

Abstract
Ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia Rad3-related (ATR) and the Mre11/Rad50/Nbs1 complex ensure genome stability in response to DNA damage. However, their essential role in DNA metabolism remains unknown. Here we show that ATM and ATR prevent accumulation of DNA double-strand breaks (DSBs) during chromosomal replication. Replicating chromosomes accumulate DSBs in Xenopus laevis egg extracts depleted of ATM and ATR. Addition of ATM and ATR proteins to depleted extracts prevents DSB accumulation by promoting restart of collapsed replication forks that arise during DNA replication. We show that collapsed forks maintain MCM complex but lose Pol ɛ, and that Pol ɛ reloading requires ATM and ATR. Replication fork restart is abolished in Mre11 depleted extracts and is restored by supplementation with recombinant human Mre11/Rad50/Nbs1 complex. Using a novel fluorescence resonance energy transfer-based technique, we demonstrate that ATM and ATR induce Mre11/Rad50/Nbs1 complex redistribution to restarting forks. This study provides direct biochemical evidence that ATM and ATR prevent accumulation of chromosomal abnormalities by promoting Mre11/Rad50/Nbs1 dependent recovery of collapsed replication forks.
Keywords: ATM and ATR, FRET, Mre11/Rad50/Nbs1 complex, replication fork restart, Xenopus laevis
Introduction
Cells preserve genome integrity by maintaining the correct number and the proper structure of chromosomes throughout the cell cycle. Surveillance mechanisms, called checkpoints, counteract deleterious effects of endogenous and environmental sources of DNA damage by delaying cell cycle progression in the presence of DNA damage and coordinating DNA damage repair.
PI-3-like kinases, including ataxia-telangiectasia mutated (ATM) and Rad3-related (ATR) play central roles in DNA damage checkpoints. ATM is defective in ataxia-telangiectasia disease (A-T), which is characterized by cancer susceptibility, radio sensitivity and neurological defects (Shiloh and Kastan, 2001).
Following DNA damage, ATM becomes activated and leads to cell cycle arrest by promoting p53 stabilization in G1 (Morgan and Kastan, 1997) and by inhibiting origin firing at G1/S boundary inducing downregulation of Cdk2 activity (Costanzo et al, 2000).
ATR kinase, instead, responds to single stranded gaps in DNA. ATR is activated following recruitment through its interacting partner ATRIP onto RPA coated single-strand DNA (Zou and Elledge, 2003). Experiments with conditional knockout mice have shown that ATR is an essential gene. Cells lacking ATR undergo chromosome fragmentation in S-phase and subsequent cell death in the absence of apparent exogenous sources of DNA damage (Brown and Baltimore, 2000).
Cells are highly sensitive even to partial ATR deficiency, as shown by the Seckel syndrome phenotype (O'Driscoll et al, 2003). ATR is essential to maintain chromosome integrity by promoting stabilization of replication forks. Cells with a mutant allele of Mec1, the ATR ortholog in budding yeast, accumulate chromosomal breaks (Cha and Kleckner, 2002). Mec1 probably prevents replication fork collapse by controlling Rad53 kinase that is required to stabilize stalled replication forks in the presence of hydroxyurea (Lopes et al, 2001; Sogo et al, 2002; Tercero et al, 2003). Recently, an extensive crosstalk between ATR and ATM in response to DNA damage has been demonstrated (Jazayeri et al, 2006).
The Mre11/Rad50/Nbs1 complex is another major player in genome stability. The complex is among the first proteins to bind to double-strand breaks (DSBs) in vivo and it is required to activate the ATM-dependent response (Carson et al, 2003; Uziel et al, 2003; Costanzo et al, 2004). Mutations in Mre11 lead to ataxia-telangiectasia like disease (ATLD), a syndrome similar to A-T, implying that ATM and Mre11 function in the same pathway (Stewart et al, 1999).
We have previously shown that Xenopus Mre11 is required to prevent DSB accumulation during DNA replication (Costanzo et al, 2001). This indicates that Mre11 is not only required to respond to DSBs but also to resolve aberrant structures that arise during normal DNA replication and therefore could lead to formation of DSBs.
Recently, we have demonstrated that ATM and ATR function during unperturbed chromosomal replication in Xenopus egg cell free extract (Shechter et al, 2004).
Using this system, we now provide novel biochemical evidence that ATM and ATR prevent accumulation of chromosomal breaks during DNA replication by directly promoting Mre11-dependent repair and recovery of collapsed replication forks.
Results
ATM and ATR prevent DSB accumulation in replicating chromosomes
To determine whether ATM and ATR are required to prevent DSB accumulation during DNA replication, we performed the TUNEL assay that detects DSBs (Costanzo et al, 2001; Li et al, 2004). We monitored DSB accumulation on chromosomes and nuclei replicated for 120 min in normal and ATM and ATR deficient Xenopus egg cell free extracts, in the presence and in the absence of replication inhibitors such as Geminin, which prevents assembly of the replication complex or Roscovitine, which blocks the activity of Cyclin-dependent kinases (Cdks) required for origin firing.
To inhibit ATM and ATR activity we treated extracts with 5 mM caffeine, an ATM and ATR inhibitor. Chromosomes isolated from caffeine-treated extracts showed a significant increase in the TUNEL labeling measured as incorporation of α-32P-labeled dGTP in the presence of terminal transferase (TdT) (Figure 1A and C). The presence of DSBs was confirmed by detection of TUNEL positive nuclei isolated from caffeine-treated extracts and analyzed by deconvolution microscopy (Figure 1B). In contrast, chromosomal DNA replicated in untreated extracts showed no TUNEL labeling (Figure 1A–C). Caffeine might directly bind DNA and induce DSBs. To verify that DSB accumulation was due to the absence of active ATM and ATR, we incubated genomic DNA in interphase extracts that had been depleted of ATM and ATR. Biotinylated Xenopus Nbs1 C-terminal peptide (Falck et al, 2005; You et al, 2005) and specific polyclonal antibodies recognizing ATR were used to deplete Xenopus ATM and Xenopus ATR, respectively (Figure 1D). Biotinylated Xenopus Nbs1 C-terminal peptide specifically binds and depletes Xenopus ATM but does not bind Xenopus ATR (Figure 1D and Supplementary Figure 1). In the absence of ATM and ATR, we detected DSB accumulation during DNA replication (Figure 1A). Quantitative analysis showed that the number of DSBs was higher in genomes replicated in the absence of both ATM and ATR (Figure 1C). A similar number of DSBs accumulated in the presence of caffeine (Figure 1C), suggesting DSBs induced by caffeine are mostly due to inhibition of ATM and ATR activity. TUNEL performed on chromatin digested with different restriction endonucleases to produce a defined number of DSBs allowed us to establish that the TUNEL assay was in a linear range. The number of DSBs in ATR and ATM depleted extracts was estimated to be 100 per chromosome (not shown).
Figure 1.
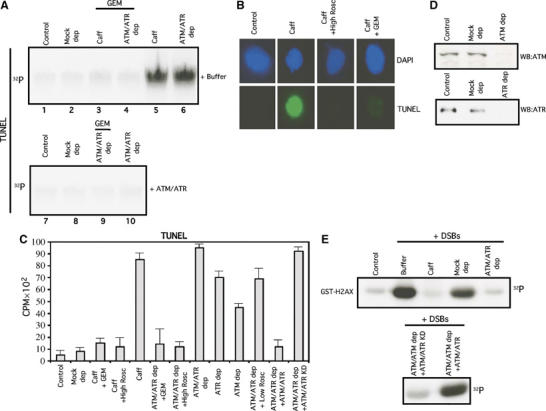
ATM and ATR prevent DSB accumulation during chromosomal DNA replication. (A) DSBs are detected by the TUNEL assay measuring incorporation of α-32P-labeled dGTP into genomic DNA in the presence of TdT (see Supplementary Materials and methods). Upper panel: Extracts supplemented with Buffer (+Buffer). Lower panel: Extracts supplemented with recombinant Flag-ATM and Flag-ATR (+ATM/ATR). Labeling of postreplicative chromatin isolated from untreated extracts (Control, lanes 1 and 7), mock-depleted extracts (Mock dep, lanes 2 and 8), an extract supplemented with 5 mM caffeine (Caff) and 4 ng/μl of Geminin (GEM) (lane 3), ATM and ATR depleted extracts (ATM and ATR dep) treated with 4 ng/μl of Geminin (lanes 4 and 9), an extract treated with 5 mM caffeine (Caff, lane 5) and ATM and ATR depleted extracts (ATM/ATR dep, lanes 6 and 10). (B) TUNEL assay on intact nuclei. Postreplicative nuclei stained with DAPI (upper panels) or with Fluorescein-dUTP in the presence of TdT (lower panels) (see Supplementary Materials and methods). Nuclei were isolated from an untreated extract (Control), an extract treated with 5 mM caffeine (Caff), an extract treated with 5 mM caffeine and 500 μM Roscovitine (Caff+High Rosc) or an extract treated with 5 mM caffeine and 4 ng/μl of Geminin (Caff+GEM). (C) Quantification of the TUNEL assay. Labeling of postreplicative chromatin isolated from a control extract (Control), a mock-depleted extracts (Mock dep), an extract supplemented with 5 mM caffeine and 4 ng/μl of Geminin (Caff+GEM), an extract supplemented with 5 mM caffeine and 500 μM Roscovitine (Caff+High Rosc), an extract supplemented with 5 mM caffeine (Caff), an ATM and ATR depleted extract treated with 4 ng/μl of Geminin (ATM/ATR dep+GEM), an ATM and ATR depleted extract supplemented with 500 μM Roscovitine (ATM/ATR dep+High Rosc), an ATM and ATR depleted extract (ATM/ATR dep), an ATR depleted extract (ATR dep), an ATM depleted extract (ATM dep), an ATM and ATR depleted extract supplemented with 5 μM Roscovitine (ATM/ATR dep+Low Rosc), an ATM and ATR depleted extract supplemented with Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR) and an ATM and ATR depleted extract supplemented with catalytically inactive Flag-ATM-KD and Flag-ATR-KD proteins (ATM/ATR dep+ATM/ATR KD). (D) Immune and peptide mediated depletion of ATM and ATR. Western blot analysis with anti-Xenopus ATM (upper panel) and anti-Xenopus ATR (lower panel) of control extracts (Control), mock depleted extracts (Mock dep), an extract depleted with biotinylated C-terminal peptide of Xenopus Nbs1 (ATM dep) and an extract depleted with anti-ATR antibodies (ATR dep). (E) Reconstitution of ATM and ATR activity in extracts. The activity of ATM and ATR protein kinases was monitored by incorporation of 32P from γ-32P-labeled ATP into GST fused to the C-terminal peptide of histone H2AX (GST-H2AX). 32P incorporation into GST- H2AX in the absence or in the presence of linear DNA at 50 ng/μl (+DSBs) was monitored in an extract incubated with buffer (Buffer), with 5 mM caffeine (Caff), in a mock depleted extract (Mock dep), in an ATM and ATR depleted extract (ATM/ATR dep), in an ATM and ATR depleted extract supplemented with catalytically inactive Flag-ATM and Flag-ATR proteins (ATM/ATR dep+ATM/ATR KD) and in an ATM and ATR depleted extract supplemented with recombinant Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR).
ATR depletion led to a higher number of DSBs than ATM depletion (Figure 1C). No DSBs were detected in mock-depleted extracts (Figure 1A and C). To confirm that DSB accumulation was due to the absence of ATM and ATR, we supplemented ATM and ATR depleted extracts with recombinant human ATM and ATR proteins purified from H293T cells overexpressing Flag-ATM and Flag-ATR. In the presence of recombinant ATM and ATR, replicating chromosomes did not accumulate DSBs during chromosomal DNA replication (Figure 1A and C). Importantly, catalytically inactive ATM and ATR were unable to prevent DSB accumulation in ATM and ATR depleted extracts (Figure 1C).
To verify that we could efficiently remove and reconstitute ATM and ATR activity in extracts, we measured ATM and ATR activity in an in vitro kinase assay using the C-terminal peptide of histone H2AX fused to GST (GST-H2AX) as substrate (Costanzo and Gautier, 2004; Costanzo et al, 2004). The addition of DNA containing DSBs to extracts stimulated GST-H2AX phosphorylation (Figure 1E). GST-H2AX phosphorylation was abrogated by caffeine or ATM and ATR depletion and restored by addition of recombinant ATM and ATR (Figure 1E). Catalytically inactive ATM and ATR, instead, were unable to restore GST-H2AX phosphorylation (Figure 1E).
We then asked whether DSB accumulation was dependent upon chromosomal DNA replication. We performed the TUNEL assay on chromatin incubated in ATM and ATR depleted or caffeine-treated extracts in the presence of Geminin or with a high dose of Roscovitine that completely suppresses DNA replication (Supplementary Figure 2). In the absence of chromosomal DNA replication, DSBs were drastically reduced (Figure 1A–C), indicating that DSBs arise spontaneously from replicating DNA in the absence of ATM and ATR.
To exclude that the TUNEL assay was labeling active replication intermediates, we monitored the kinetic of DNA replication completion. At the concentration of nuclei used for the TUNEL assay DNA replication was efficiently completed in 120 min in nondepleted extracts. In caffeine treated and ATM and ATR depleted extracts replication was completed slightly faster as shown by the kinetic of α-32P-labeled GTP incorporation (Supplementary Figure 3).
Inhibition of ATM and ATR during chromosomal replication leads to unscheduled origin firing (Marheineke and Hyrien, 2004; Shechter et al, 2004). To determine whether DSB accumulation was the result of an increased number of firing origins, we treated ATM and ATR depleted extracts with a low dose of Roscovitine that decreased the number of firing origins to the level of undepleted extracts (Supplementary Figure 2). Downregulation of origin firing by Roscovitine reduced DSB accumulation in ATM and ATR depleted extracts by 25% (Figure 1C), suggesting that DSB accumulation in the absence of ATM and ATR depends only partially on increased origin firing.
To confirm that we were detecting mostly DSBs and not single strand breaks (SSBs) or other replication intermediates such as reversed or collapsed forks, DNA isolated from untreated, caffeine treated or ATM and ATR depleted extracts was subjected to pulse field gel electrophoresis (PFGE), which has been successfully used to monitor DSB accumulation during DNA replication (Saleh-Gohari et al, 2005; Sorensen et al, 2005). Using PFGE we uncovered the presence of a significant amount of fragmented DNA derived from chromosomes replicated in caffeine treated or ATM and ATR depleted extracts (Supplementary Figure 4A). DNA fragmentation was suppressed in ATM and ATR depleted extracts supplemented with recombinant active ATM and ATR but not with catalytically inactive ATM and ATR. In addition, a high dose of Roscovitine or Geminin inhibited the accumulation of DSBs during DNA replication in ATM and ATR depleted extracts. DSBs in the presence of caffeine were further detected by the less sensitive neutral comet assay shown in Supplementary Figure 4B and C (see legend of Supplementary Figure 4B and C). These results represent additional and independent evidence showing that DSBs were accumulating during DNA replication.
ATM and ATR inhibit formation of DSBs that arise from collapsed replication forks
To determine whether active ATM and ATR are required to stabilize impaired replication forks, we monitored DSB accumulation during DNA replication in undepleted extracts and ATM and ATR deficient extracts challenged with DNA damaging agents. At 60 min (min) after addition of nuclei, when replication was still active (Supplementary Figure 3), egg extracts were supplemented either with Camptothecin (CPT), a Topoisomerase I inhibitor, or Mitomycin C (MMC), a potent crosslinking agent, in order to impair progression of replication forks (McHugh et al, 2000; Pommier, 2004). These treatments led to a caffeine-resistant replication arrest (Figure 3B). As most of the replication origins had fired at the time of CPT, and MMC addition DNA replication inhibition was dependent upon the physical block to replication fork progression induced by high doses of CPT or MMC and not by the DNA damage checkpoint.
Figure 3.
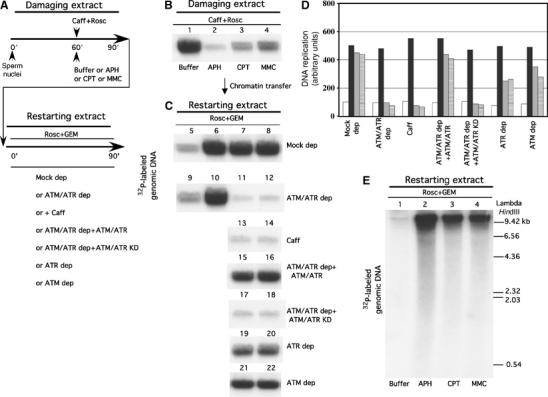
ATM and ATR promote restart of collapsed replication forks. (A) Experimental approach to dissect the role of ATM and ATR in the restart of collapsed replication forks. Chromatin replication is initiated in a first interphase extract (damaging extract). Replication fork progression is then blocked by addition of APH, CPT or MMC 60 min after nuclei addition to extracts in the presence of 5 mM caffeine and 500 μM Roscovitine. At 90 min after nuclei addition chromatin with damaged forks is isolated and incubated in a second interphase extract (restarting extract) made incompetent for origin firing and origin assembly by addition of 500 μM Roscovitine and 4 ng/μl Geminin. Different restarting extracts were used as stated in the figure. (B) DNA replication in damaging extracts supplemented at 60 min after nuclei addition with 5 mM caffeine, 500 μM Roscovitine and buffer (Buffer, lane 1), 40 μM APH (lane 2), 55 μM CPT (lane 3) or 300 μM MMC (lane 4). Replication was pulse labeled with α-32P-labeled dGTP from 60 min to 90 min from nuclei addition. (C) DNA replication in restarting extracts. Chromatin preincubated in damaging extracts was isolated after 90 min and then transferred to restarting extracts that contained 500 μM Roscovitine and 4 ng/μl Geminin. Replication was monitored with α-32P-labeled dGTP for 90 min. DNA was then isolated and run on neutral agarose gel. The different panels show replication of chromatin isolated from damaging extracts treated with Buffer (lanes 5 and 9), 40 μM APH (lanes 6 and 10), 55 μM CPT (lanes 7, 11, 13, 15, 17, 19, 21) or 300 μM MMC (lanes 8, 12, 14, 16, 18, 20, 22) and transferred to restarting extracts. Restarting extracts were mock depleted (Mock dep), ATM and ATR depleted (ATM/ATR dep), treated with 5 mM caffeine (Caff), ATM and ATR depleted and supplemented with recombinant Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR), ATM and ATR depleted and supplemented with catalytically inactive Flag-ATM and Flag-ATR proteins (ATM/ATR dep+ATM/ATR KD), ATR depleted (ATR dep) or ATM depleted (ATM dep). Note that the amount of DNA loaded on the gel in panel C is two-fold more than the amount of DNA loaded on the gel in panel B (see Supplementary Materials and methods). (D) Quantification of DNA replication in restarting extracts. Extracts were supplemented with buffer (white bars), 40 μM APH (black bars), 55 μM CPT (gray bars) or 300 μM MMC (striped bars) treated chromatin. Graph shown represents a typical experiment. (E) Nascent single strand DNA synthesis in restarting extracts. Chromatin was preincubated in damaging extracts treated with buffer (Buffer, lane 1), 40 μM APH (lane 2), 55 μM CPT (lane 3) or 300 μM MMC (lane 4), isolated and then transferred to restarting extracts. Replication was monitored for 60 min in the presence of α-32P-labeled dGTP. DNA was isolated and run on an alkaline agarose gel.
Both agents CPT and MMC induced DSB accumulation in normal extracts. However, in CPT- or MMC-treated extracts supplemented with caffeine or deprived of ATM and ATR DSB accumulation was highly increased (Figure 2A). Geminin or a high dose of Roscovitine added at 0 min prevented CPT- or MMC-induced DSBs (Figure 2B and C), demonstrating that DSB accumulation was dependent on chromosomal DNA replication. Addition of a low dose of Roscovitine that was able to inhibit the increased origin firing in the absence of ATM and ATR (Supplementary Figure 2) affected only partially DSB accumulation following CPT or MMC addition (Figure 2D). This implies that deregulated origin firing plays a marginal role in DSB accumulation. Taken together, these results suggest that ATM and ATR directly prevent DSB accumulation arising from collapsed replication forks.
Figure 2.
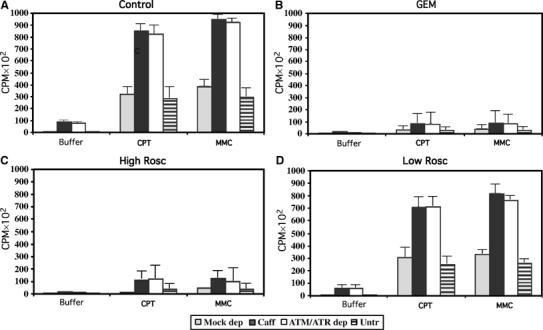
ATM and ATR inhibit DSB accumulation in the presence of damaged replication forks. DSBs detected by TUNEL labeling of postreplicative chromatin isolated after 120-min incubation in control extracts (A, Control), in extracts supplemented at 0 min with 4 ng/μl of Geminin (B, GEM), 500 μM Roscovitine (C, High Rosc) or 5 μM Roscovitine (D, Low Rosc). Extracts were supplemented at 60 min from nuclei addition with buffer (Buffer), with 55 μM CPT or with 300 μM MMC. Extracts were mock depleted (gray bars), supplemented with 5 mM caffeine (black bars), ATM and ATR depleted (white bars) or untreated (striped bars).
ATM and ATR promote restart of collapsed replication forks
To determine whether defective restart of collapsed replication forks caused DSB accumulation, we developed an assay where chromatin replication was initiated in a first extract (damaging extract) in which replication forks were stalled or collapsed by addition of either Aphidicolin (APH), CPT or MMC at 60 min after nuclei addition (see scheme in Figure 3A). This extract was also supplemented at 60 min after nuclei addition with a high dose of Roscovitine to block ongoing origin firing and with caffeine to inhibit ATM and ATR activity. After a brief incubation, chromatin bearing damaged forks was isolated and transferred to a new extract (restarting extract) made incompetent for origin firing and for new origin assembly by addition of Roscovitine and Geminin. APH, CPT or MMC treatment led to replication arrest in damaging extracts (Figure 3B). In the restarting extract, DNA replication partially resumed from damaged forks (Figure 3C and D). However, addition of caffeine or depletion of ATM and ATR completely impaired restart of replication forks damaged by CPT or MMC (Figure 3C and D). In contrast, replication forks stalled by APH were able to recover even in the absence of ATM and ATR or in presence of caffeine (Figure 3C and D). To test whether ATM and ATR were directly involved in restart of damaged replication forks, ATM and ATR depleted restarting extracts were supplemented with recombinant ATM and ATR. In this case, ATM and ATR restored normal DNA replication promoting replication fork restart (Figure 3C and D). Importantly, addition of ATR and ATM mutant proteins lacking catalytic activity was unable to promote fork restart (Figure 3C and D).
Intriguingly, extract depleted of either ATR or ATM alone was partially capable of supporting replication restart of CPT and MMC damaged chromatin (Figure 3C and D). This indicates that ATM and ATR have both overlapping and nonredundant roles in the recovery of damaged forks containing complex DNA lesions. As expected, chromatin isolated from an extract that was not supplemented with damaging agents did not replicate in restarting extracts due to the absence of stalled or collapsed replication forks from which replication could resume (Figure 3C and D). In addition, no replication was observed from sperm nuclei directly incubated in restarting extracts (not shown). To verify whether DNA replication observed in restarting extracts was due to re-establishment of functional replication forks, we monitored nascent DNA strands in restarting extracts. Using alkaline gel electrophoresis, we showed that high and low molecular weight single-strand DNA molecules could be isolated from restarting extracts containing undamaged chromatin or APH-, CPT- or MMC-treated chromatin (Figure 3E). This suggested that lagging and especially leading strand synthesis were efficiently taking place, being unlikely that DNA repair synthesis could generate the long nascent single-strand DNA molecules observed. To formally prove that α-32P-labeled dGTP incorporation was due to recovered replication forks and not to gap filling or repair synthesis, we performed BrdUTP density substitution (Li and Blow, 2005). Chromatin derived from damaging extracts supplemented with buffer, APH, CPT or MMC was incubated in restarting extracts in the presence of α-32P-labeled dGTP and BrdUTP. After 90 min, DNA was isolated and run on CsCl equilibrium gradients. Fractions from CsCl gradients revealed the presence of a single heavy light (HL) peak due to semiconservative DNA synthesis (Supplementary Figure 5), demonstrating that efficient DNA replication was taking place in restarting extracts.
ATM and ATR stabilize Pol ɛ binding to replication forks
In order to understand the mechanisms underlying ATM and ATR dependent recovery of collapsed forks, we monitored chromatin binding of MCM complex, Cdc45 and Pol ɛ in damaging and restarting extracts in the presence and in the absence of active ATM and ATR. Western blot analysis of chromatin isolated from damaging extracts after incubation with caffeine and APH, CPT or MMC showed that MCM7 and Cdc45 were regularly bound to chromatin (Figure 4A). APH treatment did not impair Pol ɛ binding in damaging extracts (Figure 4A). However, Pol ɛ binding to chromatin was lost in damaging extracts supplemented with CPT or MMC (Figure 4A). In ATM and ATR depleted restarting extracts, Pol ɛ reloading was impaired and was restored by the addition of purified recombinant ATM and ATR proteins (Figure 4B).
Figure 4.
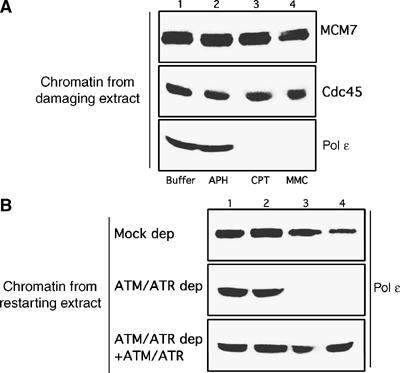
ATM and ATR promote reloading of Pol ɛ onto replication forks. (A) Western blot analysis of chromatin isolated from damaging extracts supplemented at 60 min from nuclei addition with 5 mM Caffeine, 500 μM Roscovitine+buffer (Buffer, lane 1), 40 μM APH (lane 2), 55 μM CPT (lane 3) or 300 μM MMC (lane 4) using anti-MCM7 antibodies (MCM7), anti-Cdc45 antibodies (Cdc45) or anti-Pol ɛ antibodies (Pol ɛ). (B) Western blot analysis using anti-Pol ɛ antibodies of chromatin isolated from damaging extracts that had been incubated at 60 min after sperm nuclei addition with 5 mM caffeine and 500 μM Roscovitine+buffer (Buffer, lane 1), 40 μM APH (lane 2), 55 μM CPT (lane 3) or 300 μM MMC (lane 4) and transferred to a mock depleted extract (Mock dep), an ATM and ATR depleted restarting extract (ATM/ATR dep) or an ATM and ATR depleted extract supplemented with recombinant Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR).
ATM and ATR promote Mre11 dependent DNA damage repair and fork restart
To test whether the Mre11 complex was required for the ATM and ATR dependent fork restart, we isolated chromatin replicated in Mre11 depleted damaging extracts supplemented with APH, CPT or MMC, and we incubated it in Mre11 depleted restarting extracts. Replication recovery of chromatin treated with APH was as efficient in Mre11 depleted extracts as in mock-depleted extracts (Figure 5A, lane 1 and 3C, lane 6). In contrast, no recovery was observed for CPT and MMC treated chromatin in Mre11 depleted restarting extracts (Figure 5A and B). Remarkably, addition of recombinant Mre11/Rad50/Nbs1 complex to Mre11 depleted extracts restored replication restart of CPT and MMC damaged chromatin (Figure 5A and B). ATM and ATR addition to Mre11 depleted extracts was not able to restore replication of Mre11 depleted restarting extracts (not shown). To exclude that poor restart was a consequence of inefficient ATM activation in the absence of Mre11 we added an excess of active ATM, purified from irradiated HEK293 T cells, to Mre11 depleted repairing extracts together with recombinant ATR. ATR and active ATM were not able to restore normal DNA replication (Figure 5A and B) demonstrating that ATM and ATR require Mre11/Rad50/Nbs1 complex to promote DNA damage repair and fork restart.
Figure 5.
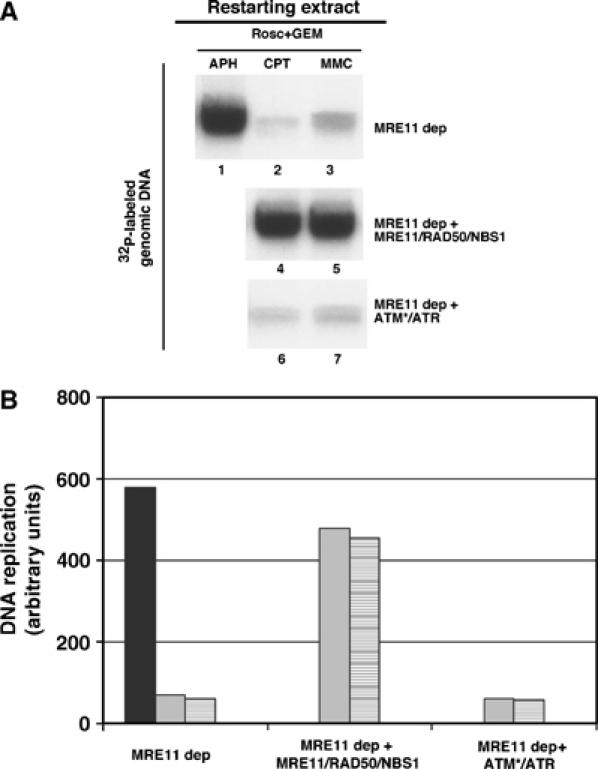
Mre11 complex is required for ATM and ATR-dependent fork restart. (A) DNA replication in restarting extracts. Chromatin was isolated from Mre11 depleted damaging extracts treated with 5 mM caffeine and 500 μM Roscovitine+40 μM APH (lane 1), 55 μM CPT (lanes 2, 4 and 6) or 300 μM MMC (lanes 3, 5 and 7) and then transferred to restarting extracts that were Mre11 depleted (Mre11 dep), Mre11 depleted supplemented with 500 nM of recombinant Mre11/Rad50/Nbs1 complex (Mre11 dep+Mre11/Rad50/Nbs1) or Mre11 depleted supplemented with Flag-ATR and active Flag-ATM proteins (Mre11 dep+ATM* and ATR). Asterisk (*) indicates that ATM has been isolated from irradiated cells (see Supplementary Materials and methods). Replication was monitored for 90 min in the presence of α-32P-labeled dGTP. DNA was then isolated and run on an agarose gel. (B) Quantification of DNA replication in Mre11 depleted restarting extracts. Extracts were supplemented with 40 μM APH (black bars), 55 μM CPT (gray bars) or 300 μM MMC (striped bars) treated chromatin. Graph shown represents a typical experiment.
ATM and ATR promote re-localization of Mre11 complex to restarting forks
To investigate the molecular mechanism underlying Mre11/Rad50/Nbs1 complex function in ATM and ATR dependent recovery of damaged forks, we designed a fluorescence resonance energy transfer (FRET) based experiment. This experiment allowed us to probe the proximity of Mre11/Rad50/Nbs1 complex to recovering forks. To overcome FRET detection problems such as auto-fluorescence and expected long distance between molecules, we used the phosphorescent FRET donor compound Europium (Eu3+). Phosphorescence bypasses short-lived auto-fluorescence (Chen et al, 2002) and excites FRET acceptor compound such as Cy5. We used Cy5 labeled dCTP (Cy5-dCTP) as acceptor compounds (Chen et al, 2002). Eu3+, excited at 340 nm, emits delayed fluorescence, known as phosphorescence, at 618 nm (Figure 6B). Energy transfer between Eu3+ and Cy5 within 10 nm can be measured as an increased emission of Cy5 at 665 nm (Chen et al, 2002). In our FRET-based assay, sperm nuclei were incubated in Mre11 depleted damaging extracts. At 60 min from nuclei addition, APH, CPT or MMC were added to these extracts to induce replication arrest. Damaging extracts were supplemented with Cy5-dCTP to label replicating forks just prior to stalling or collapsing. Cy5-dCTP labeled chromatin was then isolated at 90 min and incubated in restarting extracts containing Geminin and Roscovitine. These restarting extracts were then supplemented with Eu3+ labeled Mre11 complex (Eu3+-Mre11) at 0 min and at 45 min after chromatin addition. Eu3+ labeling did not impair Mre11 complex function in repair assays (not shown). Chromatin containing Eu3+-Mre11 was isolated 15 min after addition of Eu3+-Mre11 complex to restarting extracts and emission at 665 and 618 nm was monitored in a fluorometer following excitation at 340 nm. The normalized ratio of the emission at 665 nm (Cy5) and at 618 nm (Eu3+) is shown in Figure 6C. Emission at 618 nm, which reflects the amount of Mre11 bound to the chromatin, did not vary significantly among samples containing chromatin isolated from restarting extracts supplemented at 0 or at 45 min with Eu3+-Mre11 (not shown). Instead, the emission at 665 nm of CPT or MMC damaged chromatin incubated in restarting extract was clearly increased at the early time point (Figure 6C). Emission at 665 nm was the result of FRET occurring between Eu3+-Mre11 complex and Cy5-dCTP labeled chromatin. FRET between Eu3+-Mre11 complex and Cy5 labeled damaged chromatin during early recovery of collapsed replication forks indicated that Eu3+-Mre11 complex was present at less that 10 nm from the Cy5 labeled DNA strands processed into restarting replication forks (Figure 6B). In contrast, no FRET was measured between Cy5 labeled chromatin damaged by CPT or MMC and Eu3+-Mre11 complex added at 45 min to restarting extracts, when fork had already progressed and moved away from Cy5 labeled DNA (Figure 6C). This showed that Mre11 complex was specifically recruited to damaged forks only at the early time point when fork recovery was occurring. Importantly, FRET between Cy5-dCTP and Eu3+-Mre11 was detected only on damaged chromatin incubated in the presence of active ATM and ATR (Figure 6C). Depletion of ATM and ATR or caffeine treatment inhibited recruitment of Mre11 complex to restarting forks. Consistently, addition of recombinant ATM and ATR proteins to ATM and ATR depleted extracts restored Eu3+-Mre11 complex localization to the site of fork recovery (Figure 6C). These experiments demonstrated that ATM and ATR promoted Mre11 redistribution to restarting forks. No FRET was detected in the presence of Cy5-labeled chromatin treated with APH (Figure 6C). This indicated that Eu3+-Mre11 did not localize to replication forks recovering from an APH induced replication block. To exclude spectroscopic interferences in FRET detection, we measured the emission at 665 and 618 nm in the presence of Eu3+-Mre11 complex and chromatin that did not contain Cy5-dCTP or in the presence of Cy5 labeled chromatin and unlabeled Mre11 complex. As FRET was not detected in both cases, we were able to exclude spectroscopic artifacts (Figure 6C).
Figure 6.
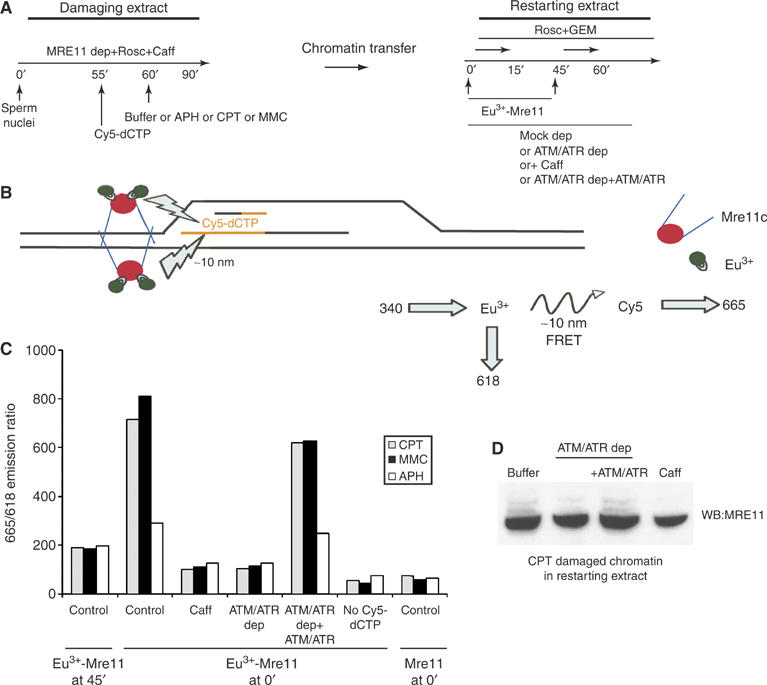
ATM and ATR promote redistribution of Mre11 complex to restarting replication forks. (A) Chromatin replication is initiated in damaging extracts. At 55 min after addition of sperm nuclei extracts are supplemented with Cy5-dCTP. At 60 min after nuclei addition extracts are supplemented with 40 μM APH, 55 μM CPT or 300 μM MMC. Cy5-dCTP labeled chromatin is then isolated and incubated in restarting extracts that were mock depleted (Mock dep), treated with 5 mM caffeine (Caff), ATM and ATR depleted (ATM/ATR dep) or ATM and ATR depleted supplemented with recombinant Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR). Eu3+-Mre11 labeled complex is added to restarting extract at 0 min or at 45 min from chromatin addition. Chromatin is isolated after 15 min and subjected to fluorometry. (B) Chromatin isolated from restarting extract is excited at 340 nm. Emission is monitored at 618 and 665 nm. Emission at 665 nm is the result of FRET between Eu3+-Mre11 localized in the range of ∼10 nm from Cy5-dCTP labeled DNA. (C) 665/618 nm emission ratio of chromatin preincubated in Mre11 depleted damaging extracts treated with CPT (gray bars), MMC (black bars) or APH (white bars). Cy5-dCTP labeled chromatin was isolated and transferred to restarting extracts supplemented with Europium labeled Mre11 complex at 0 (Eu3+-Mre11 at 0′) or at 45 min (Eu3+-Mre11 at 45′) from chromatin addition. As control nonlabeled Mre11 complex was added at 0 min from chromatin addition (Mre11 at 0′). Chromatin was then re-isolated from restarting extracts 15 min after addition of Eu3+-Mre11 and 665/618 nm emission ratio was measured in a fluorometer. Restarting extracts were mock depleted control, supplemented with caffeine (Caff), ATM and ATR depleted (ATM/ATR dep) or ATM and ATR depleted and supplemented with Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR). As a control, CPT, MMC or APH treated chromatin that did not contain Cy5-dCTP was transferred to restarting extracts supplemented with Eu3+-Mre11 complex at 0 min from chromatin addition (No Cy5-dCTP). (D) Western blot analysis of chromatin bound Mre11. Chromatin was preincubated in Mre11 depleted damaging extract containing 55 μM CPT and transferred to restarting extracts that were mock depleted buffer, ATM and ATR depleted (ATM/ATR dep), ATM and ATR depleted+Flag-ATM and Flag-ATR (ATM/ATR dep+ATM/ATR) or supplemented with 5 mM caffeine (Caff). For Western blot anti-Xenopus Mre11 antibodies were used.
Interestingly, Western blot analysis showed that Mre11 is present in its phosphorylated active forms on recovering forks (Figure 6D). Depletion of ATM and ATR or caffeine treatment abolished binding of phoshorylated Mre11 to CPT treated chromatin incubated in restarting extracts. This suggests that ATM and ATR promote directly or indirectly Mre11 phosphorylation that might be required for recovery of damaged replication forks.
Discussion
ATM and ATR preserve chromosome integrity
In this work, we have established cell-free systems to study the role of ATM, ATR and Mre11/Rad50/Nbs1 complex in DNA repair and replication fork stability. Using these systems we have demonstrated that ATM and ATR are required to prevent DSB accumulation during DNA replication by promoting restart of collapsed replication forks.
ATR but not ATM deficiency leads to increased expression during somatic cell cycle of fragile sites, specific chromosome locations in which DSBs can occur in mammalian cell lines (Casper et al, 2002). Our biochemical analysis reveals that ATM and ATR are partially redundant in protecting replicating chromosomes during embryonic DNA replication. Although ATR plays a more relevant role than ATM in this process, the absence of both ATM and ATR results in the accumulation of more DSBs than single ATM or ATR deficiency. It is possible that ATM deficiency leads to DSB accumulation mainly during embryonic and not somatic DNA replication.
The higher efficiency of ATR in preventing DSBs that we have documented might account for the differences in the severity of the phenotypes associated to the lack of ATR or ATM during development of vertebrate organisms.
Consistent with our results, recent findings demonstrate the existence of a crosstalk between ATM and ATR. Indeed, both ATM and ATR are required to respond to DSBs during S-phase, and ATM is required for ATR function in the presence of DSBs (Yoo et al, 2004; Jazayeri et al, 2006). DSB accumulation in the absence of ATM could alternatively be explained by an impaired ATR function.
ATM and ATR promote repair and restart of damaged replication forks
To examine whether ATM and ATR are involved in repairing and restarting disrupted replication forks, we have developed an assay to monitor recovery of stalled and collapsed replication forks. In this assay, replicating forks collapsed by CPT or MMC in the absence of active ATM and ATR are transferred to extracts lacking damaging agents in which origin firing and origin assembly is inhibited (restarting extracts). In restarting extracts, ATM and ATR get activated by damaged DNA and promote recovery of collapsed forks. Instead, restarting extracts lacking ATM and ATR do not support fork recovery. These data suggest a model in which ATM and ATR are normal components of replication forks that allow progression of DNA replication by promoting continuous repair and restart of collapsed forks. This model is consistent with the recent electron microscopy analysis of replication intermediates in checkpoint deficient cells (Lopes et al, 2006) that suggest the requirement for an active DNA damage checkpoint to restart DNA replication on damaged templates. It is possible that fork restart might require DNA recombination events stimulated by ATM and ATR, which are known to promote recombination mediated DNA repair (Sancar et al, 2004; Sorensen et al, 2005).
The DSBs that we observe might be secondary to nuclease processing of stalled or collapsed forks that cannot restart in the absence of ATM and ATR. Alternatively, DSBs might form when forks encounter SSBs in the parental strand. SSBs are commonly generated directly by reactive oxygen species and indirectly by repair of apurinic sites (Caldecott, 2004). Such unrepaired SSBs might generate DSBs during DNA replication leading to spontaneous fork collapse (Caldecott, 2003) requiring ATM and ATR to restart.
Intriguingly, ATM and ATR are not necessary in our experimental conditions to promote resolution of replication forks stalled by APH, similar to the findings of Blow and co-workers (Luciani et al, 2004). APH inhibits DNA polymerization and induces accumulation of extended regions of single-stranded DNA (Byun et al, 2005). However, APH does not induce extensive DSB accumulation during DNA replication in Xenopus egg extract (Li et al, 2004). This might be due to specific features of embryonic DNA replication such as a different chromatin organization or a high density of replication origins that could prevent replication fork collapse in the presence of APH and allow a rapid fork restart without any requirement for DNA repair.
Together, these observations strongly suggest that ATM and ATR are required to restart collapsed rather than transiently stalled forks.
ATM and ATR promote reloading of the replicative polymerase Pol ɛ
The behavior of the proteins involved in the assembly of the replication apparatus such as the MCM complex and Cdc45 on collapsed replication forks in vertebrates is poorly understood. Replication proteins are disassembled from collapsed forks in lower eukaryotes in the absence of a functional checkpoint (Sogo et al, 2002; Lucca et al, 2004). However, MCM7 was not removed from damaged chromatin in egg extract even in the absence of ATM and ATR. This difference could be explained by the observation that in vertebrates multiple MCM complexes are loaded on the chromatin for every active origin (Edwards et al, 2002). This redundancy might help to restart collapsed forks without reassembling a new replication complex.
Cdc45, instead, is not abundant in Xenopus extracts. In this case a different and yet unknown mechanism must be invoked to explain Cdc45 permanence on damaged forks.
Differently from MCM7 and Cdc45, Pol ɛ is lost from replication forks damaged by CPT or MMC but not by APH in the absence of ATM and ATR. Pol ɛ is required for efficient DNA replication, DNA repair, and cell cycle checkpoint control in eukaryotic cells. Xenopus egg extracts depleted of Pol ɛ are unable to assemble a proper DNA synthesis machinery at the fork (Waga et al, 2001) and Mec1 has been shown to be required for Pol ɛ presence on stalled replication forks (Cobb et al, 2003). Our findings indicate that ATM and ATR promote reloading of Pol ɛ onto recovering replication forks revealing a fundamental link between ATM, ATR and Pol ɛ in maintaining replication fork stability in high eukaryotes. Consistent with these results, checkpoint deficient cells have been shown to be defective in re-establishing DNA synthesis on damaged leading strand templates (Lopes et al, 2006).
As MCM7 and MCM2 are phosphorylated by ATR (Cortez et al, 2004; Yoo et al, 2004), it is tempting to speculate that this phosphorylation promotes reloading of Pol ɛ.
The essential function of Mre11 complex downstream of ATM and ATR
Mre11 is required for ATM activation in the presence of DSBs (Carson et al, 2003; Uziel et al, 2003; Costanzo et al, 2004). The Mre11/Rad50/Nbs1/ complex is essential for life, whereas cells tolerate ATM deficiency (Petrini et al, 1995; Dolganov et al, 1996; Xiao and Weaver, 1997; Haber, 1998; Luo et al, 1999; Kang et al, 2002; Uziel et al, 2003). This implies that Mre11 has other essential functions. Mre11 has been shown to localize to stalled replication forks suggesting a possible function during DNA replication (Mirzoeva and Petrini, 2003). We have previously shown that Mre11 is required to prevent DSBs during normal chromosomal DNA replication. Intriguingly, Mre11 is phosphorylated in a caffeine-sensitive manner during S-phase in Xenopus eggs extract (Costanzo et al, 2001). Our results suggest a model in which Mre11, ATM and ATR promote the recapture of the broken DNA molecules at collapsed replication forks favoring the reassembly of new functional forks, perhaps through DNA recombination mediated events. This hypothesis is consistent with previous findings showing that Mre11 complex is capable of tethering DNA fragments (de Jager et al, 2001; Hopfner et al, 2001; Costanzo et al, 2004). The FRET-based experiment supports this model by showing that ATM and ATR stimulate redistribution of Mre11 complex to restarting replication forks and suggest that ATM and ATR prevent DSB accumulation during DNA replication by coordinating Mre11/Rad50/Nbs1 complex functions (Figure 7).
Figure 7.
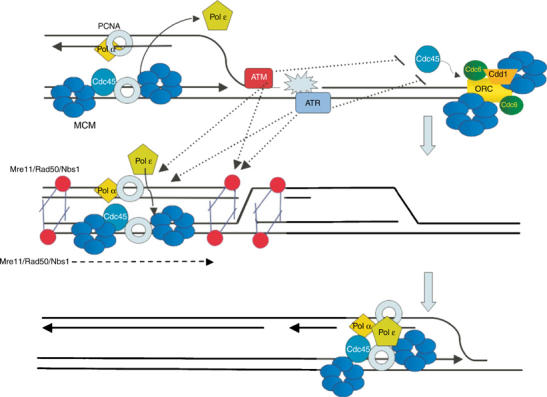
Multiple roles for ATM, ATR and Mre11 complex in promoting genome stability during DNA replication: ATM and ATR prevent DSB accumulation during normal or challenged DNA replication by regulating origin firing; by coordinating redistribution of Mre11 complex to collapsed replication forks; by stimulating restart of the collapsed replication fork and by promoting reloading of Pol ɛ onto recovering forks.
Thus, the essential role of Mre11 complex in vertebrates might be attributed to the restart of collapsed replication forks. The common features observed in the phenotypes of A-T, ATLD and Seckel syndrome might be explained by the inefficient restart of collapsed replication forks during embryonic DNA replication, a process that requires ATM, ATR and the Mre11 complex.
Materials and methods
Xenopus laevis egg extracts
Interphase egg extracts were prepared as described (Murray, 1991) from cytostatic factor (CSF) arrested egg extracts and released from mitosis by addition of 0.4 mM CaCl2 in the presence of 100 μg/ml of Cycloheximide.
Antibodies
Rabbit polyclonal Xenopus anti-ATR antibodies were raised by Harlan Sera Lab UK against a GST-tagged fragment corresponding to amino acids 2551–2654 of XATR. IgG were separated from crude serum with AffiBlue columns (Biorad). High-affinity IgG were then selected on affinity columns prepared coupling GST-ATR fragment to CNBR activated Sepharose (Amersham). Antibodies against Xenopus ATM and Xenopus Mre11 were previously described (Costanzo et al, 2000; Costanzo et al, 2001). MCM7 antibody was obtained from Santa Cruz. Anti-Cdc45 was a gift from Johannes Walter. Anti-Pol ɛ p60 subunit was a gift from Dr Akio Sugino.
Supplementary Material
Supplementary Figure Legends and Materials and Method
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank K Cimprich and M Kastan for the Flag-ATR and Flag-ATM constructs. We thank T Paull for Mre11/Rad50/Nbs1 baculovirus. In addition, we thank S Boulton and T Hunt for comments on the manuscript. This work was funded by Cancer Research UK.
References
- Brown EJ, Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402 [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19: 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW (2003) XRCC1 and DNA strand break repair. DNA Repair (Amst) 2: 955–969 [DOI] [PubMed] [Google Scholar]
- Caldecott KW (2004) DNA single-strand breaks and neurodegeneration. DNA Repair (Amst) 3: 875–882 [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD (2003) The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 22: 6610–6620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW (2002) ATR regulates fragile site stability. Cell 111: 779–789 [DOI] [PubMed] [Google Scholar]
- Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602–606 [DOI] [PubMed] [Google Scholar]
- Chen XC, Hentz NG, Hubbard F, Meier TI, Sittampalam S, Zhao G (2002) Development of a fluorescence resonance energy transfer assay for measuring the activity of Streptococcus pneumoniae DNA ligase, an enzyme essential for DNA replication, repair, and recombination. Anal Biochem 309: 232–240 [DOI] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Glick G, Elledge SJ (2004) Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA 101: 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Gautier J (2004) Xenopus cell-free extracts to study DNA damage checkpoints. Methods Mol Biol 241: 255–267 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Paull T, Gottesman M, Gautier J (2004) Mre11 assembles linear DNA fragments into DNA damage signaling complexes. PLoS Biol 2: E110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, Carroll D, Gautier J (2001) Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell 8: 137–147 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J (2000) Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell 6: 649–659 [DOI] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C (2001) Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 8: 1129–1135 [DOI] [PubMed] [Google Scholar]
- Dolganov GM, Maser RS, Novikov A, Tosto L, Chong S, Bressan DA, Petrini JH (1996) Human Rad50 is physically associated with human Mre11: identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol Cell Biol 16: 4832–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC (2002) MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem 277: 33049–33057 [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Haber JE (1998) The many interfaces of Mre11. Cell 95: 583–586 [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA (2001) Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 105: 473–485 [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8: 37–45 [DOI] [PubMed] [Google Scholar]
- Kang J, Bronson RT, Xu Y (2002) Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J 21: 1447–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Blow JJ (2005) Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J 24: 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Kim SM, Lee J, Dunphy WG (2004) Absence of BLM leads to accumulation of chromosomal DNA breaks during both unperturbed and disrupted S phases. J Cell Biol 165: 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- Lucca C, Vanoli F, Cotta-Ramusino C, Pellicioli A, Liberi G, Haber J, Foiani M (2004) Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23: 1206–1213 [DOI] [PubMed] [Google Scholar]
- Luciani MG, Oehlmann M, Blow JJ (2004) Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J Cell Sci 117: 6019–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH (1999) Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA 96: 7376–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marheineke K, Hyrien O (2004) Control of replication origin density and firing time in Xenopus egg extracts: role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem 279: 28071–28081 [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA (2000) Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol 20: 3425–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH (2003) DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res 1: 207–218 [PubMed] [Google Scholar]
- Morgan SE, Kastan MB (1997) p53 and ATM: cell cycle, cell death, and cancer. Adv Cancer Res 71: 1–25 [DOI] [PubMed] [Google Scholar]
- Murray AW (1991) Cell cycle extracts. Methods Cell Biol 36: 581–605 [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Petrini JH, Walsh ME, DiMare C, Chen XN, Korenberg JR, Weaver DT (1995) Isolation and characterization of the human MRE11 homologue. Genomics 29: 80–86 [DOI] [PubMed] [Google Scholar]
- Pommier Y (2004) Camptothecins and topoisomerase I: a foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: importance of DNA replication, repair and cell cycle checkpoints. Curr Med Chem Anti-Cancer Agents 4: 429–434 [DOI] [PubMed] [Google Scholar]
- Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T (2005) Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 25: 7158–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85 [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655 [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Kastan MB (2001) ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 83: 209–254 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T (2005) The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7: 195–201 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99: 577–587 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S, Masuda T, Takisawa H, Sugino A (2001) DNA polymerase epsilon is required for coordinated and efficient chromosomal DNA replication in Xenopus egg extracts. Proc Natl Acad Sci USA 98: 4978–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Weaver DT (1997) Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res 25: 2985–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HY, Shevchenko A, Dunphy WG (2004) Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem 279: 53353–53364 [DOI] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P (2005) ATM activation and its recruitment to damaged DNA require binding to the C Terminus of Nbs1. Mol Cell Biol 25: 5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends and Materials and Method
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5