SYBR Green qPCR SuperMix (Invitrogen), according to the manufacturer's instructions. All experiments included a standard curve. All samples were analysed in duplicates, and the mean and standard deviation were calculated. Primers are described in Supplementary data 8.
Analysis of cell cycle distribution
Cells were treated 30 min with BrdU (20 μM). Cells were then trypsinised, fixed with ethanol, and incubated for 30 min in 4 N HCl, 0.5% Triton. Cells were then extensively washed in PBS supplemented with 1% bovine serum albumin, incubated for 1 h with anti-BrdU antibody (BD Biosciences) and then 30 min with an FITC-conjugated secondary antibody (Sigma). Cells were then stained with propidium iodide (BD Pharmingen) and analysed by flow cytometry (FACScalibur) on FL1 (anti-BrdU) and FL3 (propidium iodide) to determine their cell cycle distribution.
Supplementary Material
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4
Supplementary Information 5
Supplementary Information 6
Supplementary Information 7
Supplementary Information 8
Acknowledgments
We thank Drs S Khochbin and B Amati for materials and C Chailleux for technical help with siRNA silencing and chromatin immunoprecipitations. S Tyteca was supported by a studentship from the French Ministry of Science and G Legube by a studentship from the ‘Ligue Nationale Contre le Cancer'. DT's group is supported by a grant from the ‘Ligue Nationale Contre le Cancer' as an ‘équipe labellisée'.
References
- Ard PG, Chatterjee C, Kunjibettu S, Adside LR, Gralinski LE, McMahon SB (2002) Transcriptional regulation of the mdm2 oncogene by p53 requires TRRAP acetyltransferase complexes. Mol Cell Biol 22: 5650–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell 110: 55–67 [DOI] [PubMed] [Google Scholar]
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell 8: 1243–1254 [DOI] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R (2004) A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428: 431–437 [DOI] [PubMed] [Google Scholar]
- Brady ME, Ozanne DM, Gaughan L, Waite I, Cook S, Neal DE, Robson CN (1999) Tip60 is a nuclear hormone receptor coactivator. J Biol Chem 274: 17599–17604 [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR (2004) A new class of C. elegans synMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev Cell 6: 563–576 [DOI] [PubMed] [Google Scholar]
- Chan HM, Narita M, Lowe SW, Livingston DM (2005) The p400 E1A-associated protein is a novel component of the p53 → p21 senescence pathway. Genes Dev 19: 196–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Col E, Caron C, Chable-Bessia C, Legube G, Gazzeri S, Komatsu Y, Yoshida M, Benkirane M, Trouche D, Khochbin S (2005) HIV-1 Tat targets Tip60 to impair the apoptotic cell response to genotoxic stresses. EMBO J 24: 2634–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16: 979–990 [DOI] [PubMed] [Google Scholar]
- Doyon Y, Selleck W, Lane WS, Tan S, Cote J (2004) Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol 24: 1884–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B (2003) MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V, Lane WS, Nakatani Y, Livingston DM (2001) The p400 complex is an essential E1A transformation target. Cell 106: 297–307 [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain [In Process Citation]. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC (2000) Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol 2: E65–E67 [DOI] [PubMed] [Google Scholar]
- Hlubek F, Lohberg C, Meiler J, Jung A, Kirchner T, Brabletz T (2001) Tip60 is a cell-type-specific transcriptional regulator. J Biochem (Tokyo) 129: 635–641 [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27 [DOI] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102: 463–473 [DOI] [PubMed] [Google Scholar]
- Kamine J, Elangovan B, Subramanian T, Coleman D, Chinnadurai G (1996) Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology 216: 357–366 [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR III, Abmayr SM, Washburn MP, Workman JL (2004) Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306: 2084–2087 [DOI] [PubMed] [Google Scholar]
- Legube G, Linares LK, Lemercier C, Scheffner M, Khochbin S, Trouche D (2002) Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. EMBO J 21: 1704–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D (2004) Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem 279: 44825–44833 [DOI] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA 101: 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW (2004) The p53 response to DNA damage. DNA Repair (Amst) 3: 1049–1056 [DOI] [PubMed] [Google Scholar]
- Minsky N, Oren M (2004) The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell 16: 631–639 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC (1994) Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9: 1799–1805 [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69: 1237–1245 [DOI] [PubMed] [Google Scholar]
- Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188: 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Morales V, Magnaghi-Jaulin L, Harel-Bellan A, Richard-Foy H, Trouche D (2000) RbAp48 belongs to the histone deacetylase complex that associates with the retinoblastoma protein. J Biol Chem 275: 9797–9804 [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85 [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419: 729–734 [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 102: 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, Livingston DM, Amati B (2004) E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol 24: 4546–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 13: 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley RT, Cote J (2003) The MYST family of histone acetyltransferases. Curr Top Microbiol Immunol 274: 203–236 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4
Supplementary Information 5
Supplementary Information 6
Supplementary Information 7
Supplementary Information 8