

Abstract
The present study tested the hypothesis that increasing epoxyeicosatrienoic acids by inhibition of soluble epoxide hydrolase (sEH) would lower blood pressure and ameliorate renal damage in salt-sensitive hypertension. Rats were infused with angiotensin and fed a normal-salt diet or an 8% NaCl diet for 14 days. The sEH inhibitor, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA), was given orally to angiotensin-infused animals during the 14-day period. Plasma AUDA metabolite levels were measured, and they averaged 10±2 ng/mL in normal-salt angiotensin hypertension and 19±3 ng/mL in high-salt angiotensin hypertension on day 14 in the animals administered the sEH inhibitor. Mean arterial blood pressure averaged 161±4 mm Hg in normal-salt and 172±5 mmHg in the high-salt angiotensin hypertension groups on day 14. EH inhibitor treatment significantly lowered blood pressure to 140±5 mm Hg in the normal-salt angiotensin hypertension group and to 151±6 mm Hg in the high-salt angiotensin hypertension group on day 14. The lower arterial blood pressures in the AUDA-treated groups were associated with increased urinary epoxide-to-diol ratios. Urinary microalbumin levels were measured, and ED-1 staining was used to determine renal damage and macrophage infiltration in the groups. Two weeks of AUDA treatment decreased urinary microalbumin excretion in the normal-salt and high-salt angiotensin hypertension groups and macrophage number in the high-salt angiotensin hypertension group. These data demonstrate that sEH inhibition lowers blood pressure and ameliorates renal damage in angiotensin-dependent, salt-sensitive hypertension.
Keywords: kidney, inflammation, endothelium-derived factors, albuminuria
Although treatment of hypertension has significantly advanced in recent decades, a chronic elevation in blood pressure still results in progressive renal damage, as evidenced by the escalating incidence of end-stage renal disease (ESRD).1,2 The development of hypertension after long-term administration of angiotensin has many of the same renal and vascular changes that are associated with human essential hypertension.3,4 Likewise, animal models of angiotensin-dependent hypertension demonstrate a further elevation in blood pressure when fed a high-salt diet or salt sensitivity.3-5 High dietary salt also increases the susceptibility to kidney damage in hypertensive patients and in angiotensin-dependent hypertensive rats.3,4 These parallels between patients with essential hypertension and the angiotensin infusion model make this an extremely useful model to evaluate early changes that occur in the kidney that ultimately result in ESRD.
Cytochrome P450 epoxygenase metabolites are involved in the long-term regulation of blood pressure and in the response of the kidney to a high-salt diet.6-8 Similarly, salt-sensitive hypertension is associated with an inability of the kidney to properly increase epoxygenase levels.8-10 Recent studies have provided evidence that increasing epoxygenase levels have renal- and cardiovascular-protective actions.11-14 Inhibition of the enzyme, soluble epoxide hydrolase (sEH), which metabolizes epoxides to less active diols, has been one approach to increase epoxide actions. Although sEH inhibition lowers blood pressure in angiotensin-infused rats fed a normal-salt diet,12,15 the blood pressure-lowering effects in angiotensin-dependent, salt-sensitive hypertension remain unknown. In previous studies, we used the sEH inhibitor 1-cyclohexyl-3-dodecylurea (CDU) that had to be injected once daily to acquire plasma CDU levels that were sufficient to inhibit the enzyme.12,15 Unfortunately, CDU has poor solubility and a high melting point that limit the use of this urea-based sEH inhibitor.16-18 We have recently designed an sEH inhibitor that is an adamantine derivative that possesses increased water solubility compared with the cyclohexyl derivative CDU.16 Thus, the purpose of the present study was to determine the effects of the newly developed sEH inhibitor administered orally to angiotensin-infused hypertensive rats fed a normal- or a high-salt diet.
Methods
Animals and Blood Pressure Measurement
The Medical College of Georgia Animal Care and Use Committee approved the experimental procedures, and all animal studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats were divided into experimental groups; 2 groups were subjected to sham surgery, 2 groups received angiotensin and vehicle treatment, and 2 groups received angiotensin and 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA). Angiotensin was infused at a continuous rate with a minipump (65 ng/min), and rats were fed either normal chow or a high-salt diet containing 8% NaCl and allowed access to food and water ad libitum. AUDA was dissolved in a drinking water solution containing β-cyclodextrin (0.5 g/L) that was sonicated for 1 hour. AUDA (25 mg/L) was administered in the drinking water for the 2-week period.
Telemetry transmitters were implanted and data collected as previously described.15 Mean arterial pressure as well as heart rate was obtained. The Biotelemetry Core at the Medical College of Georgia provided assistance with these studies.
Measurement of Oxylipid and AUDA Levels
The levels of the arachidonic acid metabolites, epoxyeicosatrienoic (EET) and dihydroxyeicosatrienoic (DHETE) acids, and the linoleic acid metabolites, epoxyoctadecamononoic (EPOME) and dihydroxyoctadecamononoic (DHOME) acids, were measured in the urine as described previously.19 Rats were housed in metabolic cages that separated urine from food and feces for the final 24 hours of the study. Urine was collected in a tube containing 5 mg of triphenylphosphine and cooled on dry ice. Samples were stored at -80°C until assayed. Samples were extracted, separated by reverse-phase, high-performance liquid chromatography, and analyzed by negative-mode electrospray ionization and tandem mass spectroscopy as previously described.15,19
Blood was collected into tubes containing heparin and centrifuged to obtain plasma. Plasma was stored at -80°C until assayed. For plasma sample AUDA measurements, a 100-μL aliquot was diluted with 200 μL of distilled water and mixed by vortexing before internal standard addition. Samples were then mixed, followed by extraction with 500 μL of ethyl acetate. The samples were centrifuged at 6000 rpm for 5 minutes, and the ethyl acetate was collected. The organic extraction was repeated; the extracts were combined, evaporated to dryness under dry N2 gas, and reconstituted in 50 μL of methanol; and a 5-μL aliquot was analyzed. Mass spectrometry analysis was performed by positive-mode electrospray ionization on a Micromass Ultima triple quadrupole mass spectrometer with multireaction monitoring.18
Measurement of Urinary Electrolytes, Cyclooxygenase Metabolites, and Albumin
Ion-selective electrodes were used to determine urinary electrolyte concentrations. The prostaglandin (PG) and thromboxane (TX) levels in urine were measured by enzyme immunoassays for 6-keto-PGF1α, PGF2α, PGE2, and TXB2. Urinary albumin excretion was assessed as an index for renal injury. Albumin levels were measured by a competitive enzyme immunoassay.
Evaluation of Renal Inflammatory Cell Infiltration
At the end of the AUDA treatment period, kidneys were immediately fixed in 10% buffered formalin solution and embedded in paraffin for light microscopic evaluation. Sections were cut at a thickness of 2 to 3 μm for immunohistochemistry. Kidney sections were stained with a monoclonal ED-1 antibody (Serotec) that recognizes rat monocytes/macrophages, and hematoxylin was used as the counter-stain. ED-1-positive cells were counted in the cortex and medulla, and the number of monocytes/macrophages per area was determined. Analysis was performed with the observer masked to the treatment groups.
Statistical Analysis
All data are presented as mean±SEM. The significance of differences between groups for blood pressure and heart rate data were evaluated with an ANOVA for repeated measures, followed by Duncan’s multiple range post hoc tests. An ANOVA with the Newman-Keuls multiple range test was used for statistical comparison between groups for body weight, urinary excretion rates, and ED-1 levels. A probability value > 0.05 was considered significant.
Results
Blood pressure and heart rate were assessed by telemetry, and the results are presented in Figures 1 and 2. Consistent with previous reports,5,15 blood pressure was significantly increased in angiotensin-infused rats fed a normal-salt diet (ANG-HT). Administration of the sEH inhibitor AUDA was started on day 0 and was continued for 14 days. AUDA treatment significantly (P > 0.05) lowered arterial blood pressure in the ANG-HT group (Figure 1, top panel). As expected, heart rate declined initially and returned to levels similar to control values by day 10 after the start of the angiotensin infusion. AUDA did not alter heart rate over the 14-day treatment period (Figure 1, bottom panel).
Figure 1.
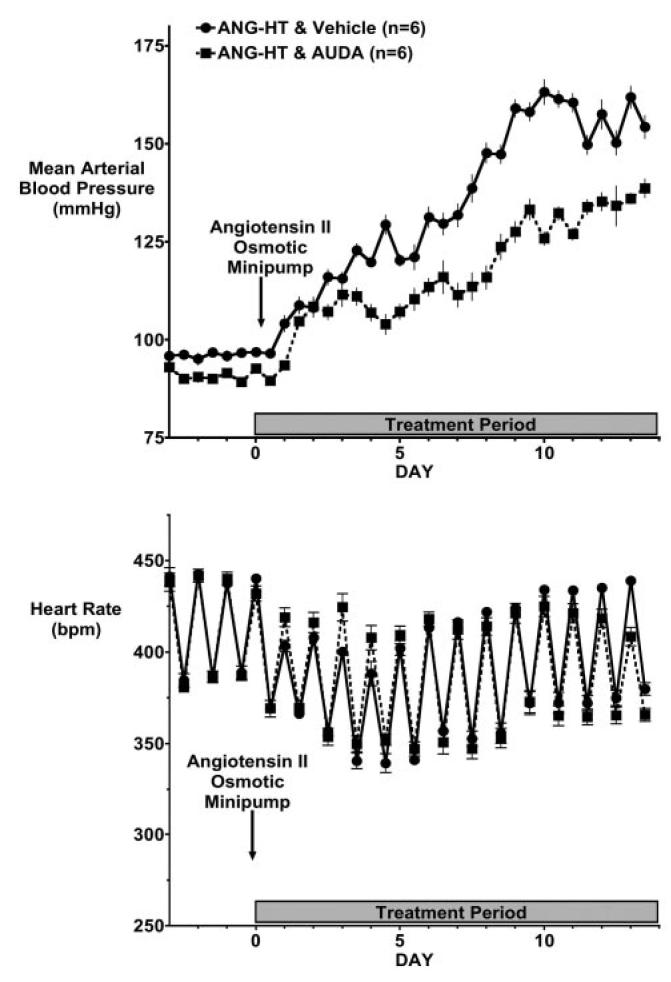
Effect of EH inhibition on blood pressure and heart rate in angiotensin-infused rats fed a normal salt diet (ANG-HT). Data represent day and night 12-hour mean blood pressures (A) and heart rate (B) in ANG-HT animals treated with vehicle or AUDA. Values are mean±SEM. Other abbreviations are as defined in text.
Figure 2.
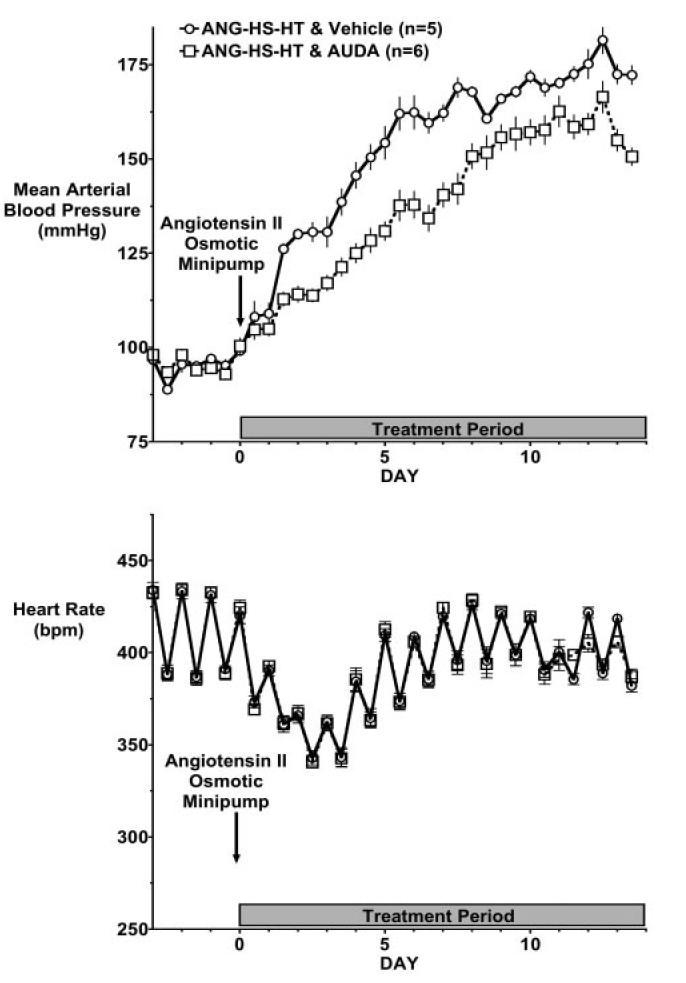
Effect of EH inhibition on blood pressure and heart rate in angiotensin-infused rats fed a high-salt diet (ANG-HS-HT). Data represent day and night 12-hour mean blood pressures (A) and heart rate (B) in ANG-HS-HT animals treated with vehicle or AUDA. Values are mean†SEM. Other abbreviations are as defined in text.
A high-salt diet (HS) resulted in a further increase in blood pressure in angiotensin-infused rats. EH inhibition significantly (P > 0.05) lowered blood pressure in the ANG-HS-HT group (Figure 2, top panel). Similar to the findings in the ANG-HT group, heart rate was not different in ANG-HS-HT rats treated with AUDA (Figure 2, bottom panel).
Body weight and urinary electrolytes were determined at the end of the 2-week period (supplemental Table I). Body weight did not differ between groups on day 14. Urine volume significantly increased in the ANG-HT group (P > 0.05), but this increase was not significantly altered in animals receiving AUDA. As expected, animals fed an HS diet had significant increases in urine volume, sodium excretion, and chloride excretion (P > 0.05). EH inhibition did not significantly alter urinary water or electrolyte excretion in the ANG-HS-HT group.
Plasma and urine AUDA levels were measured to confirm proper EH inhibitor treatment. AUDA levels in the plasma averaged 10±2 ng/mL in the ANG-HT group and 19±3 ng/mL in the ANG-HS-HT group after 14 days of treatment. Higher plasma AUDA levels in rats fed an HS diet can be attributed to increased water intake during the treatment period. Urinary AUDA excretion averaged 38±10 ng/d in the ANG-HT group and 121±61 ng/d in the ANG-HS-HT group treated with AUDA.
Urinary arachidonic and linoleic acid metabolite levels at the end of the 2-week angiotensin infusion period are presented in Figure 3. The EET-DHETE and EPOME-DHOME ratios were decreased in angiotensin-infused animals on a normal or an HS diet, with the exception of the EPOME-DHOME ratio in the ANG-HS-HT group. AUDA treatment significantly increased these ratios but had a greater effect on the EET-DHETE ratio (P > 0.05). The increase in the epoxide-diol ratio was due to a decreased urinary excretion of the diol and increased epoxide excretion in the ANG-HT group administered AUDA (supplemental Table II). On the other hand, AUDA administration to the ANG-HS-HT group significantly decreased diol excretion in the face of maintained or slightly decreased epoxide excretion.
Figure 3.

Urinary excretion ratio of EET and DHETE and the linoleic acid metabolites EPOME and DHOME. Data represent the daily urinary excretion rate in normal-salt rats (n=7), angiotensin-infused rats fed a normal-salt diet (ANG-HT, n=6), ANG-HT rats treated with AUDA, high-salt rats (n=9), angiotensin-infused rats fed a high-salt diet (ANG-HS-HT, n=9), and ANG-HS-HT rats treated with AUDA (n=7). Values are mean±SEM. *Significant difference between normal-salt and ANG-HT group; †significant difference between ANG-HT and ANG-HT & AUDA-treated group. Other abbreviations are as defined in text.
We also determined the effect of sEH inhibition on urinary excretion of cyclooxygenase (COX) metabolites in angiotensin-dependent hypertension. Figure 4 presents the urinary COX metabolite levels in the ANG-HT group. Urinary PGE2 levels significantly increased in the ANG-HT group compared with the control rats receiving a normal-salt diet. Two weeks of AUDA treatment significantly increased urinary 6-keto PGF1α, and there was a tendency for decreased PGE2 levels in the ANG-HT AUDA-treatment group. Angiotensin infusion and an HS diet resulted in a different COX metabolite profile (Figure 5). Urinary PGE2 levels significantly increased and PGF2α levels significantly decreased in the ANG-HS-HT group. Intriguingly, AUDA treatment significantly increased urinary 6-keto PGF1α and TXB2 levels and significantly decreased urinary PGE2 levels in the ANG-HS-HT group. These findings suggest that AUDA has effects on COX metabolite production in angiotensin salt-sensitive hypertension.
Figure 4.

Urinary excretion of COX metabolites in angiotensin-infused rats fed a normal-salt diet (ANG-HT). Data represent the daily urinary excretion rate in normal-salt (n=8), ANG-HT (n=9), and ANG-HT rats treated with AUDA (n=7). Values are mean±SEM. *Significant difference between normal-salt and ANG-HT group; †significant difference between ANG-HT and ANG-HT & AUDA-treated group. Other abbreviations are as defined in text.
Figure 5.

Urinary excretion of COX metabolites in angiotensin-infused rats fed a high-salt diet (ANG-HS-HT). Data represent the daily urinary excretion rate in high-salt (n=10), ANG-HS-HT (n=10), and ANG-HS-HT rats treated with AUDA (n=7). Values are mean±SEM. *Significant difference between high-salt and ANG-HS-HT group or ANG-HS-HT and AUDA group; †significant difference between ANG-HT and ANG-HT & AUDA-treated group. Other abbreviations are as defined in text.
Urinary albumin excretion was measured as an index of renal injury. Two weeks of angiotensin infusion increased urinary albumin excretion in rats fed a normal-salt (0.8±0.2 vs 8.5±2.2 mg/d) or an HS diet (0.9±0.1 vs 32.1±4.2 mg/d). Fourteen days of AUDA treatment significantly decreased urinary albumin excretion in the ANG-HT (4.4±0.6 mg/d) and ANG-HS-HT (5.8±2.0 mg/d) groups. We also assessed macrophage infiltration by assessing ED-1 staining in kidney histologic sections. ED-1-positive cells were significantly increased in the ANG-HS-HT group, and sEH inhibition reduced the number of ED-1-positive cells to that observed in control rats fed an HS diet (Figure 6). Though not significant, there was a trend for increased ED-1-positive cells in the ANG-HT group fed a normal-salt diet that was decreased by AUDA treatment. Overall, the macrophage infiltration and urinary albumin excretion rates suggest that AUDA treatment decreased renal injury in angiotensin salt-sensitive hypertension.
Figure 6.

Comparison of ED-1-positive cell numbers in the kidney glomeruli sections from angiotensin-infused rats fed a high-salt diet (ANG-HS-HT). A, High-salt rat; B, ANG-HS-HT rat; and C, ANG-HT and AUDA-treated rat. Black arrows indicate ED-1-positive cells identified by a blinded observer. The area of kidney represented in each panel is 127 μm2. Bottom graphs depict ED-1-positive cells per area. *Significant difference between the high-salt group and the ANG-HS-HT group; †significant difference between ANG-HS-HT and ANG-HS-HT & AUDA-treated group. Other abbreviations are as defined in text.
Discussion
Angiotensin hypertension has many of the same renal and vascular alterations that are detected in human essential hypertension.3,4 Chronic angiotensin infusion with a high-salt diet causes a further elevation in arterial blood pressure and increased risk for end-organ damage.3,4 We have established along with others that an inability to increase kidney EET levels in response to increased sodium intake is a contributing factor to salt-sensitive hypertension.8-10 In addition, we have previously demonstrated that increasing epoxide-diol ratios with sEH inhibitors can lower blood pressure and provide renal and vascular protection in angiotensin hypertension.12,15 In the present study, we used a newly developed sEH inhibitor with improved properties and evaluated arterial blood pressure and renal injury in angiotensin salt-sensitive hypertension. Arterial blood pressure was decreased in ANG-HS-HT rats given the sEH inhibitor AUDA. In addition, we demonstrated that AUDA treatment decreased renal damage as assessed by decreased urinary microalbumin levels and decreased ED-1-positive cells in ANG-HS-HT.
The possible use of sEH inhibitors as therapy for cardiovascular diseases including hypertension has garnered attention since the initial description of antihypertensive actions in the spontaneously hypertensive rat.20 Although these cyclohexyl ureas were potent inhibitors of the rat and mouse sEH enzyme, this first generation of inhibitors had poor solubility and low in vivo availability.16-18 Morisseau et al17 reported that 1,3-dialkylureas with a polar acid group incorporated on the 13th atom from the carbonyl of the urea are as active as nonfunctionalized hydrophobic inhibitors. Within the same series of compounds, the adamantane group led to increased potency with the recombinant human sEH and no loss of potency with the murine enzyme.17 Surprisingly, most of the adamantane derivatives were more water soluble when compared with the cyclohexyl compounds. Two previous reports have demonstrated that AUDA has antihypertensive effects when administered in drinking water.21,22 The results of the present study also demonstrate an antihypertensive action of orally administered AUDA in ANG-HT and ANG-HS-HT in the rat.
The elevation in blood pressure was attenuated by AUDA treatment in ANG-HT and ANG-HS-HT in the rat. This finding is in agreement with previous studies that demonstrated that sEH inhibition decreases blood pressure in angiotensin hypertension in the rat or mouse.12,15,20-22 This blood pressure effect may be more generalized because sEH inhibition lowers blood pressure in spontaneously hypertensive rats and deoxycorticosterone acetate salt-sensitive hypertensive rats.20,22 We also demonstrated that blood pressure was lowered to the same extent in angiotensin-infusion hypertension irrespective of dietary salt intake. Interestingly, the actions of AUDA appear to be due to a peripheral action of the sEH inhibitor. Sellers et al23 recently reported that intracerebroventricular delivery of AUDA actually increased blood pressure and heart rate in the SHR. On the other hand, AUDA injected intravenously into angiotensin hypertensive mice caused a decrease in blood pressure and heart rate during a 2-hour period.21 The decrease in heart rate during the initial days of angiotensin infusion is consistent with reports that heart rate decreases and cardiac output decreases or remains constant in this model of hypertension24,25 Although an intravenous bolus infusion of AUDA had effects on heart rate, we failed to observe an effect on heart rate with either chronic intraperitoneal injection of CDU or oral administration of AUDA in angiotensin-hypertensive rats. This lack of effect on heart rate in the present study could possibly be due to the slower increase in plasma AUDA levels that occurs when AUDA is administered in the drinking water compared with an intravenous injection. Thus, the route of administration and pharmokinetic properties of sEH inhibitors can alter their cardiovascular actions.
Another aspect of the present study was to measure urinary arachidonic acid and linoleic acid metabolites. As we previously demonstrated with chronic CDU treatment,12,15 AUDA increased the urinary EET-DHETE and EPOME-DHOME ratios in angiotensin hypertension. This finding supports the postulate that sEH inhibition and increasing epoxide-diol ratios are likely responsible for the renal and cardiovascular functional changes produced in angiotensin hypertension. We have consistently found that the epoxide-diol ratio rather than epoxide level alone is a better indicator of sEH inhibitor effectiveness and correlates very well with improved vascular function and decreased renal damage.
A novel facet of this study was the evaluation of urinary COX metabolites in animals treated with AUDA. One consistent finding in angiotensin hypertension was that urinary PGE2 excretion increased and that this increase was attenuated or returned to control levels after AUDA treatment. In addition, sEH inhibition increased 6-keto-PGF1α in angiotensin hypertension. Alterations in TXB2 excretion in AUDA-treated rats are puzzling because TXB2 excretion was not altered in ANG-HT but was significantly elevated in ANG-HS-HT. At this time, we do not have an explanation for this difference observed in TXB2 excretion between the two AUDA-treated groups, and we cannot provide any speculation based on other findings. In any case, the finding of increased PGI2 and decreased PGE2 in angiotensin hypertension rats treated with AUDA was a unexpected result, and these COX metabolite changes could be contributing to renal protection.
There is considerable controversy concerning the contribution of COX metabolites to blood pressure control, renal function, and glomerular injury in angiotensin-dependent hypertension. On the one hand, studies have demonstrated that the generation of COX-2 metabolites can contribute to increased renin secretion and angiotensin generation in renovascular hypertension.26-28 In contrast, COX-2 or nonselective COX inhibitors have been shown to have no effect or actually increase blood pressure in angiotensin-dependent hypertension models, including the 2-kidney, 1-clip model of hypertension.29-32 Additionally, increased TX levels and defective PGI2 buffering capacity have been implicated in altered renovascular and blood pressure regulation in angiotensin-dependent hypertension.33,34 We observed increased urinary excretion of PGE2 in angiotensin-hypertensive rats. Interestingly, elevated PGE2 levels and PGE2 type 1 receptors have been associated with renal disease in hypertensive patients and animal models of hypertension.35,36 Elevated PGE2 levels could also be a consequence of the increased macrophage infiltration associated with angiotensin salt-sensitive hypertension. The finding of decreased macrophage infiltration in AUDA-treated, hypertensive animals suggests that the anti-inflammatory actions of sEH inhibition could account for decreased urinary PGE2 levels. Likewise, increased PGI2 levels in AUDA-treated hypertensive animals could provide vascular protection. The exact mechanisms responsible for decreased PGE2 and increased PGI2 levels in angiotensin hypertensive rats treated with AUDA remains unknown.
The present findings also demonstrate that AUDA treatment protects the kidney from damage that occurs during the progression of angiotensin salt-sensitive hypertension. Angiotensin salt-sensitive hypertensive rats treated with AUDA had decreased urinary albumin levels and decreased numbers of ED-1-positive cells. Interestingly, C-reactive protein and microalbuminuria are strong predictors of increased risks for cardiovascular events and ESRD in human hypertensive patients.37 Previous reports have demonstrated that angiotensin-infused rats have renovascular and tubulointerstitial injury that is augmented by a high-salt diet.3,4,38 Salt-sensitive hypertension has also been linked to an inability of the kidney to increase EET generation.10,39 We provided additional evidence in this study that increasing epoxide-diol ratios ameliorated the renal injury associated with angiotensin salt-sensitive hypertension. We cannot separate the direct effects of increasing EET levels versus blood pressure effects on kidney damage because blood pressure decreased in angiotensin hypertensive animals treated with AUDA. Even though blood pressure was lowered to the same extent in normal- and high-salt angiotensin hypertension, AUDA did have a greater effect on the decrease in ED-1 and albumin excretion in high-salt-diet angiotensin hypertension. We have similar findings in type 2 diabetic Goto-Kakizaki rats, which demonstrate the ability of AUDA to afford renal protection independent of lowering blood pressure.40 Collectively, there are sufficient experimental findings to support the claim that sEH inhibition reduces hypertension-induced renal damage.
Perspectives
We determined the effect of AUDA on blood pressure and renal injury in angiotensin salt-sensitive hypertension. The sEH inhibitor, AUDA, when administered orally for 2 weeks, decreased blood pressure and slowed the progression of renal injury in angiotensin-hypertensive animals on a normal- or high-salt diet. We also demonstrated that AUDA increased urinary epoxide-diol ratios and caused changes in COX metabolite levels. Therefore, manipulation of endogenous lipids by orally active sEH inhibitors has beneficial actions in angiotensin salt-sensitive hypertension.
Acknowledgments
The authors thank Laura Townsend and Hiram Ocasio for technical assistance. This work was supported by National Institutes of Health grants HL-59699 and HL074167 and an American Heart Association Established Investigator Award to J.D. Imig; National Institutes of Environmental Health Sciences grant R37 ES02710 and Superfund Basic Research Program P42 ES04699 to B.D. Hammock; and an American Heart Association Southeast Affiliate Postdoctoral Fellowship to X. Zhao.
References
- 1.Foley RN, Parfrey PS, Harnett JD, Kent GM, Murray DC, Barre PE. Impact of hypertension on cardiomyopathy, morbidity and mortality in end-stage renal disease. Kid Int. 1996;49:1379–1385. doi: 10.1038/ki.1996.194. [DOI] [PubMed] [Google Scholar]
- 2.Brenner BM. The epidemic of cardiovascular disease in end-stage renal disease. Curr Opin Nephrol Hypertens. 1999;8:365–369. doi: 10.1097/00041552-199905000-00015. [DOI] [PubMed] [Google Scholar]
- 3.Cowley AW, Roman RJ. The role of the kidney in hypertension. JAMA. 1996;275:1581–1589. [PubMed] [Google Scholar]
- 4.Simon G, Abraham G. Angiotensin II administration as an experimental model of hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management. Raven Press; New York: 1995. pp. 1423–1435. [Google Scholar]
- 5.Sasser JM, Pollock JS, Pollock DM. Renal endothelin in chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol. 2002;283:R243–R248. doi: 10.1152/ajpregu.00086.2002. [DOI] [PubMed] [Google Scholar]
- 6.Roman RJ. P-450 Metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 7.Imig JD. Eicosanoid regulation of the renal vasculature. Am J Phys Renal Physiol. 2000;279:F965–F981. doi: 10.1152/ajprenal.2000.279.6.F965. [DOI] [PubMed] [Google Scholar]
- 8.Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest. 1994;94:2414–2420. doi: 10.1172/JCI117608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Messer-Letienne I, Bernard N, Roman RJ, Sassard J, Benzoni D. Cytochrome P-450 arachidonate metabolite inhibition improves renal function in Lyon hypertensive rats. Am J Hypertens. 1999;12:398–404. doi: 10.1016/s0895-7061(98)00256-8. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X, Pollock DM, Inscho EW, Zeldin DC, Imig JD. Decreased renal CYP2C enzymes and impaired vasodilation are associated with salt-sensitive hypertension. Hypertension. 2003;41:709–714. doi: 10.1161/01.HYP.0000047877.36743.FA. [DOI] [PubMed] [Google Scholar]
- 11.Imig JD, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular responsiveness to angiotensin II in hypertension is ameliorated by the sulfonamide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. doi: 10.1097/00004872-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 13.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Hallar H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 16.Kim IH, Morisseau C, Watanabe T, Hammock BD. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem. 2004;47:2110–2212. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 17.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy D, Hammock BD. Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol. 2002;63:1599–1608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe T, Morisseau C, Newman JW, Hammock BD. In vitro metabolism of the mammalian soluble epoxide hydrolase inhibitor, 1-cyclohexyl-3-dodecyl-urea. Drug Metab Dispos. 2003;31:846–853. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
- 19.Newman JW, Wantanabe T, Hammock BD. The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J Lipid Res. 2002;43:1563–1578. doi: 10.1194/jlr.d200018-jlr200. [DOI] [PubMed] [Google Scholar]
- 20.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker P, Graham L, Engler M, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 21.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 22.Loch D, Hammock B, Brown L. Soluble epoxide hydrolase inhibition in DOCA-salt hypertensive rats prevents vascular remodeling and dysfunction. Cardiovasc J S Afr. 2004;15:S9. [Google Scholar]
- 23.Sellers KW, Sun C, Diez-Freire C, Waki H, Morisseau C, Falck JR, Hammock BD, Paton JF, Raizada MK. Novel mechanism of brain epoxide hydrolase-mediated blood pressure regulation in the spontaneously hypertensive rat. FASEB J. 2005;19:626–628. doi: 10.1096/fj.04-3128fje. [DOI] [PubMed] [Google Scholar]
- 24.Kreiger JE, Roman RJ, Cowley AW. Hemodynamic and blood volume in angiotensin II salt-dependent hypertension in dogs. Am J Physiol Heart Circ Physiol. 1989;257:H1402–H1412. doi: 10.1152/ajpheart.1989.257.5.H1402. [DOI] [PubMed] [Google Scholar]
- 25.Granger JP, Blaine EH, Stacy DL, La Rock MJ. Effects of long-term increased in plasma ANP on angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 1990;258:H1427–H1431. doi: 10.1152/ajpheart.1990.258.5.H1427. [DOI] [PubMed] [Google Scholar]
- 26.Lin L, Balazy M, Pagano PJ, Nasjletti A. Expression of prostaglandin H2-mediated mechanism of vascular contraction in hypertensive rats: relation to lipoxygenase and prostacyclin synthase activities. Circ Res. 1994;74:197–205. doi: 10.1161/01.res.74.2.197. [DOI] [PubMed] [Google Scholar]
- 27.Hartner A, Goppelt-Struebe M, Hilgers KF. Coordinate expression of cyclooxygenase-2 and renin in the rat kidney in renovascular hypertension. Hypertension. 1998;31:201–205. doi: 10.1161/01.hyp.31.1.201. [DOI] [PubMed] [Google Scholar]
- 28.Wang JL, Cheng HF, Harris RC. Cyclooxygenase-2 inhibition decreases renin content and lowers blood pressure in a model of renovascular hypertension. Hypertension. 1999;34:96–101. doi: 10.1161/01.hyp.34.1.96. [DOI] [PubMed] [Google Scholar]
- 29.Hartner A, Cordasic N, Goppelt-Struebe M, Veelken R, Hilgers KF. Role of macula densa cyclooxygenase-2 in renovascular hypertension. Am J Physiol Renal Physiol. 2003;284:F498–F502. doi: 10.1152/ajprenal.00136.2002. [DOI] [PubMed] [Google Scholar]
- 30.Richter CM, Godes M, Wagner C, Maser-Gluth C, Herzfeld S, Dorn M, Priem F, Slowinski T, Bauer C, Schneider W, Neumayer HH, Kurtz A, Hocher B. Chronic cyclooxygenase-2 inhibition does not alter blood pressure and kidney function in renovascular hypertensive rats. J Hypertens. 2004;22:191–198. doi: 10.1097/00004872-200401000-00029. [DOI] [PubMed] [Google Scholar]
- 31.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110:61–69. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng ZJ, Finckenberg P, Louhelainen M, Merasto S, Tikkanen I, Vapaatalo H, Mervaala EMA. Cardiovascular and renal effects of cyclooxygenase inhibition in transgenic rats harboring mouse renin-2 gene (TGR[mREN2]) Eur J Pharmacol. 2003;461:159–169. doi: 10.1016/s0014-2999(03)01307-4. [DOI] [PubMed] [Google Scholar]
- 33.Kawada N, Dennehy K, Solis G, Modlinger P, Hamel R, Kawada JT, Aslam S, Moriyama T, Imai E, Welch WJ, Wilcox CS. TP receptors regulate renal hemodynamics during angiotensin II slow pressor response. Am J Physiol Renal Physiol. 2004;287:F753–F759. doi: 10.1152/ajprenal.00423.2003. [DOI] [PubMed] [Google Scholar]
- 34.Chatziantoniou C, Ruan X, Arendshorst WJ. Interactions of cAMP-mediated vasodilators with angiotensin II in rat kidney during hypertension. Am J Physiol Renal Physiol. 1993;265:F573–F582. doi: 10.1152/ajprenal.1993.265.6.F845. [DOI] [PubMed] [Google Scholar]
- 35.Suganami T, Mori K, Tanaka I, Mukoyama M, Sugawara A, Makino H, Muro S, Yahata K, Ohuchida S, Maruyama T, Narumiya S, Nakao K. Role of prostaglandin E receptor EP1 subtype in the development of renal injury in genetically hypertensive rats. Hypertension. 2003;42:1183–1190. doi: 10.1161/01.HYP.0000101689.64849.97. [DOI] [PubMed] [Google Scholar]
- 36.Arrazola A, Diez J. Enhanced renal PGE2 in hypertensives with increased red cell Na-Li countertransport. Am J Physiol Heart Circ Physiol. 1991;261:H134–H139. doi: 10.1152/ajpheart.1991.261.1.H134. [DOI] [PubMed] [Google Scholar]
- 37.Bakris G. Inclusion of albuminuria in hypertension and heart guidelines. Kid Int. 2004;92:S124–S125. doi: 10.1111/j.1523-1755.2004.09231.x. [DOI] [PubMed] [Google Scholar]
- 38.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension. 1999;33:1012–1019. doi: 10.1161/01.hyp.33.4.1013. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X, Pollock DM, Zeldin DC, Imig JD. Salt-sensitive hypertension after exposure to angiotensin is associated with an inability to upregulate renal epoxygenases. Hypertension. 2003;42:775–780. doi: 10.1161/01.HYP.0000085649.28268.DF. [DOI] [PubMed] [Google Scholar]
- 40.Olearczyk JJ, Mitchell BC, Imig JD. Inhibition of soluble epoxide hydrolase protects the kidney from the development of nephropathy in hypertensive Goto-Kakizaki rats. FASEB J. 2005;25:A581. doi: 10.1042/CS20080039. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]