

Abstract
In mammals, four different protein kinases, heme-regulated inhibitor, double-stranded RNA-dependent protein kinase (PKR), general control non-derepressible-2 (GCN2) and PKR-like endoplasmic reticulum kinase, regulate protein synthesis in response to environmental stresses by phosphorylating the α-subunit of the initiation factor 2 (eIF2α). We now report that mammalian GCN2 is specifically activated in vitro upon binding of two nonadjacent regions of the Sindbis virus (SV) genomic RNA to its histidyl-tRNA synthetase-related domain. Moreover, endogenous GCN2 is activated in cells upon SV infection. Strikingly, fibroblasts derived from GCN2−/− mice possess an increased permissiveness to SV or vesicular stomatitis virus infection. We further show that mice lacking GCN2 are extremely susceptible to intranasal SV infection, demonstrating high virus titers in the brain compared to similarly infected control animals. The overexpression of wild-type GCN2, but not the catalytically inactive GCN2-K618R variant, in NIH 3T3 cells impaired the replication of a number of RNA viruses. We determined that GCN2 inhibits SV replication by blocking early viral translation of genomic SV RNA. These findings point to a hitherto unrecognized role of GCN2 as an early mediator in the cellular response to RNA viruses.
Keywords: initiation factor, protein kinase, translational control, virus
Introduction
In eukaryotic cells, protein synthesis is mainly regulated at the level of initiation of mRNA translation. The reversible phosphorylation of the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) is a well-characterized mechanism of translational control in response to a wide variety of cellular stresses, including nutrient starvation, iron deficiency, heat shock, UV irradiation and viral infection (de Haro et al, 1996; Dever, 2002). Four different eIF2α kinases have been identified that specifically phosphorylate eIF2α on Ser-51. All known eIF2α kinases share a conserved kinase domain linked to unique regulatory regions. This feature confers on each kinase the ability to respond to a specific stress stimulus, thus ensuring that cells may become sensitized to a wide variety of stress-induced signals (Dever, 2002). Thus, heme-regulated inhibitor (HRI) is activated both by heme deficiency and under conditions of heat shock and oxidative stress (Lu et al, 2001). Double-stranded RNA-dependent protein kinase (PKR) is induced by interferon (IFN) and activated by double-stranded RNA (dsRNA) during viral infection (Kaufman, 2000). General control non-derepressible-2 (GCN2) is an eIF2α kinase that is activated by amino acid or serum deprivation and UV irradiation (Berlanga et al, 1999; Hinnebusch, 2000; Deng et al, 2002). The fourth eIF2α kinase, PKR-like endoplasmic reticulum (ER) kinase (PERK, also known as PEK), is activated by unfolded proteins in the ER (Shi et al, 1998; Harding et al, 1999).
It is well known that eIF2α phosphorylation can regulate both gene-specific and general translation. Thus, the eIF2α kinases phosphorylate eIF2α, leading to the inhibition of eIF2B activity. This generates low levels of ternary complex eIF2–GTP–Met-tRNAiMet, resulting in reduced general translation and increased translation of the transcription factors, GCN4 in yeast and ATF4 in mammals, which activate expression of their target genes involved in the stress response (Hinnebusch, 1997; Harding et al, 2000).
GCN2 was originally characterized in Saccharomyces cerevisiae as being required for amino-acid control of GCN4 mRNA translation. It is activated in amino-acid-starved cells through binding of uncharged tRNA to a region homologous to histidyl-tRNA synthetase (HisRS), located at the C-terminus of the kinase domain (Wek et al, 1989). In this case, eIF2α phosphorylation enhances the translation of GCN4, a transcriptional activator of genes involved in the biosynthesis of amino acids (Hinnebusch, 2000). A GCN2 ortholog identified in mammals (Berlanga et al, 1999; Sood et al, 2000) is also activated under conditions of amino-acid starvation (Harding et al, 2000). Moreover, the above mechanism for transcriptional activation of gene expression seems to be conserved throughout eukaryotic evolution. Thus, upon amino-acid deprivation, mammalian GCN2 stimulates the translation of the transcription factor ATF4, which activates the transcription of CHOP, a downstream target gene that is itself a transcription factor that controls expression of a set of stress-induced target genes (Harding et al, 2000). Although the GCN2 gene is not essential for cell viability, mice devoid of GCN2 show a poor adaptation to amino-acid deprivation (Zhang et al, 2002). Finally, the fact that mammalian GCN2 is also activated in response to specific stress signals such as serum deprivation (Berlanga et al, 1999) or UV irradiation (Deng et al, 2002) suggests the possibility that this eIF2α kinase might be involved in other stress-induced pathways.
Numerous studies have indicated that PKR plays a key role in IFN-mediated host defense against some viruses (Balachandran et al, 2000; Stojdl et al, 2000). In contrast, a number of studies have shown that PKR is not the only mechanism to antagonize virus infection, although the nature of PKR redundancy is not known (Abraham et al, 1999; Zhou et al, 1999). Thus, PKR has been shown to be nonessential in the innate antiviral response of animals challenged with encephalomyocarditis virus, vaccinia virus (VV), Sindbis virus (SV) and influenza virus (Yang et al, 1995; Abraham et al, 1999). These results provide strong evidence for the existence of alternative antiviral pathways (Abraham et al, 1999).
Here, we report that mammalian GCN2 is activated by the SV RNA genome. We propose that mammalian GCN2 is involved in an innate antiviral response pathway in the host defense against RNA viruses.
Results
Viral RNA activates mammalian GCN2
We first studied the activation of mammalian GCN2 by uncharged tRNAs, as occurs with its yeast counterpart (Wek et al, 1989), in a kinase activity assay using [γ-32P]ATP and rabbit reticulocyte eIF2 as a substrate. A significant increase in the phosphorylation of both GCN2 itself and eIF2α was detected in the presence of increasing concentrations of uncharged tRNA (Figure 1A). This treatment did not significantly affect PKR (Figure 1A). By contrast, the synthetic dsRNA poly(I)–poly(C), a well-known activator of PKR, was devoid of significant GCN2 activation capacity (Figure 1B). A mutation in the m2 motif of the HisRS domain in yeast GCN2 strongly decreased the binding to tRNA, thus making the kinase unable to phosphorylate eIF2α in response to amino-acid deprivation (Wek et al, 1995). The GCN2-m2 mutant protein possessed reduced autophosphorylation and eIF2α kinase activities, both in the absence or presence of uncharged tRNA (Figure 1A). Therefore, uncharged tRNA also activates mammalian GCN2, its HisRS-related domain being required for such activation.
Figure 1.
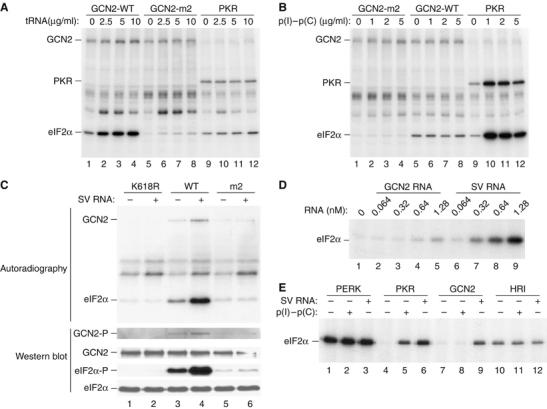
SV RNA stimulates GCN2-mediated phosphorylation of eIF2α. Purified wild-type GCN2 (GCN2-WT), the HisRS mutant (GCN2-m2) and PKR were assayed for their ability to phosphorylate eIF2α in the absence or the presence of increasing concentrations of uncharged bovine liver tRNA (Sigma) (A) or of poly(I)–poly(C) (Sigma) (B), as indicated. Phosphoproteins were analyzed by SDS–PAGE and autoradiography. Note an unknown phosphoprotein above the eIF2α band (A), which seems to be the consequence of the unspecific copurification of a kinase activity bound to TALON affinity resin. (C) In vitro eIF2α kinase assay of purified GCN2-WT, GCN2-K618R or GCN2-m2, in the absence or presence of SV RNA. Proteins were resolved into 10% SDS–PAGE and transferred to an immobilon-P membrane. The membrane was exposed to autoradiography (upper panels) and then probed with different antisera to detect eIF2α phosphorylated at serine 51 (eIF2α-P), total eIF2α and phosphorylated, and total GCN2 (lower panels) as indicated. (D) Kinase reactions were performed in the presence of the indicated concentrations of SV RNA or GCN2 RNA (as a negative control) and analyzed as described in (A). (E) Phosphorylation of eIF2α by Drosophila PERK, or mammalian PKR, GCN2 and HRI in the absence or presence of either poly(I)–poly(C) (1 μg/ml) or SV RNA (2.5 μg/ml, 0.64 nM). The analysis was carried out as described in (A).
Surprisingly, genomic SV RNA directly activated GCN2, enhancing both kinase autophosphorylation and eIF2α phosphorylation (Figure 1C). Moreover, this enhancement was not observed when the catalytically inactive GCN2-K618R variant was tested (Figure 1C). The finding that the GCN2-m2 mutant protein also possessed reduced autophosphorylation and eIF2α phosphorylation in response to SV RNA (Figure 1C) suggested that SV RNA activates GCN2 through binding to the m2 motif of its HisRS domain.
To examine the specificity of GCN2 activation by SV RNA, the effect of increasing concentrations of SV RNA or a control RNA derived from the GCN2 gene itself was assayed. A concentration of SV RNA as low as 1.28 nM was sufficient to yield a substantial activation of GCN2 (Figure 1D). In contrast, control RNA was nearly inactive at any of the concentrations tested. To test whether activation by SV RNA is a specific feature of GCN2, we examined the response of the four eIF2α kinases (GCN2, PERK, PKR, HRI) to this RNA as well as to poly(I)–poly(C). The subsequent in vitro kinase assay revealed that PERK and HRI phosphorylated eIF2α in the absence of activators (Figure 1E). Interestingly, whereas both SV RNA and poly(I)–poly(C) activated PKR, GCN2 activity was induced exclusively by SV RNA, suggesting that the presence of a dsRNA alone does not account for the activation of GCN2 and underscores the role of SV RNA as a novel activator of mammalian GCN2.
A bipartite sequence located at the 5′-terminus of SV RNA promotes the activation of GCN2
To determine the region of SV RNA responsible for the GCN2 activation, subgenomic RNAs encompassing different regions of 5′-terminus SV RNA were assayed. A construct bearing the first 2288 nucleotides (nts) was as active as the full-length SV RNA (Figure 2A–C), suggesting that all the elements necessary for GCN2 activation are contained within this region. Progressive deletions from the 3′-end of this RNA fragment led us to identify an element located between nucleotides 1920 and 2162 that was necessary to achieve full activation of GCN2 (Figure 2A). The fragment comprising nts 942–2288, which includes this putative activator element, did not bring about the same degree of activation as RNA nts 1–2288 (Figure 2B), suggesting that another sequence apart from nts 1920 to 2162 is also necessary. Additional experiments allowed us to define a new fragment (nts 502–1099) as relevant for GCN2 activation. Therefore, we cloned two fragments (nts 502–1099 and nts 1920–2168) separately or in tandem. Although these fragments were unable to activate GCN2 separately, the two joined fragments of RNA achieved a degree of activation similar to that obtained with fragment nts 1–2288 or full-length SV RNA (Figure 2C). These regions were named GAR for GCN2-Activating RNA. The activation by GAR was abolished when the RNA was first denatured by boiling at 100°C for 5 min, showing that the secondary structure of GAR is essential for activating GCN2. These results are summarized in Figure 2D.
Figure 2.
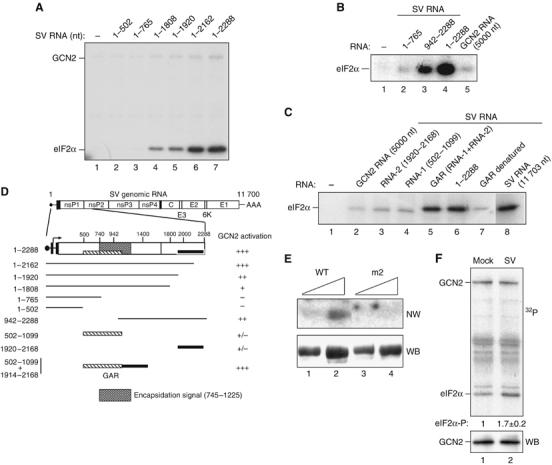
Activation of GCN2 by SV RNA involves two nonadjacent regions at the 5′-end of the viral RNA and the HisRS domain of GCN2. (A) The 5′-end-containing RNAs encompassing the indicated nucleotides of SV RNA were assayed for their ability to activate GCN2 as described in Figure 1. (B) Activation of GCN2 by the indicated fragments of viral RNA. GCN2 RNA was assayed as a negative control. (C) Fragments 1920–2168 (RNA-2) and 502–1099 (RNA-1) were tested alone or in combination, upon joining them by DNA recombinant technology (GAR construct; GCN2 Activating RNA). The heat-denaturated GAR fragment was also assayed, as well as native SV RNA 1–2288 and a full-length RNA (1–11 703). All RNAs were used at the same concentration (0.64 nM). In (B) and (C), only eIF2α phosphorylation is shown. (D) Schematic diagram depicting regions of SV genomic RNA involved in activation of GCN2 according to data from (A) to (C). GCN2 activation by the different RNA fragments is expressed as % of activity with respect to those obtained with full-length SV RNA: (+++), 75–100% activity, (++), 50–75% activity, (+), 25–50% activity and (+/−) less than 25% activity. Data are expressed as the mean of at least three independent experiments. GAR (hatched and black boxes) and the encapsidation signal in the SV genome (shadow box) are also drawn. (E) GCN2-(WT) and the HisRS mutant GCN2-m2 were purified as described in Materials and methods. Two different amounts of proteins were resolved in a 7.5% SDS–PAGE, transferred to a nitrocellulose membrane and probed with a 32P-labeled GAR fragment in a Northwestern blot assay (NW, top panel). The amount of loaded protein was monitored by reprobing the membrane with anti-myc antibody (WB, bottom panel). (F) MEFs were mock-infected or infected with SV at an MOI=50, and cell lysates were prepared at 2 h.p.i. and subjected to immunoprecipitation with anti-GCN2 antibody. Immune complexes were assayed for their ability to phosphorylate eIF2α (top panel). The amount of GCN2 was monitored by immunoblot using anti-MGCN2 antibody (bottom panel). The amount of 32P incorporated in eIF2α was quantified in a Phosphorimager BAS-1500 (Fujifilm). A representative experiment out of three that yielded similar results is shown. Numbers under the top panel represent the increase in eIF2α phosphorylation in immune complexes from SV-infected cells compared to those from mock-infected ones. Values indicate the mean±s.d.
Since GCN2 binds directly to uncharged tRNA through the HisRS domain, which is, in turn, essential for responding to SV RNA, we tested whether wild-type (WT) GCN2 and the mutant GCN2-m2 can bind the GAR sequence. As shown in Figure 2E, the WT GCN2, but not the mutant GCN2-m2, protein bound the GAR sequence. We therefore propose that the two nonadjacent regions at the 5′-end of SV RNA form a defined structure (GAR) that binds to the HisRS-related domain of GCN2 to promote kinase activation.
Additionally, we showed that endogenous GCN2 becomes activated in mouse embryo fibroblasts (MEFs) following SV infection (Figure 2F). Despite the existence of some basal levels of GCN2 activity in uninfected cell extracts, a consistent increase in the phosphorylation of eIF2α (about two-fold) was observed in SV-infected MEFs at 2 h postinfection (h.p.i.). This enhanced eIF2α kinase activity was definitively attributed to GCN2 activation since anti-GCN2 antibodies specifically precipitated an eIF2α kinase activity from WT, but not from GCN2−/− MEFs (see Supplementary Figure S1). Furthermore, under similar conditions, the in vitro eIF2α kinase activity of PKR immune complexes showed an increase only in SV-infected GCN2−/− MEFs, when the virus had already started replication and E1 protein had significantly accumulated (see Supplementary Figure S2). These data demonstrate that GCN2, but not PKR, could be activated, leading to phosphorylation of eIF2α, early during SV infection of WT cells.
SV infection of mouse cells lacking GCN2
To test the effect of the GCN2 gene product on SV replication in cultured cells, we used immortalized MEFs derived from GCN2−/− and control animals. Cells were infected at high (Figures 3A and 4B) or low (Figures 3B and 4A) multiplicities of infection (MOI) and metabolically labeled to analyze viral protein synthesis at various times postinfection. An extensive time course analysis of WT and GCN2−/− MEFs infected with SV at high MOI showed that an early, exponentially increasing phase of viral protein synthesis was followed by a late phase in which the rate of viral protein synthesis became constant and maximal in GCN2−/− cells. Strikingly, the kinetics of SV protein synthesis in WT MEFs was delayed about 8 h (Figure 3A). Thus, the E1 viral protein was first clearly detected in GCN2−/− and control cells at 4 and 12 h.p.i., respectively (Figure 3A, bottom). To confirm the increased permissiveness of GCN2−/− MEFs to SV infection, cells were infected with a lower MOI, and protein synthesis was monitored at longer times. In SV-infected GCN2−/− cells, there was a steadily increasing synthesis of SV proteins as well as a significant reduction in cellular translation at 6 h.p.i. (Figures 3B and 4A). At late times, there was a rapid decrease of viral protein synthesis due to lysis of the cells (Figure 3B). However, in MEFs derived from control mice, no viral proteins were detected, even at late times (Figure 3B).
Figure 3.
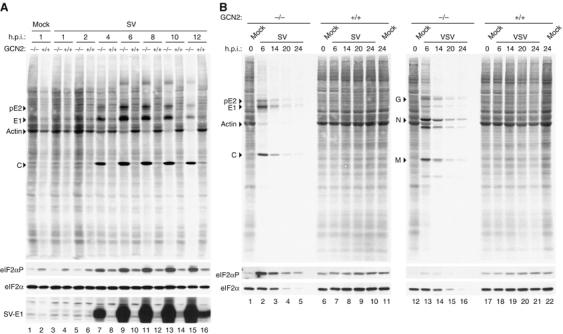
Cells were not infected (Mock) or infected with either SV at an MOI of 50 (A) or the indicated viruses at an MOI of 10 (SV) or 5 (VSV) (B) and metabolically labeled with [35S]Met-Cys for 30 min at the indicated h.p.i. Cells were lysed in a sample buffer and equivalent amounts of total protein were subjected to 12% SDS–PAGE, transferred to nitrocellulose membranes and subjected to autoradiography (upper panels). The membranes were then probed with specific antisera for the detection of SV-E1 protein, eIF2α-P and total eIF2α (bottom panels) as indicated. The position of the main viral protein bands of each virus as well as that of the actin band in mock-infected cells is also indicated.
Figure 4.
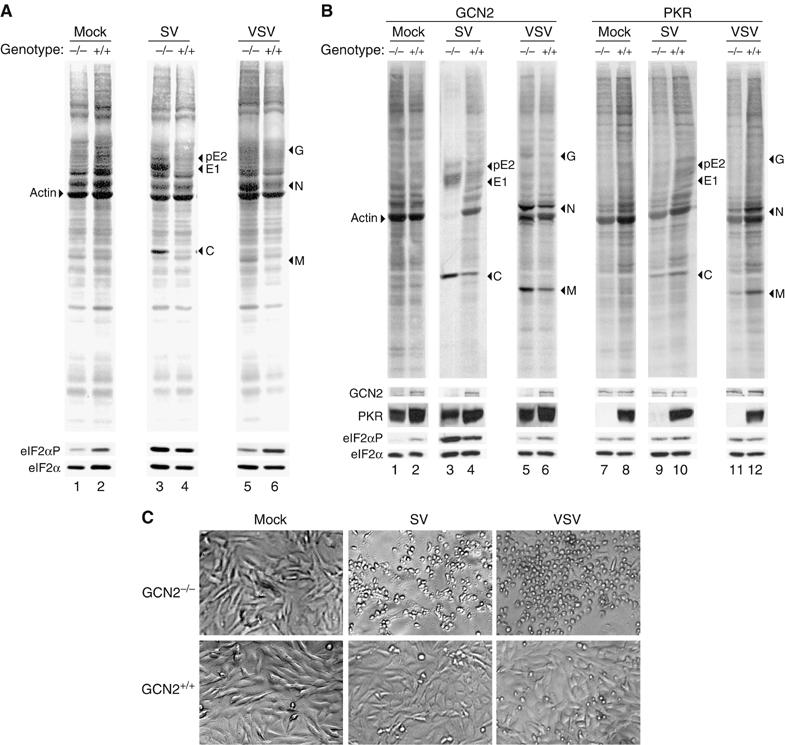
Cells were not infected (Mock) or infected with the indicated viruses at an MOI of either 10 (SV) and 5 (VSV) (A) or 50 (SV) and 25 (VSV) (B). Viral protein synthesis was analyzed at 6 h.p.i. as described in Figure 3 (upper panels). The membranes were then probed with specific antisera for detection of GCN2, PKR, eIF2α-P and total eIF2α (bottom panels) as indicated. (C) Result of GCN2 depletion on the cytopathic effect produced by the indicated viruses. Cells were infected as in (A) and phase-contrast micrographs of mock- or virus-infected cell monolayers were taken at 18–20 h.p.i.
To determine whether the permissiveness of GCN2−/− MEFs is restricted to SV, we performed the same analysis for vesicular stomatitis virus (VSV) infection. As occurred with SV, VSV protein synthesis was largely increased in GCN2−/− MEFs compared to WT MEFs (Figures 3B and 4A and B). It is interesting to note that both SV and VSV showed a marked cytopathic effect in GCN2−/− as compared to GCN2+/+ MEFs (Figure 4C). This finding most probably reflects the increased ability of SV and VSV to replicate in GCN2−/− cells.
It was recently reported that eIF2α phosphorylation initiates cytoprotective responses and that activation of these pathways can single handedly promote a stress-resistant preconditioned state (Lu et al, 2004). Thus, we examined the effect of GCN2 absence on other viruses such as VV and influenza virus (FLU). Remarkably, in contrast to SV and VSV, the pattern of VV and FLU protein synthesis in both GCN2−/− and GCN2+/+ MEFs at 6 h.p.i. was virtually identical (see Supplementary Figure S3), supporting the idea that GCN2 does not protect the cultured MEFs against these viruses. We conclude that the GCN2 genotype of cultured MEFs modulates their susceptibility to infection by some, but not all, viruses.
MEFs derived from PKR−/− and control mice were also assayed for their permissiveness to either SV or VSV infection. In the absence of IFN treatment, the synthesis of either SV or VSV proteins did not significantly increase in PKR-deficient fibroblasts. In fact, a slight decrease in the viral proteins was observed in PKR−/− compared to PKR+/+ MEFs (Figure 4B), due to a reduction in the rate of protein synthesis in PKR−/− compared to PKR+/+ MEFs. Equivalent levels of total eIF2α confirm that similar amounts of total protein are present in each lane (Figure 4B). We conclude that the elimination of PKR did not increase SV or VSV replication compared to control cells.
Although we found similar levels of phosphorylated eIF2α (eIF2α-P) in mock-infected WT and GCN2−/− MEFs when cells were grown in normal medium, the increased amount of eIF2α-P observed in mock-infected WT MEFs probably reflects the activation of GCN2 due to the low concentration of methionine during the metabolic labeling (see Supplementary Figure S4). PKR-null MEFs also contained levels of eIF2α-P similar to that of control cells. A slight increase in the amount of eIF2α-P was observed in the SV (or VSV)-infected GCN2+/+ MEFs compared to the levels found in mock-infected cells of the same genotype, which reflects GCN2 activation and the low level of replication of SV in those cells. The substantial increase in eIF2α-P observed in GCN2−/− cells infected with SV is indicative of a highly productive viral replication in these cells (Figures 3 and 4). These results suggest that PKR activation accounts for most of the eIF2α phosphorylation observed in SV-infected GCN2−/− cells.
Susceptibility of mice lacking GCN2 to SV infection
To test the role of GCN2 antiviral activity against SV infection in vivo, GCN2−/− and control (GCN2−/+) mice were infected intranasally (i.n.) with SV, and virus replication in the brains was analyzed. In the 129SvEv strain, SV replicates within the first 7 days of infection and subsequently declines due to the immune system-mediated clearance of virus. As early as 3 days after infection, GCN2−/− mice showed significant titers of SV in their brains, while control animals had no detectable virus (Figure 5A). On days 3 to 4 postinfection, brain titers in GCN2−/− animals were around 4 logs higher than those seen in control mice. From day 5 onward, amounts of virus were significantly increased in the brains of control animals and both GCN2−/− and control mice showed equally high levels (106 plaque-forming unit (PFU)/ml) of virus by day 7 (Figure 5A). Thus, the kinetics of SV replication was accelerated in GCN2−/− animals, while the kinetics of SV protein synthesis in GCN2−/− MEFs was accelerated as well.
Figure 5.
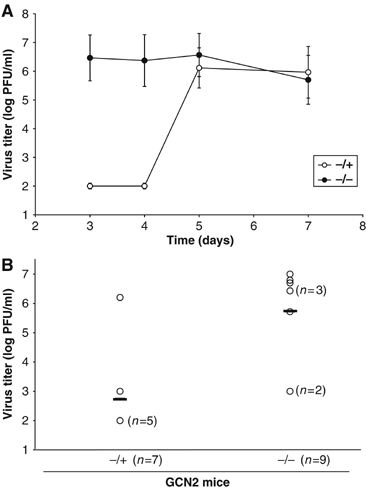
SV-infected GCN2−/− mice show high brain titers compared with control (GCN2−/+) mice. GCN2−/− and control mice were infected i.n. with 1 × 106 PFU and killed on different days after inoculation. Brain homogenates (3 ml) were titered by plaque assay on cell monolayers, as described under Materials and methods. (A) Viral titers at different days after inoculation. Two or three animals were used for each data point. Individual titers did not vary by more than one log. Data are representative of three independent experiments, and error bars represent s.d. of the mean values. (B) Viral titers at day 3 after inoculation. Viral titers below 102 PFU are not detectable by this method. Open circles represent individual titers, and the horizontal line corresponds to the mean of all individual titers (−/+=2.74±1.57; −/−=5.73±1.59). The differences in viral titers found between the two groups of mice are statistically significant (P<0.01).
To better determine the extent of viral infection at the early stages and obtain statistical values, a higher number of mice of each genotype were infected i.n. with 1 × 106 PFU and killed on day 3 postinfection for measurement of virus titers in the brain. As before, a significant majority of GCN2−/− mice showed high brain titers (more than 106 PFU/ml), whereas most control animals had no detectable virus (less than 102 PFU/ml) (Figure 5B). Collectively, our data show that GCN2 is critical for protection against SV at early times of infection.
The overexpression of GCN2 blocks SV replication
To further corroborate our conclusions, we analyzed the effect of GCN2 overexpression on virus replication in cultured cells. 3T3-derived cell lines that overexpress WT GCN2 or the K618R-inactive mutant were obtained. The genotype of these cell lines was confirmed by RT–PCR and by Western blot analyses, showing a GCN2 expression of 50–80% over the endogenous protein.
We then analyzed the replication of several viruses, including RNA viruses such as SV, Semliki forest virus (SFV), VSV and the DNA virus VV, in cells stably transfected with GCN2 constructs. As shown in Figure 6A, the ability of SV to form plaques was severely reduced in cells overexpressing WT GCN2. In contrast, the replication of VV was not hampered in those cells. For VSV and SFV, GCN2 had some effect on both the number and the size of lysis plaques. As expected, no effect was observed when the viruses were plaqued on cells overexpressing the K618R mutant. To better quantitate the effect of GCN2 overexpression on virus replication, the viral yields produced for each virus in a single cycle of infection was measured. As shown in Table I, SV viral titers were reduced more than 2 logs in cells overexpressing WT GCN2. The replication of SFV and VSV were also affected, but to a lesser extent (about a 1 log reduction).
Figure 6.
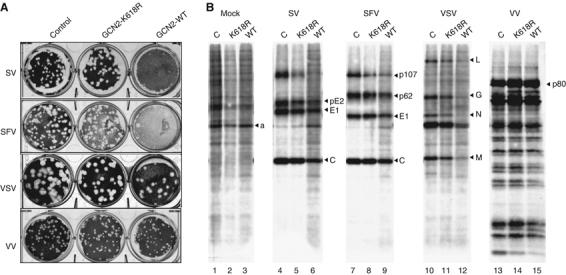
(A) NIH 3T3 cells bearing an empty vector (control), or expressing either the wild-type (GCN2-WT) or the K618R mutant form (GCN2-K618R) of GCN2 were infected with dilutions of the indicated viruses. After 3 days, lysis plaques were visualized. (B) NIH 3T3 cells were infected with viruses at an MOI of 10 PFU/cell and labeled with [35S]Met-Cys for 1 h at 5 h.p.i., except for vaccinia infections, where the labeling period started at 16 h.p.i. Cells were lysed in a sample buffer and equivalent amounts of total protein were analyzed by SDS–PAGE followed by autoradiography. C stands for cells bearing an empty vector.
Table 1.
Viral yields in a single cycle of infection
| 3T3 clone | Viral titer (log PFU/ml)a |
||
|---|---|---|---|
| SFV | SV | VSV | |
| Control | 9.02±0.35 | 8.20±0.15 | 7.74±0.12 |
| GCN2 (K618R) | 9.13±0.48 | 8.30±0.35 | 7.40±0.02 |
| GCN2 WT |
8.00±0.42 |
6.10±0.21 |
6.00±0.01 |
| Cells were infected with the indicated virus at an MOI of 5 and viral yields were titered in 3T3 cells. | |||
| aValues represent the mean±s.d. of three experiments using two different clones from both WT and mutant GCN2-expressing 3T3 cells. | |||
Given the role of GCN2 as a translational regulator, we speculated whether GCN2 overexpression could be inhibiting virus replication by blocking translation of viral mRNAs. Indeed, the synthesis of viral proteins was diminished, and the shutoff of host protein synthesis associated with infection by SV was attenuated in cells overexpressing WT GCN2 (Figure 6B), suggesting that the replication of these RNA viruses is, in fact, delayed. In agreement with data from Figure 6A, the synthesis of VV proteins was not impaired in cells overexpressing WT GCN2. Again, the antiviral effect observed was mainly due to the kinase activity of GCN2, since only minor effects were observed in cells overexpressing the K618R mutant.
GCN2 inhibits SV replication by blocking early viral translation of genomic RNA
To determine which step in the SV viral cycle is affected by GCN2 activation, we studied the translation of the nonstructural proteins (nsPs) from the incoming viral RNA, the first step that must occur in order to generate the proteins necessary to assemble a replication complex. To do so, we used an SV variant, designated Toto1101/Luc (Bick et al, 2003), which expresses Firefly luciferase as an in-frame fusion within nsP3 (Figure 7A) and allows for simple quantification of the levels of proteins generated by translation of incoming as well as replicated genomic RNA. We performed a time-course analysis of WT and GCN2−/− MEFs transfected with Toto1101/Luc RNA, and we found that translation of incoming viral RNA was reduced in control cells compared to that in GCN2−/− MEFs (Figure 7B). Moreover, in the presence of ribavirin, an inhibitor of viral RNA synthesis, protein synthesis did not significantly change until at least 6 h.p.i., most probably reflecting the reduced ability of SV to replicate in WT MEFs.
Figure 7.
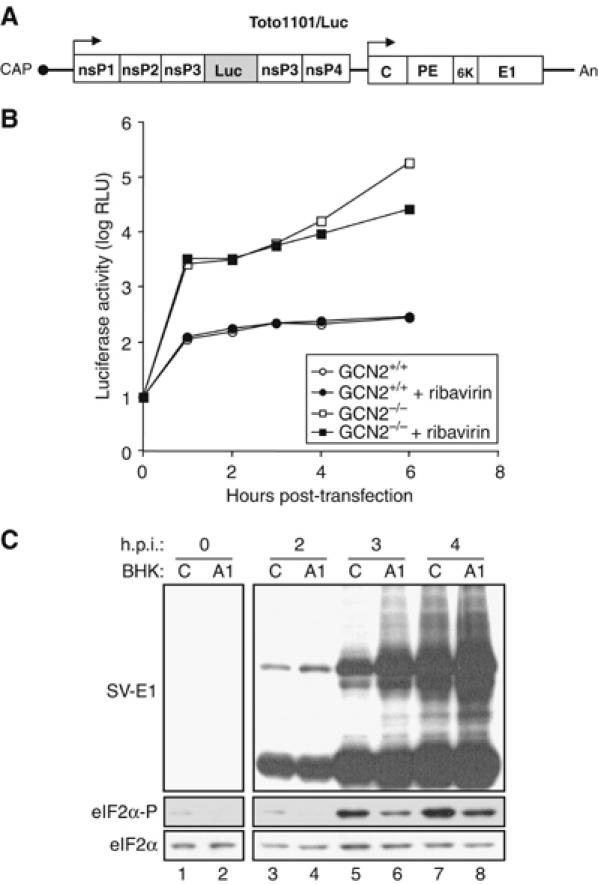
GCN2 blocks translation of incoming SV RNA by phosphorylating eIF2α. (A) Schematic representation of luciferase-expressing SV variant Toto1101/Luc. Solid lines represent UTRs, and the location of the cap and poly(A) tail (An) are indicated. The approximate locations of the nsPs, nsP1, nsP2, nsP3 and nsP4, and structural protein capsid (C), glycoproteins (PE and E1) and the 6 kDa protein (6K) are shown by open boxes. The shaded box within nsP3 corresponds to the Firefly luciferase encoding region. (B) MEFs derived from GCN2−/− and control mice were transfected with Toto1101/Luc RNA and at the indicated times luciferase activity was measured. Where indicated, cells were treated with 0.5 mM of ribavirin. Data are representative of three independent experiments. RLU, relative light units. (C) Control (C) and A1-transduced (A1) BHK cells were infected with SV (MOI=1). At the indicated h.p.i., cell lysates were analyzed by immunoblot with specific antisera for the detection of SV-E1 protein, eIF2α-P and total eIF2α as indicated.
To further examine the correlation between eIF2α phosphorylation and SV replication, we measured the impact of lowering the level of eIF2α-P in BHK cells overexpressing an active fragment of GADD34 (A1), the regulatory subunit of an eIF2α-specific holophosphatase complex, on the SV viral cycle. As predicted, the A1-transduced BHK cells showed reduced levels of eIF2α-P under basal conditions or when stressed (see Supplementary Figure S5). Interestingly, the viral cycle is accelerated in A1-transduced, compared to control, cells infected with SV, as shown by the earlier accumulation of structural viral proteins (Figure 7C).
Our data indicate that GCN2 blocks the synthesis of nsPs from SV RNA, thus preventing the replication of SV. Moreover, the observed translational inhibition of nsPs results mainly from the increased levels of eIF2α-P produced by the activation of GCN2 by viral genomic RNA.
Discussion
In this paper, we show that the mammalian GCN2 plays a novel role in the antiviral response to certain RNA viruses. Indeed, we have presented a large body of evidence that implicates GCN2 in the antiviral response of the cell. First, GCN2 binds and is activated in vitro by viral RNA sequences. Second, GCN2 is activated upon infection with SV. Third, cells devoid of GCN2 show a higher permissiveness for SV (or VSV) infection than control cells. The specific role of GCN2 is further supported by the fact that PKR-null cells did not show this increased susceptibility to promote SV (or VSV) replication. Furthermore, the antiviral effect attributed to GCN2 does not reflect the cytoprotective role of eIF2α phosphorylation, recently proposed for PERK in mammalian cells (Lu et al, 2004).
Fourth, and more importantly, mice lacking GCN2 show a significant increase in susceptibility to SV replication over control mice, demonstrating the physiological relevance of this mechanism. Fifth, overexpression of GCN2 leads to the suppression of SV (or VSV) replication, inhibits SV protein synthesis and significantly decreases viral production. Interestingly, these effects involve the enzymatic activity of GCN2, since overexpression of the catalytic inactive GCN2-K618R mutant did not show any effect on viral replication. Finally, we provide experimental evidence showing that GCN2 prevents replication of SV by phosphorylating eIF2α, thereby blocking early viral translation of genomic SV RNA. We consider these results to be compelling evidence that GCN2 plays a key role in host defense against viral infection.
Our results demonstrate for the first time that, in addition to uncharged tRNA, murine GCN2 is activated in vitro by viral RNA sequences. We mapped two short noncontiguous sequences (GAR) in SV RNA involved in GCN2 activation that seem to fold in a common structure. Owing to the apparent complexity of GAR, we do not yet know which specific elements of GAR are directly involved in GCN2 activation. Notably, one of the fragments of GAR (nts 502–1099) matched a previously defined sequence at nts 745–1225, involved in the encapsidation of SV RNA, which can be folded in a cloverleaf-type structure (Weiss et al, 1994; Frolova et al, 1997). Our studies also demonstrate that GCN2 directly binds SV RNA through its HisRS-related domain, which also binds uncharged tRNA.
Interestingly, we observed that other viral RNA genomes, including those for poliovirus and human immunodeficiency virus (HIV-1), also activated GCN2 in vitro (manuscript in preparation). We found important differences between GCN2 and PKR. Thus, GCN2, but not PKR, is activated by uncharged tRNA, and conversely, PKR, but not GCN2, is activated by the synthetic dsRNA poly(I)–poly(C). Furthermore, GCN2−/− MEFs show much more permissiveness for SV infection than control or PKR−/− MEFs. Our results fully agree with recent reports concluding that PKR does not play a role in alphavirus replication (Ryman et al, 2002; Ventoso et al, 2006). These experiments indicate that there may be at least two pathways leading to an antiviral state, one dependent on PKR (elicited by IFN or poly(I)–poly(C)) and another dependent on GCN2 and independent of IFN or poly(I)–poly(C), which prevents replication of SV in the early stages of the viral replicative cycle. Finally, our data indicate that other RNA viruses such as VSV or SFV show a similar susceptibility to GCN2, although to a lesser extent.
What is the molecular mechanism by which GCN2 inhibits viral replication? The most obvious explanation is that it does so by phosphorylating eIF2α, thereby blocking viral translation. Previous reports showed that PKR is activated upon infection with SV (Saito, 1990), and it has been suggested that the PKR-dependent inhibition of translation is not the only and, most likely, not the major pathway mediating translational shutoff during SV infection (Gorchakov et al, 2004). Our data are consistent with those results but further indicate that PKR activation induced by SV infection is more evident in cells devoid of GCN2, probably because PKR activation in these cells, but not in normal cells, occurs early in the infection as a consequence of the appearance of replicative forms of the viral RNA. We have shown that GCN2 is activated in SV-infected cells, and that a fraction of phosphorylated eIF2α in these cells can most probably be ascribed to the GCN2 activity. Moreover, our data indicate that GCN2 prevents replication of SV in the early stages of the viral replicative cycle by inhibiting translation of the incoming SV RNA through eIF2α phosphorylation. Therefore, we believe that infection-induced GCN2-mediated eIF2α phosphorylation plays an important role in host resistance to infection, and that GCN2 is an important component of innate cell ability to respond to RNA viruses. This is consistent with the fact that the GAR structure is only present in the genomic RNA, which is translated early to produce the nonstructural and replicase proteins necessary for viral RNA synthesis and the subsequent production of subgenomic 26S mRNA encoding the structural proteins (C, E1 and E2) (Strauss and Strauss, 1994), whose translation appears to be resistant to eIF2α phosphorylation (Ventoso et al, 2006).
What is the physiological relevance of this novel GCN2-dependent pathway involved in antiviral defense? The importance of GCN2 in preventing SV infection was underscored by the fact that mice lacking this eIF2α kinase show high titers in the brain, while no detectable virus was obtained in the brain of control animals at early times. This viral replication does not contribute to the death of these mice, in good agreement with previous reports showing that resistance to fatal SV infection in mice is age-dependent. Thus, at 4–5 weeks of age, most mice strains no longer develop a fatal disease (Thach et al, 2000). Our results clearly indicate that the replication of SV and VSV is inhibited by GCN2 activity. Interestingly, both viruses have a marked tropism for the brain (Ryman et al, 2000), where murine GCN2 is highly expressed (Berlanga et al, 1999). Additionally, during embryogenesis Drosophila melanogaster GCN2 mRNA is dynamically expressed in several tissues, but at later stages this expression becomes restricted to a few cells of the central nervous system (CNS) (Santoyo et al, 1997). All these observations underscore the plausible role of GCN2 in translational control and its potential physiological significance, especially in host defense against RNA viruses that infect the CNS.
Materials and methods
Cells, virus and plasmids
MEF cell lines established from WT (+/+) and knockout (−/−) GCN2 (Harding et al, 2003), and PKR (Abraham et al, 1999) animals have been described. MEFs, NIH 3T3 and HEK 293T cell lines were grown in Dubecco's modified Eagle's medium containing 10% (v/v) fetal calf serum.
The plasmid encoding a constitutively active GADD34 C-terminal protein fragment was expressed in BHK cells by transduction of the A1 retrovirus, as described previously (Novoa et al, 2001). Cells were selected by culture in normal growth medium supplemented with puromycin (5 μg/ml).
Plasmid pT7-SV (Strauss and Strauss, 1994) bearing infectious SV cDNA was used to synthesize full-length and subgenomic RNAs by in vitro transcription. RNAs derived from internal sequences of the SV genome were obtained from the following constructs: pGEM-SV942-2288, the SmaI–BglII fragment from the pT7-SV plasmid, was subcloned into the pGEM-1 plasmid (Promega) using SmaI and BamHI enzymes; pGEM-SV1920-2168, the EcoRI-NheI fragment from pT7-SV, was subcloned into pGEM-1 using EcoRI and XbaI enzymes; and pGEM-SV502-1099+1914–2168, the SspI–MscI fragment (nts 502–1099) from pT7-SV, was subcloned into pGEM-1 digested with SmaI. The fragment MscI–BglII (nts 1914–2288) from pT7-SV was then placed after the first fragment using MscI and BamHI enzymes, generating GAR (GCN2 Activating RNA) cDNA. pcMGCN2-wt (Berlanga et al, 1999) and pcMHRI-WT (Berlanga et al, 1998) plasmids have been described. Human PKR cDNA (kindly provided by Dr J Gil) and D. melanogaster PERK cDNA (Pomar et al, 2003) were used as a template for PCR amplification and for cloning of the corresponding open-reading frames in a pcDNA3.1/Myc-His vector (Invitrogen) (pcPKR and pcPERK). pcMGCN2-K618R and pcMGCN2-m2 were obtained from pcMGCN2-wt using the QuickChangeTM site-directed mutagenesis kit (Stratagene). In pcMGCN2-K618R, lysine 618 was replaced by arginine. In pcMGCN2-m2, phenylalanine 1142 and arginine 1143 were replaced by leucine and isoleucine, respectively. Plasmid pToto1101/Luc has been described (Bick et al, 2003).
For transient transfections, 293T cells were plated on 60-mm dishes. Plasmids (5 μg/dish) were transfected by the calcium phosphate method (Graham and van der Eb, 1973) or using Lipofectamine™ and Plus™ Reagents (Invitrogen), according to the manufacturer's instructions. For stable transfections, NIH 3T3 cells in 100-mm dishes were transfected with pcDNA3.1/Myc-His, pcMGCN2-wt and pcMGCN2-K618R (8 μg/dish). Stably transfected cells were selected using growth medium containing 0.4 mg/ml of G418 (Gibco Life Technologies).
Stably transfected NIH 3T3 cell clones expressing WT or K618R mutant GCN2 cDNAs were infected with different viruses. SV, SFV and VSV were amplified in NIH 3T3 cells. VV was grown in HeLa cells. Plaque assay of viruses was carried out as described before (Ventoso and Carrasco, 1995).
Metabolic labeling of cells
Cells (5 × 105) were labeled with 50 μCi/ml (1 Ci=37 GBq, 1000 Ci/mmol) of [35S]Met/Cys mixture (Promix, Amersham) for 0.5–1 h in a Met/Cys-free growth medium and lysed in a sample buffer as described previously (Ventoso et al, 1998). Equivalent amounts of cell extracts were analyzed by SDS–PAGE/fluorography and exposed to X-ray films (Kodak).
In vitro synthesis of RNAs
Plasmids were used to synthesize full-length and subgenomic RNAs by in vitro transcription using a T7 RNA polymerase kit (Promega). Transcription reactions were treated with DNAase RQ1. The integrity of RNAs was confirmed by agarose gel electrophoresis. RNAs encompassing nts 1–2288, 1–2162, 1–1920, 1–1808, 1–765 and 1–502 were obtained by in vitro transcription from a pT7-SV plasmid linearized with BglII, BspHI, EcoRI, NheI, SmaI and SspI enzymes, respectively. GAR was obtained after in vitro transcription from a pGEM-SV (502–1099+1914–2168) plasmid linearized with NheI enzyme.
Affinity purification, immunoprecipitation, immunoblotting and eIF-2α kinase assays
Transiently transfected cells were washed once with phosphate-buffered saline containing 90 mM sodium fluoride, 17.5 mM sodium molybdate and 17.5 mM β-glycerophosphate and lysed in lysis buffer (20 mM Tris–HCl (pH 8.0), 200 mM NaCl, 10% (v/v) glycerol, 1% (v/v) Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 2 mM β-glycerophosphate, 1 mM tetrasodium diphosphate and a protease inhibitor cocktail (Complete®, Boehringer Mannheim)). Cell debris was removed by centrifugation. Cell lysates were incubated with TALON™ metal affinity resin (Clontech) for 30 min at 4°C. Metal affinity resin was washed twice with lysis buffer containing 10 mM imidazole, twice with lysis buffer containing 0.5 M NaCl and 10 mM imidazole, and twice with buffer H (20 mM HEPES/KOH, pH 7.6, 50 mM KCl, 1 mM MgCl2, 5% (v/v) glycerol) containing 10 mM imidazole before elution in buffer H containing 100 mM imidazole. After elution, dithiothretiol was added to adjust its concentration to 1 mM.
Mock- or virus-infected cells were lysed, at the indicated h.p.i., on lysis buffer as described above. Cell lysates were subjected to immunoprecipitation as described previously (Berlanga et al, 1998) using anti-MGCN2 antibody. After extensive washing, immune complexes were resuspended in buffer H, containing 1 mM DTT, and assayed for their ability to phosphorylate eIF2α.
Affinity-purified proteins or immune complexes were assayed for their ability to phosphorylate eIF2α in a total volume of 20 μl, without or with RNAs at various concentrations, for 30 min at 30°C in kinase buffer (20 mM Tris–HCl (pH 7.6), 2.5 mM MgCl2, 2.5 mM Mg (OAc)2, 0.25 mg/ml BSA, 50 μM ATP including purified rabbit reticulocyte eIF2 (0.5 μg) and 3 μCi of [γ-32P]ATP (3000 Ci/mmol)). Incubations were terminated by the addition of SDS sample buffer, and phosphoproteins were analyzed by SDS–PAGE on a 10% polyacrylamide gel (28.5:1 (w/w) acrylamide/bisacrylamide) and by autoradiography. When indicated, proteins were transferred to Immobilon-P membranes for further Western blot analysis using different specific antisera: rabbit anti-eIF2α phospho-Ser-51 (Research Genetics), mouse anti-eIF2α (Scorsone et al, 1987), rabbit anti-MGCN2 (Berlanga et al, 1999), rabbit anti-GCN2 phospho-Thr898 (Harding et al, 2000), rabbit anti-Tik 100A recognizing mouse PKR (Abraham et al, 1999), rabbit anti-E1 SV (Sanz et al, 2003) and mouse anti-myc (Invitrogen).
Northwestern blot assay
GCN2-(WT) and GCN2-m2 expressed in HEK 293T cells were bound to TALON™ metal affinity resin and then eluted using an SDS–PAGE loading buffer. Different volumes of the eluted proteins were resolved in a 7.5% SDS–PAGE, transferred to nitrocellulose membrane and probed with a 32P-labeled GAR fragment, as described previously (Wek et al, 1995). A radiolabeled GAR probe was produced by an in vitro transcription reaction in which 95% of the UTP was replaced by 50 μCi of [α-32P]UTP in the nucleotide mix.
Experimental infection of mice
The GCN2-deficient allele deletes exon 12, which encodes the ATP-binding loop of the kinase domain, and therefore, it lacks all kinase activity. A detailed description of the targeting strategy and allele structure has been published (Maurin et al, 2005). The mice used here were from a 129SvEv background. WT 129SvEv mice were obtained from Taconic, Germantown, NY. Control mice (GCN2−/+) were obtained by crossing GCN2−/− and WT animals.
129SvEv mice (4–6 weeks old) were used throughout. Anesthetized mice were infected i.n. with 1 × 106 PFU of SV diluted in 10 μl of PBS applied to the nares of each animal. Mice were killed, and the brains were aseptically removed and snap-frozen on liquid nitrogen. Specimens were homogenized in 3 ml of PBS on ice, and titers were determined on NIH 3T3 cell monolayers as described above.
Determination of luciferase activity
Cells were washed once with PBS, lysed using passive lysis buffer and luciferase activity was determined with the luciferase assay system (Promega) following the manufacturer's recommendations. Luciferase activity was measured on a Monolight 2010 luminometer (Analytical Luminescence Laboratory).
Supplementary Material
Supplementary Information
Acknowledgments
We thank Mónica Elías for useful experimental work during the initial stage of this work. We thank Charles M Rice and Isabel Novoa for plasmid pToto1101/Luc and plasmids to generate A1-transduced BHK cell lines, respectively. We are grateful to Miguel A Sanz and JC Bell for providing us biological reagents and cells. We also thank José Alcalde and Colin Lister for excellent technical assistance. This work was supported in part by Grants PM98-0128 and BMC2002-03933 from the DGICYT (to C de H), USPHS (United States Public Health Service) Grants DK47119 and ES08681 (to DR) and by an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular ‘Severo Ochoa'. JJB was a recipient of postdoctoral fellowships from the Comunidad Autónoma de Madrid and the Ministerio de Educación y Ciencia.
References
- Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC (1999) Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem 274: 5953–5962 [DOI] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN (2000) Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13: 129–141 [DOI] [PubMed] [Google Scholar]
- Berlanga JJ, Herrero S, de Haro C (1998) Characterization of the hemin-sensitive eukaryotic initiation factor 2α kinase from mouse nonerythroid cells. J Biol Chem 273: 32340–32346 [DOI] [PubMed] [Google Scholar]
- Berlanga JJ, Santoyo J, de Haro C (1999) Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2α kinase. Eur J Biochem 265: 754–762 [DOI] [PubMed] [Google Scholar]
- Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR (2003) Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J Virol 77: 11555–11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haro C, Méndez R, Santoyo J (1996) The eIF-2α kinases and the control of protein synthesis. FASEB J 10: 1378–1387 [DOI] [PubMed] [Google Scholar]
- Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N (2002) Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol 12: 1279–1286 [DOI] [PubMed] [Google Scholar]
- Dever TE (2002) Gene-specific regulation by general translation factors. Cell 108: 545–556 [DOI] [PubMed] [Google Scholar]
- Frolova E, Frolov I, Schlesinger S (1997) Packaging signals in alphaviruses. J Virol 71: 248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I (2004) PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol 78: 8455–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52: 456–467 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (1997) Translational regulation of yeast GCN4. A window on factors that control initiator–tRNA binding to the ribosome. J Biol Chem 272: 21661–21664 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (2000) Mechanism and regulation of initiator methionyl–tRNA binding to ribosomes. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 185–243. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Kaufman RJ (2000) The double-stranded RNA-activated protein kinase PKR. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 503–527. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Lu L, Han AP, Chen JJ (2001) Translation initiation control by heme-regulated eukaryotic initiation factor 2α kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: 7971–7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP (2004) Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J 23: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin AC, Jousse C, Averous J, Parry L, Bruhat A, Cherasse Y, Zeng H, Zhang Y, Harding HP, Ron D, Fafournoux P (2005) The GCN2 kinase biases feeding behavior to maintain amino acid homeostasis in omnivores. Cell Metab 1: 273–277 [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153: 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomar N, Berlanga JJ, Campuzano S, Hernández G, Elías M, de Haro C (2003) Functional characterization of Drosophila melanogaster PERK eukaryotic initiation factor 2α (eIF2α) kinase. Eur J Biochem 270: 293–306 [DOI] [PubMed] [Google Scholar]
- Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE (2000) Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol 74: 3366–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman KD, White LJ, Johnston RE, Klimstra WB (2002) Effects of PKR/RNaseL-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol 15: 53–76 [DOI] [PubMed] [Google Scholar]
- Saito S (1990) Enhancement of the interferon-induced double-stranded RNA-dependent protein kinase activity by Sindbis virus infection and heat-shock stress. Microbiol Immunol 34: 859–870 [DOI] [PubMed] [Google Scholar]
- Santoyo J, Alcalde J, Méndez R, Pulido D, de Haro C (1997) Cloning and characterization of a cDNA encoding a protein synthesis initiation factor-2α (eIF-2α) kinase from Drosophila melanogaster. Homology to yeast GCN2 protein kinase. J Biol Chem 272: 12544–12550 [DOI] [PubMed] [Google Scholar]
- Sanz MA, Rejas MT, Carrasco L (2003) Individual expression of Sindbis virus glycoproteins. E1 alone promotes cell fusion. Virology 305: 463–472 [DOI] [PubMed] [Google Scholar]
- Scorsone KA, Panniers R, Rowlands AG, Henshaw EC (1987) Phosphorylation of eukaryotic initiation factor 2 during physiological stresses which affect protein synthesis. J Biol Chem 262: 14538–14543 [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18: 7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC (2000) A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2α. Genetics 154: 787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Abraham N, Knowles S, Marius R, Brasey A, Lichty BD, Brown EG, Sonenberg N, Bell JC (2000) The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol 74: 9580–9585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG (1994) The alphaviruses: Gene expression, replication, and evolution. Microbiol Rev 58: 491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach DC, Kimura T, Griffin DE (2000) Differences between C57BL/6 and BALB/cBy mice in mortality and virus replication after intranasal infection with neuroadapted Sindbis virus. J Virol 74: 6156–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventoso I, Barco A, Carrasco L (1998) Mutational analysis of poliovirus 2Apro. Distinct inhibitory functions of 2Apro on translation and transcription. J Biol Chem 273: 27960–27967 [DOI] [PubMed] [Google Scholar]
- Ventoso I, Carrasco L (1995) A poliovirus 2A(pro) mutant unable to cleave 3CD shows inefficient viral protein synthesis and transactivation defects. J Virol 69: 6280–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, Esteban M (2006) Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev 20: 87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Geigenmüller-Gnirke U, Schlesinger S (1994) Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res 22: 780–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jackson BM, Hinnebusch AG (1989) Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc Natl Acad Sci USA 86: 4579–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC (1995) The histidyl-tRNA synthetase-related sequence in the eIF-2α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol 15: 4497–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schäfer R, Kumar A, Williams BR, Aguet M, Weissmann C (1995) Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J 14: 6095–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, Jefferson LS, Cavener DR (2002) The GCN2 eIF2α kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Paranjape JM, Der SD, Williams BR, Silverman RH (1999) Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology 258: 435–440 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information