

Abstract
The electronic and vibrational structure of β-carotene's early excited states are examined using femtosecond time-resolved stimulated Raman spectroscopy. The vibrational spectrum of the short-lived (∼160 fs) second excited singlet state (S2,1Bu+)of β-carotene is obtained. Broad, resonantly enhanced vibrational features are observed at ∼1100, 1300, and 1650 cm-1 that decay with a time constant corresponding to the electronic lifetime of S2. The temporal evolution of the vibrational spectra are consistent with significant population of only two low-lying excited electronic states (1Bu+ and 2Ag-) in the ultrafast relaxation pathway of β-carotene.
Introduction
Carotenoids play an essential role in photosynthetic light harvesting complexes as photoprotectors and as accessory pigments carrying the major light harvesting burden in the wavelength region from 450 to 550 nm.1,2 Because the photon energy is stored as electronic excitation of the carotenoid before being transferred to chlorophy11, understanding this process requires exact knowledge of the electronic excited-state structure of carotenoids.
Hudson and Kohler's discovery of low-lying optically forbidden 2Ag- excited singlet state in short polyenes more than tirty years ago3,4 and the simultaneous theoretical rationalization of this phenomenon5 are responsible for the most commonly accepted picture of the electronic states of carotenoids (Figure 1). Subsequent theoretical modeling predicted an additional lowlying (1Bu-) excited state located between the 1Bu+ and 2Ag- states for linear polyenes containing more than five double bonds.6 Sashima et al.7 were the first to suggest experimental confirmation of this prediction in resonance Raman excitation profiles on all-trans spheroidene and subsequently other carotenoids including β-carotene.8 The notion that a third electronic state plays a role in the excited-state dynamics was also suggested by recent high-time-resolution transient absorption measurements on β-carotene and lycopene.9
Figure 1.
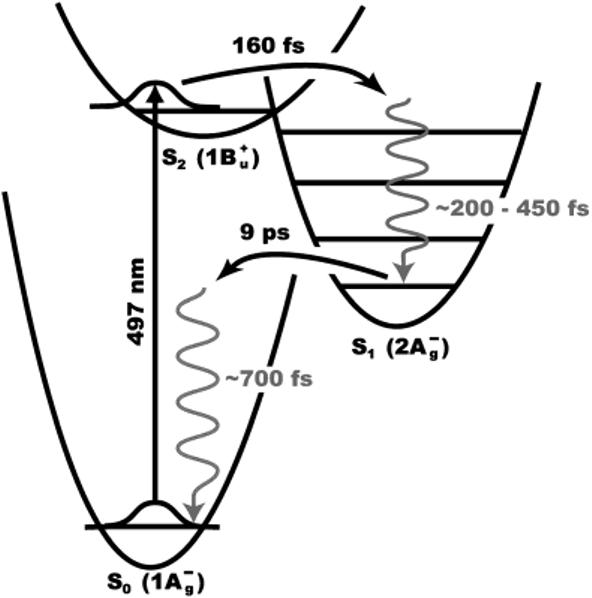
Energy-level diagram of the low-lying electronic states of β-carotene. In addition to the well-known S2 and S1 excited states, another low-lying excited state (1Bu-) between S2 and S1 has been proposed.6-9
Ultrafast vibrational spectroscopy is ideally suited to explore the nature of the states involved in the excited-state relaxation of β-carotene due to the inherent sensitivity of vibrational features to molecular and electronic structure. For instance, the symmetric C=C stretching vibration in β-carotene differs by as much as 250 cm-1 in the ground (1Ag-) and first excited (2Ag-) states making them easily distinguishable.10 It is difficult, however, to apply vibrational techniques to the dynamics of the early 1Bu+ and proposed 1Bu- states due to the lack of time resolution, optical activity, or spectral coverage in traditional time-resolved Raman or IR absorption experiments. To address this challenge, we have been developing the technique of time-resolved femtosecond stimulated Raman spectroscopy (FSRS), which makes it possible to obtain vibrational spectra of excited electronic states that decay on the order of 100 fs with lifetime limited spectral resolution.11-15
In the present work we use FSRS to examine the temporal evolution of β-carotene's vibrational structure during the first picosecond after photoexcitation. Very broad and resonantly enhanced vibrational features are observed immediately after excitation that disappear with a time constant matching the decay of the second excited 1Bu+ electronic state of β-carotene. The vibrational features due to S2 smoothly evolve into those previously reported for the S1 (2Ag-) state. These results are consistent with the dominance of only two electronic states in the relaxation pathway of β-carotene.
Material and Methods
A ∼9 μM solution (OD497) mm-1) of recrystallized β-carotene in cyclohexane was prepared under dim light and recirculated through a 1mm path length cell at a rate sufficient to replenish the illuminated volume between shots.
The apparatus and method used to perform FSRS have been described by McCamant et al. 12,16 FSRS spectra are obtained using three ultrafast laser pulses provided by modification of the output of a regeneratively amplified Ti:sapphire oscillator. A visible actinic pump (∼30 fs, 497 nm, 50 nJ) generated by a noncollinearly phase matched optical parametric amplifier (NOPA) drives electronic excitation of β-carotene to the S2(1Bu+) state. Stimulated Raman transitions are initiated by a Raman pump pulse (800 fs, 17 cm-1, 793 nm, 50 nJ) produced by spectral filtering of the amplifier output together with a broadband continuum Raman probe pulse (<30 fs, 830-960 nm, <5 nJ) generated by focusing the output in sapphire. Both Raman probe and actinic pump pulse durations are minimized with prism compressors. The continuum is split into probe and reference paths to correct for fluctuations in the shape of the continuum. All three pulses are made collinear and focused onto the sample with spot sizes ranging from ∼50 μm for the Raman probe and actinic pump to ∼100 μm for the Raman pump. The Raman probe is separated from the other two pulses after the sample with long pass filters and focused alongside the reference onto the slit of a spectrograph where they are dispersed and imaged onto a dual diode array detector.
The instrumental time resolution is determined by the group velocity mismatch between the actinic pump and Raman probe pulses in the medium. Frequency resolved optical Kerr effect cross correlation reveals an instrument response of 115 fs with less than 10 fs chirp across the spectral range of the Raman probe continuum. The frequency resolution is dominated by the duration of the Raman pump pulse and is on the order of 18 cm-1. Stimulated Raman spectra are generated by dividing successive Raman pump-on and pump-off spectra after correcting for the dark background and fluctuations in the continuum using the reference. The observed Raman intensity thus corresponds to gain in the probe continuum and is defined as
The resulting spectra contain vibrational features due to the solvent, ground, and excited states of β-carotene as well as a broad background due to any transient absorption or emission bands that spectrally overlap with the Raman probe continuum. To obtain excited-state vibrational Raman spectra, we employ the subtraction procedure presented in Figure 2 for the +50 fs spectrum of β-carotene in cyclohexane. A spectrum containing features due to electronically excited β-carotene (D) is obtained by one-to-one subtraction of a solvent spectrum (B) and a scaled ground-state spectrum (0.8*C) from the raw data (A). The scaling of the ground-state spectrum is necessary to compensate for the decrease in ground-state population due to electronic excitation. To remove the broad baseline caused by transient dumping of electronically excited β-carotene by the Raman pump, the entire spectrum is fit to a sum of Gaussian peaks and a fifth-order polynomial baseline. Subtraction of the baseline (E) yields a background free excited-state stimulated Raman spectrum of β-carotene (F).
Figure 2.
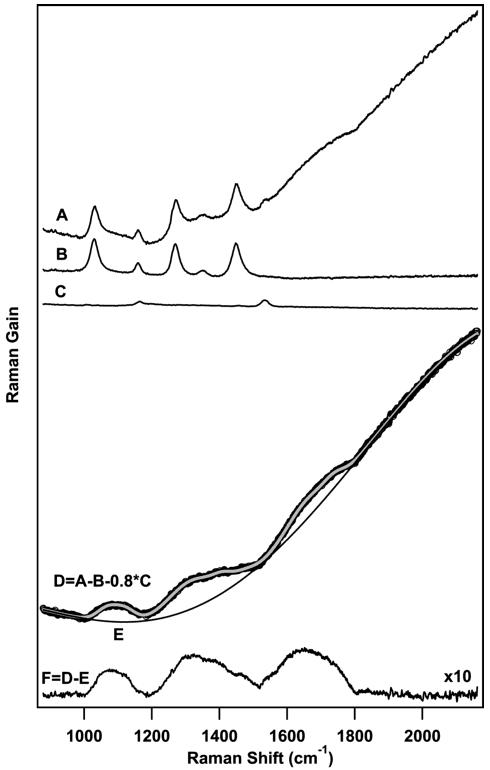
Subtraction procedure used to obtain time-resolved femto-second stimulated Raman spectra (FSRS) of β-carotene. A raw S2spectrum at +50 fs (D) is obtained by 1:1 subtraction of the solvent spectrum (B) and a scaled ground-state β-carotene spectrum (0.8*C) from the raw data (A). The resulting spectrum contains both Raman features as well as a background due to the near-infrared absorption of S2. The entire spectrum (black) is fit to a sum (indicated in gray) of Gaussian peaks and a fifth-order polynomial baseline (E) which is then subtracted to obtain a Raman spectrum of the second excited singlet state of β-carotene (F).
Results
Time-resolved femtosecond stimulated Raman spectra of β-carotene in cyclohexane are presented in Figure 3. A ground-state spectrum of β-carotene is included for comparison clearly displaying three main vibrational features: the methyl rock (1005 cm-1), the symmetric C—C stretch (1161 cm-1) and the symmetric C=C stretch (1528 cm-1). The features at +50 fs are assigned to the vibrational structure of the second excited singlet state of β-carotene (1Bu+) due to their decay with a time constant that corresponds to the NIR absorption decay as well as their resonance enhancement. A possible decomposition of the +50 fs spectrum into Gaussian peaks is indicated. The minimum fwhm of the fitted peaks was fixed at 70 cm-1 as dictated by the uncertainty principle for a transition between two vibrational levels of an excited electronic state with a ∼160 fs lifetime.11 Comparison with the ground-state spectrum in combination with group frequency arguments leads to an initial assignment of methyl rock type vibrations at 1067 and 1124 cm-1,a C—C stretching vibration at 1313 cm-1 and C=C stretching mode at 1654 cm-1. All three features rapidly decrease in intensity except for the high-frequency side of the C=C stretch, causing a visible change in the shape of the peak in the +200 fs spectrum compared to the +50 fs spectrum. At +350 fs a novel feature can be observed on the high-frequency side of the C=C stretch at ∼1760 cm-1, which subsequently blueshifts, narrows, and increases in intensity. This peak can be assigned to the symmetric C=C stretching vibration in β-carotene'S1 (2Ag-) state on the basis of previously published work.10,17 The growth, narrowing, and blue shift of the S1 peak has been studied previously and assigned to a two-step vibrational relaxation process occurring with ∼200 and 450 fs time constants.12
Figure 3.
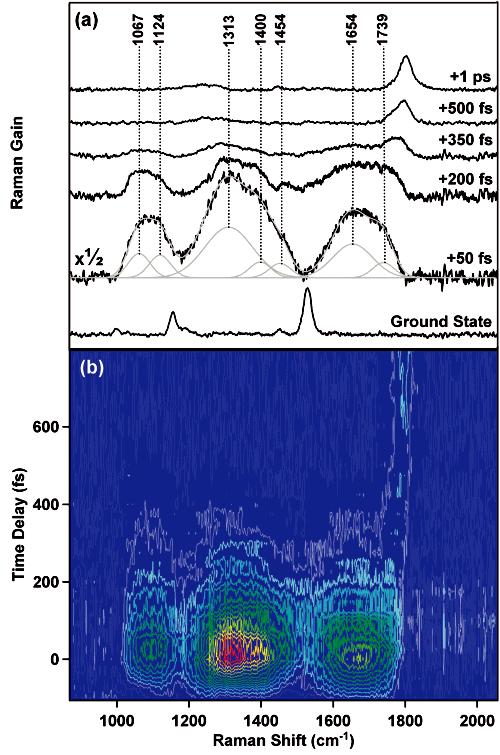
(a) Selected time-resolved femtosecond stimulated Raman spectra of β-carotene after photoexcitation at 497 nm. The spectra represent an average of 3600 Raman pump-on and Raman pump-off exposures corresponding to an acquistion time of ∼11 min per time point. (b) Contour plot of all acquired spectra with 25-fs steps between -100 and +500 fs and 50-fs steps between +500 fs and +800 fs. All spectra are averages of 100 Raman pump-on and pump-off exposures corresponding to an acquistion time of 18 s per time point or ∼20 min for the entire plot. Ground-state, solvent, and background features have been subtracted.
To completely picture the dynamics of β-carotene during the first picosecond after photoexcitation, a contour plot of FSRS spectra is presented in Figure 3b. The contour plot reveals a rapid rise in stimulated Raman intensity with a time constant determined by the instrument response of the FSRS setup between -100 and +50 fs. This rise is followed by a rapid decay of all the indicated S2 features dictated by its electronic lifetime. There is essentially no spectral evolution of the S2features except for the high-frequency side of the broad C=C band, which appears to be decaying more slowly than the remainder of the spectrum and eventually converges into a comparatively sharp, high-frequency peak belonging to S1.
The temporal evolution of the NIR transient absorption of S2 at 915 nm as well as the integrated area of its vibrational features is presented in Figure 4. Least-squares fits to the data (solid) as well as the instrument response (dotted) are included. All data were fit to a convolution of the instrument response and a variable exponential decay time. The resulting time constants for the transient absorption are 134 ± 12 fs at 895 nm, 138 ± 9 fs at 915 nm ±shown), 148 ± 7 fs at 940 nm, and 161 ± 6 fs at 960 nm. The increasing time constant with wavelength is consistent with previous observations.18 The time constants for the decay of the integrated Raman intensity are 147 ± 14 fs, 121 ± 9 fs, and 136 ±14 fs for the methyl rock, the C—C stretching, and C=C stretching vibrations, respectively. The error limits are from the statistical fitting and do not include systematic uncertainty in the baseline which is included in the pictured error bars. Experience from a number of experiments suggests that the overall error limits are on the order of ∼30-40 fs. Nevertheless, the agreement between electronic and vibrational kinetics is excellent.
Figure 4.
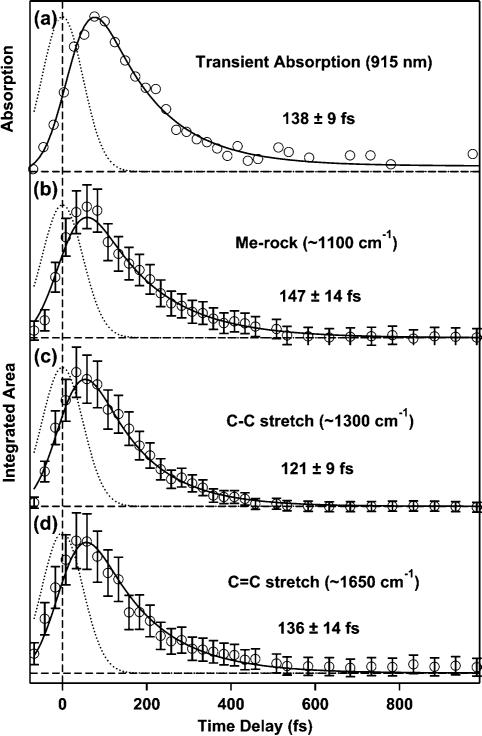
(a) Temporal evolution of the transient near-infrared absorption of the S2 state of β-carotene at 915 nm. (b)—(d) Kinetics of the integrated areas of the three major Raman bands corresponding to the S2 electronic state. Included are the instrument response (dotted lines), kinetic fits (solid lines), and lifetimes.
Discussion
Time-resolved femtosecond stimulated Raman spectra have been obtained to monitor the structural evolution of β-carotene during the first picosecond after photoexcitation at 497 nm. Very broad and intense vibrational features are observed for time delays <400 fs. Analysis of the temporal evolution of the observed Raman features reveals exponential decay with time constants in agreement with the decay of the NIR transient absorption of the second excited singlet state (1Bu+)of β-carotene, strongly suggesting that the features are due to S2. Further confirmation is given by the resonance enhancement of the S2 vibrational features in contrast to the off-resonant S1and ground-state transitions.
The vibrational spectra acquired with 25-fs time steps execute a smooth transition from the S2 to the well-known S1 pattern with no indication of an intermediate 1Bu- state, consistent with theoretical predictions for shorter polyenes.19,20 The apparent delay in the rise of S1 Raman intensity (∼200 fs) relative to the decay of S2 is associated with vibrational relaxation within S112 caused by localization of excess energy in totally symmetric vibrations such as the C=C stretch during the S2 → S1 internal conversion process.19 The suggestion that this delay may be due to a dark intermediate electronic state can be rejected for two reasons. First, it is improbable that such a state would have no detectably unique Raman intensity in the time-resolved FSRS spectra presented here. The 1Bu- state should be evidenced by the rise and subsequent decay of vibrational features distinct from those attributable to the 1Bu+ and 2Ag- states. In particular, if the transient absorption of the 1Bu- state has a NIR component, as suggested,21 the intensity of its vibrational features should be similar to those for the 1Bu+ state. Second, we would expect the existence of a third feature to be revealed in the temporal evolution of the transient absorption kinetics of β-carotene besides that attributable to vibrational relaxation in S1.22,23 It only remains to rationalize our results with the observations made recently by Cerullo et al.9 Their reported Stokes shift in the excited-state absorption on a sub-20-fs time scale is best and most simply explained by the rapid initial nuclear relaxation out of the Franck-Condon region24 along the highly displaced C=C stretch19,25,26 as well as other Franck-Condon active modes.27
Our vibrational spectra of S2 exhibit a general upshift in vibrational frequencies compared with the ground state, in agreement with theoretical predictions made for octatetraene, 28 but the frequencies appear to be significantly higher than those predicted from analysis of the ground-state absorption features 29 as well as resonance Raman excitation profiles.30 The unexpected spectral width (>100 cm-1) of these features significantly exceeds that dictated by the uncertainty principle and vibrational dephasing (∼70 cm-1). Contributions from other possible processes such as nonlinear absorption or a dispersive character of the lineshapes are unlikely in this case due to the lack of resonance enhancement.16 Instead, the observed breadth may be due to conformational heterogeneity caused by thermal population of low-frequency vibrational modes. Additionally, nonstationary effects within S2 may contribute to the observed spectral width.14 Impulsive excitation of low-frequency modes on the excited-state potential energy surface by the ultrashort actinic pump (∼30 fs) leads to nonstationary wavepackets. Significant anharmonic coupling between these low-frequency modes and the observed higher frequency transitions could lead to considerable spectral broadening.
In conclusion, our FSRS spectra exhibit intense and broad vibrational features that are due to the second (1B u+) excited state of β-carotene. A smooth transition to the features characteristic of the first excited singlet state (2A g-) is observed supporting a simple two-state relaxation model for β-carotene.
Acknowledgment
We thank Sangwoon Yoon for assistance with the experimental setup and Soo Y. Lee for helpful discussions. This work was supported by a grant from the National Science Foundation (CHE-9801651) and by the Mathies Royalty fund.
References and Notes
- (1).Frank HA, Cogdell RJ. Photochem. Photobiol. 1996;63:257–64. doi: 10.1111/j.1751-1097.1996.tb03022.x. [DOI] [PubMed] [Google Scholar]
- (2).Ritz T, Damjanovic A, Schulten K, Zhang JP, Koyama Y. Photosynth. Res. 2000;66:125–144. doi: 10.1023/A:1010750332320. [DOI] [PubMed] [Google Scholar]
- (3).Hudson BS, Kohler BE. Chem. Phys. Lett. 1972;14:299–304. [Google Scholar]
- (4).Hudson BS, Kohler BE. Annu. Rev. Phys. Chem. 1974;25:437–460. [Google Scholar]
- (5).Schulten K, Karplus M. Chem. Phys. Lett. 1972;14:305–309. [Google Scholar]
- (6).Tavan P, Schulten K. J. Chem. Phys. 1986;85:6602–6609. [Google Scholar]
- (7).Sashima T, Nagae H, Kuki M, Koyama Y. Chem. Phys. Lett. 1999;299:187–194. [Google Scholar]
- (8).Sashima T, Koyama Y, Yamada T, Hashimoto H. J. Phys. Chem. B. 2000;104:5011–5019. [Google Scholar]
- (9).Cerullo G, Polli D, Lanzani G, De Silvestri S, Hashimoto H, Cogdell RJ. Science. 2002;298:2395–2398. doi: 10.1126/science.1074685. [DOI] [PubMed] [Google Scholar]
- (10).Noguchi T, Kolaczkowski S, Arbour C, Aramaki S, Atkinson GH, Hayashi H, Tasumi M. Photochem. Photobiol. 1989;50:603–609. [Google Scholar]
- (11).Kukura P, McCamant DW, Davis PH, Mathies RA. Chem. Phys. Lett. 2003;382:81–86. [Google Scholar]
- (12).McCamant DW, Kukura P, Mathies RA. J. Phys. Chem. A. 2003;107:8208–8214. doi: 10.1021/jp030147n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McCamant DW, Kukura P, Mathies RA. Appl. Spectrosc. 2003;57:1317–1323. doi: 10.1366/000370203322554455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee SY, Zhang D, McCamant DW, Kukura P, Mathies RA. J. Chem. Phys. Theory of Femtosecond Stimulated Raman Spectroscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yoshizawa M, Hattori Y, Kobayashi T. Phys. Re V. B. 1994;49:13259–13262. doi: 10.1103/physrevb.49.13259. [DOI] [PubMed] [Google Scholar]
- (16).McCamant DW, Kukura P, Yoon S, Mathies RA. Rev. Sci. Instrum. Femto-second Broadband Stimulated Raman Spectroscopy: Apparatus and Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McCamant DW, Kim JE, Mathies RA. J. Phys. Chem. A. 2002;106:6030–6038. doi: 10.1021/jp0203595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang JP, Skibsted LH, Fujii R, Koyama Y. Photochem. Photobiol. 2001;73:219–222. doi: 10.1562/0031-8655(2001)073<0219:taftus>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (19).Garavelli M, Celani P, Bernardi F, Robb MA, Olivucci M. J. Am. Chem. Soc. 1997;119:11487–11494. [Google Scholar]
- (20).Garavelli M, Smith BR, Bearpark MJ, Bernardi F, Olivucci M, Robb MA. J. Am. Chem. Soc. 2000;122:5568–5581. [Google Scholar]
- (21).Zhang JP, Inaba T, Koyama Y. J. Mol. Struct. 2001;598:65–78. [Google Scholar]
- (22).Shreve AP, Trautman JK, Owens TG, Albrecht AC. Chem. Phys. Lett. 1991;178:89–96. [Google Scholar]
- (23).de Weerd FL, van Stokkum IHM, van Grondelle R. Chem. Phys. Lett. 2002;354:38–43. [Google Scholar]
- (24).Ye T, Gershgoren E, Friedman N, Ottolenghi M, Sheves M, Ruhman S. Chem. Phys. Lett. 1999;314:429–434. [Google Scholar]
- (25).Mantini AR, Marzocchi MP, Smulevich G. J. Chem. Phys. 1989;91:85–91. [Google Scholar]
- (26).Garavelli M, Negri F, Olivucci M. J. Am. Chem. Soc. 1999;121:1023–1029. [Google Scholar]
- (27).Furthermore, in the model by Cerullo et al. , 9 the observed emission is attributed to a breakdown of parity rendering the 1B u- ← 1A g- an allowed electronic transition. The small observed Stokes shift in β -carotene (< 300 cm -1) thus requires simultaneous pumping of both the 1B u+ and 1B u- states by their 15 fs excitation pulse (fwhm ∼ 1000 cm-1). Any transient absorption features due to 1B u- should thus be observable immediately after optical excitation rather than after a delay
- (28).Zerbetto F, Zgierski MZ, Negri F, Orlandi G. J. Chem. Phys. 1988;89:3681–3688. [Google Scholar]
- (29).Torii H, Tasumi M. J. Phys. Chem. 1990;94:227–231. [Google Scholar]
- (30).Sue J, Mukamel S. J. Opt. Soc. Am. B. 1988;5:1462–1472. [Google Scholar]