

Abstract
Mammalian hepatic carboxylesterases (CEs) play important roles in the detoxification of ester-containing pyrethroids, which are widely used for the control of agricultural pests and disease vectors such as mosquitoes. Pyrethroids and pyrethroid-like fluorescent substrates exhibit a consistent pattern of stereoselective hydrolysis by a recombinant murine hepatic CE. We sought to understand whether this pattern is maintained in other hepatic CEs and to unravel the origin of the stereoselectivity. We found that all hepatic CEs tested displayed a consistent pattern of stereoselective hydrolysis: the chiral center(s) in the acid moiety more strongly influenced stereoselective hydrolysis than the chiral center in the alcohol moiety. For cypermethrin analogues with a cyclopropane ring in the acid moiety, trans-isomers were generally hydrolyzed faster than the corresponding cis-isomers. For fenvalerate analogues without a cyclopropane ring in the acid moiety, 2R-isomers were better substrates than 2S-isomers. These general hydrolytic patterns were examined by modeling the pyrethroid-like analogues within the active site of the crystal structure of human carboxylesterase 1 (hCE1). Stereoselective steric clashes were found to occur between the acid moieties and either the catalytic Ser loop (residues 219-225) or the oxyanion hole (residues140-144). These clashes appeared to explain the stereopreference between trans- and cis-isomers of cypermethrin analogues, and the 2R- and 2S-isomers of fenvalerate analogues by hCE1. The implications these findings have on the design and use of effective pesticides are discussed.
Introduction
Carboxylesterases (CEs) play a major role in the metabolism, detoxification, and elimination of endogenous and exogenous esters (1). Various CEs are present in a wide variety of organs and tissues of many mammalian species including humans, in which the highest hydrolase activity occurs in the liver (2). These enzymes are α/β fold hydrolases, and the family shares a common mechanism of hydrolysis involving a catalytic triad composed of a serine, a histidine, and either an aspartic acid or a glutamic acid residue (3). These enzymes can hydrolyze a wide variety of ester, amide, and thioester substrates. Despite their promiscuity, however, many CEs display stereo-/enantioselective hydrolysis toward certain substrates.
Currently, pyrethroids are among the most widely used insecticides in the world. The use of pyrethroids will probably increase as organophosphates are phased out, resulting in an increased incidence of human exposure. Because of the rapid biodegradation by mammalian and human liver enzymes (e.g., esterases and monooxygenases), pyrethroids are well-known to present only low to moderate oral toxicity to mammals (4) and to be generally safe for humans (5). However, evidence has suggested that significant long-term and high level exposure to pyrethroids might cause human health problems such as suppressive effects on the immune system (6), endocrine disruption (7), with lymph node, splenic, and testicular damage as well as carcinogenesis (8, 9). Moreover, the expression of CE isozymes varies with life stage, individuals, sex, and species (1), resulting in differential susceptibility to the same pyrethroid between human infants and adults (10) or between neonatal and adult mammals (11). Stereoisomerism exists in most pyrethroid insecticides, and different stereoisomers of the same pyrethroid may exhibit remarkable differences in resistance to hydrolysis by the same CE isozyme (12, 13). This hydrolytic stability may contribute to differential toxicity. Thus, knowledge of the pyrethroid structure-hydrolytic activity relationship and the tissue distribution of CE isozymes is a critical component to the understanding of the metabolism of the natural pyrethrines and their analogues (the pyrethroids).
Optically pure pyrethroid-like fluorescent substrates are useful in elucidating the origin of stereoselective hydrolysis by different CE isozymes in mammals and/or humans. These substrates have been developed in our laboratory to facilitate such a measurement (13). Carboxylesterases hydrolyze the ester bond, releasing the cyanohydrin which quickly rearranges into the corresponding fluorescent aldehyde under neutral or basic conditions (14). A murine liver CE (NCBI accession no. BAC36707) exhibited similar stereoselective hydrolysis of pyrethroids (e.g., cypermethrin) and pyrethroid-like fluorescent substrates (14). It was not clear, however, whether other mammalian and human hepatic CEs would exhibit this stereoselectivity.
Recently, X-ray crystal structures of human and rabbit liver CEs (hCE1 and rCE) have been obtained in the Redinbo laboratory (16, 17). The origin of the stereoselective hydrolysis of cocaine by hCE1 has also been examined recently (17). This structure made it possible to study the molecular basis for stereoselective hydrolysis of these fluorescent substrates and/or pyrethroids in mammals or humans. Thus, the major objectives of this study were (1) to evaluate the stereoselectivity of CEs including recombinant and purified hCE1, commercial porcine, and rabbit liver CEs toward the stereoisomers of these fluorescent substrates (Figure 1), (2) to combine the data obtained from recombinant and purified murine liver CEs (NCBI accession nos. BAC36707 and NM_133960 (13)) and compare the differences in stereoselective of different CEs toward these fluorescent substrates, and (3) to probe the origin of stereoselectivity of the pyrethroid-like fluorescent substrates using the crystal structures of hCE1.
Figure 1.

Structural labels for different stereoisomers of cypermethrin and fenvalerate analogues.
Materials and Methods
Chemicals. All fluorescent substrates were synthesized as reported previously (13), and the chemical purity of these substrates determined by GC/MS (13) is over 99.9%. Full spectral characterization is provided separately (13). Optical purity of eight stereoisomers of cypermethrin analogues labeled as 1R cis, αR; 1R cis, αS; 1R trans, αR; 1R trans, αS; 1S cis, αR; 1S cis, αS; 1S trans, αR; and 1S trans, αS (Figure 1, 13) is 97.7%, 98.8%, 98.7%, 99.9%, 99.4%, 99.4%, 99.9%, and 98.0%, respectively. Optical purity of four stereoisomers of fenvalerate analogues labeled as 2R, αR;2R, αS;2S, αR; and 2S, αS (Figure 1, 13) is 99.9%, 99.1%, 100%, and 98%, respectively. 6-Methoxy-2-naphthaldehyde (>99% purity) was obtained from AVOCADO Research Chemical, Ltd. (Heysham, Lancaster U.K.). Porcine liver CEs (catalog no. E2884) and rabbit liver CEs (catalog no. E-9636) were purchased from Sigma Chemical Co. (St. Louis, MO). Both porcine and rabbit liver CEs are a mixture of multiple isozymes. Two recombinant mouse liver carboxylesterases (NCBI accession no. BAC 36707 and NM_133960) were purified as previously reported (12).
Preparation of hCE1. Recombinant hCE1 was purified from a baculovirus expression system [Nishi, K., et al. (manuscript in preparation)]. In brief, recombinant baculovirus was generated by coinfection of a full-length cDNA encoding hCE1 cloned into a pAcUW21 transfer vector (BD Biosciences Pharmingen, San Diego, CA) with virus DNA into Sf21 insect cells. Then, recombinant baculovirus-infected High Five insect cells were harvested and homogenized 72 h post-infection, and hCE1 was solubilized by adding 1% (final concentration) octyl-β-d-glucopyranoside. After ultracentrifugation (100 000g, 60 min), the supernatant was applied to a DEAE anion-exchange chromatography (eluted with 20 mM Tris-HCl (pH 8.0) containing 100 mM NaCl). The protein was further purified using Rotofor (Bio-Rad Laboratories, Hercules, CA) pH 5-8 and loaded onto a Superose 12 gel-filtration column (Amersham Biosciences, Piscataway, NJ), and the catalytically active fractions were collected.
Protein Concentration Determination. Protein concentration was determined according to the method of Smith et al. (18). Briefly, a solution (180 μL) of bicinchoninic acid protein assay reagents A and B (Pierce, Rockford, IL, a ratio of 50:1) was added to 20 μL of protein samples. The mixture was incubated for 30 min at 37 °C before absorbance was measured at 562 nm. On the basis of a standard curve of bovine serum albumin, absorbance was converted into protein concentration.
Fluorescent Assays. Assays were performed by the method of Huang et al. (13). In short, fluorescent assays were conducted with a Spectrafluor Plus (Tecan, Research Triangle Park, NC). Activities were measured in black 96-well polystyrene flat-bottom microtiter plates (Corning, Inc., New York, NY) at 30 °C. Substrates were prepared in ethanol (10 mM, except for fenvalerate analogues, which also contained 10% Me2SO). Reaction mixtures contained (total volume 201 μL): 20 μL of protein solution, 180 μL of 20 mM Tris/HCl buffer (pH 8.0), and 1 μL of substrate solution. Previous studies demonstrated less than 1% loss of the total of the aldehyde regardless of whether phosphate or tris buffer were used during the reaction. The reaction was initiated by the addition of 1 μL of substrate solution (final concentration 50 μM) followed by shaking for 5 s. This substrate final concentration (50 μM) was chosen because it is at least 10-fold higher than the Km(s) for most stereoisomers and we considered limitation of the substrate solubility (solubility limitation for cypermethrin and fenvalerate analogues are around 10 and 2 μM, respectively, under pH 8.0, 20 mM tris/HCl buffer containing 0.5% ethanol). Three replicates were conducted for each substrate. Fluorescence was monitored with excitation at 330 nm (band-pass, [bp] 35) and emission at 465 nm (bp 35; excitation maximum) 330 nm; emission maximum) 460 nm). Assays for chiral pyrethroid substrates were conducted with five flashes (15 cycles) to give a ∼5 min linear assay. The amount of protein in each assay varied with the substrate and was adjusted so that no more than 5% of the substrate was hydrolyzed over the reported time. A standard curve of the dependence of aldehyde fluorescence response on protein concentration was generated by adding an equivalent amount of each protein sample as used in enzymatic assays to adjust for the protein-induced aldehyde quenching.
Kinetic Assays. Kinetic assays were performed by the method of Huang et al. (13). Fluorescent assays for both BAC36707 and hCE1 were conducted according to the procedure described above, except for a 5-min incubation at 30 °C before adding substrates. The concentrations of cypermethrin analogues were 9.0, 4.5, 2.7, 1.8, 0.9, 0.45, 0.225, and 0.022 5 (μM final concentration), respectively. Three independent replicates were conducted for each concentration. Reported kinetic data (Km and Vmax) were calculated based on the Michaelis-Menten equation and three replicates.
Manual Docking Studies. Cypermethrin and fenvalerate analogues were manipulated and docked manually using O (19) into the active site of the hCE1-homatropine structure (RCSB accession code 1MX5) after all ligand, water, and carbohydrate atoms were removed from the structure for simplicity. Placement was restricted so that the ester linkages were in proximity to the active site serine 221. All molecular graphics figures were created using PyMOL (20).
Results
Biochemical Results. The results of fluorescent assays (Table 1) suggested that selective activities of CEs toward cypermethrin analogues varied greatly among different stereoisomers and among different CEs. Briefly, the two chiral centers in the acid moiety of the cypermethrin analogues produced more dramatic effects on hydrolysis than the chiral center in the alcohol moiety (cyanohydrin) for both purified CEs (hCE1, NM_133960 and BAC36707) and for a mixture of isozymes such as porcine and rabbit liver CEs. Selective activities for trans-isomers were generally higher than cis-isomers. For example, CEs, except for hCE1, exhibited highest hydrolytic activity toward the (1S trans, αS) stereoisomer, with up to 2-fold lower hydrolytic activity toward the corresponding diastereomer (1S trans, αR). However, the overall pattern of stereoselectivity for each CE displayed little difference. For this study, we consider the enzyme to show a preference toward one stereoisomer if it ishydrolyzed over 5 times faster than the least favored isomer in a single series. For example, porcine liver CEs preferred five stereoisomers (1R trans, αR;1R trans, αS; 1R cis, αS; 1S trans, αR; and 1S trans, αS) to the other three (1R cis, αR; 1R cis, αS; and 1S cis, αS). However, purified hCE1 only favored trans-isomers (1R trans, αR; 1R trans, αS;1S trans, αS; and 1S trans, αS). In addition, the highest activities varied with CEs, ranging from hCE1 (55.2 (nmol/min)/mg) to BAC36707 (327 (nmol/min)/mg).
Table 1.
.Specific Activity of Mammalian and Human Liver Carboxylesterases toward Pyrethroid-like Fluorescent Substratesa
| substrates | porcineb | rabbitb | humanc hCE1 | moused NM_133960 | moused BAC36707 |
|---|---|---|---|---|---|
| Cypermethrin Analogues | |||||
| 1R cis, αR | 2.35(0.17) | 5.67(0.49) | 0.60(0.02) | 3.94(0.37) | 19.74(2.18) |
| 1S cis, αR | 2.45(0.22) | 5.71(0.83) | 0.36(0.13) | 7.44(0.66) | 8.44(0.37) |
| 1R trans, αR | 26.8(1.8)e | 12.2(1.4) | 16.0(1.60)e | 7.63 (0.27) | 26.2(2.8)e |
| 1S trans, αR | 51.0(2.6)e | 37.5(2.6)e | 21.3(2.2)e | 85.1(3.2)e | 281(24)e |
| 1R cis, αS | 33.4(4.5)e | 7.41(0.04) | 0.64(0.06) | 20.3(0.1)e | 2.28(0.31) |
| 1S cis, αS | 6.62(0.46) | 3.73(0.37) | 0.66(0.05) | 8.69(0.85) | 6.69(1.06) |
| 1R trans, αS | 42.4(3.8)e | 55.6(7.5)e | 55.2(3.1)e | 7.34(0.76) | 14.6(1.2)e |
| 1S trans, αS | 61.2(1.8)e | 83.4(11.3)e | 14.7(1.3)e | 155(2)e | 327(25)e |
| Fenvalerate Analogues | |||||
| 2R,αR | 1.62(0.07) | 17.1(1.0)e | 0.38(0.01)e | 10.3(0.4)e | 40.3(2.6)e |
| 2R, αS | 8.34(0.35) | 15.0(0.4)e | 0.58(0.10)e | 15.2(0.3)e | 54.2(3.4)e |
| 2S,αR | 4.31 (0.32) | 0.25(0.04) | NDf | 0.63(0.05) | NDf |
| 2S, αS | 2.66(0.20) | 4.61(0.04) | 0.04(0.03) | 0.79(0.11) | NDf |
Values stand for mean activities ((nmol/min)/mg protein, SD in parentheses) based on triplicate assays.
Porcine and rabbit liver carboxylesterases are commercial from Sigma-Aldrich; they were mixtures of multiple isozymes.
Human (hCE1) carboxy-lesterase was expressed in insect cells with a recombinant bacu-lovirus expression system, then recombinant hCE1 was sequentially purified by anion-exchange chromatography, preparative electrophoresis, and gel-filtration chromatography.
Mouse: two recombinant mouse liver carboxylesterase (NCBI accession nos. NM_133960 and BAC36707) were recently purified (12), and data were presented in ref 13.
Preference was considered if one stereoisomer is hydrolyzed by one carboxylesterase over 5 times faster than the least favored isomer in a single series.
ND = not detectable under the same protein as other stereoisomers.
Similarly, stereoisomerism also greatly affected the specific activities of CEs toward the fenvalerate analogues (Table 1). In general, the stereoselectivity of porcine liver CEs was poor, but all other CEs favored the 2R form in the acid moiety, and the chiral center in the alcohol moiety did not make a significant difference in the stereoselective activity. The highest activities varied greatly with CEs, ranging from hCE1 (0.58 (nmol/min)/mg) to BAC36707 (54.2 (nmol/min)/mg).
Kinetic Analysis of Carboxylesterases toward Cypermethrin Analogues. To evaluate the stereoselectivity of CEs toward pyrethroid-like fluorescent substrates, CEs (e.g., hCE1, BAC36707) and cypermethrin analogues were chosen because these CEs could hydrolyze all stereoisomers of cypermethrin analogues. The specificity (kcat/Km, Table 2) suggested that the stereoselectivity of hCE1 and BAC36707 toward the most sensitive isomer and the least sensitive one was up to 41 (1R trans, αS versus 1S cis, αR) and 150 (1S trans, αS versus 1R cis, αS), respectively. Additionally, there was a small difference in Km for both CEs (2.6-fold for BAC36707 and 8.3-fold for hCE1), but a dramatic difference in kcat (over 300 times) for both hCE1 and BAC36707.
Table 2.
.Kinetic Constants for Carboxylesterases toward Cypermethrin Analogues
| CEs | substrate | Km (μM) | kcat( × 10-4s-1) | kcat/Km(M-1s-1) |
|---|---|---|---|---|
| hCE1 | 1S cis, αR | 2.3 ± 0.5 | 1.8 ± 0.1 | 78 ± 4 |
| 1R trans, αS | 19.0 ± 1.2 | 600 ± 30 | 3200 ± 200 | |
| BAC36707a | 1R cis, αS | 1.68 ± 0.06 | 20 ± 2 | 1200 ± 200 |
| 1S trans, αS | 4.36 ± 0.34 | 7700 ± 1300 | 180000 ± 30000 |
Data were presented in ref 13.
Structural Basis of the Stereoselective Hydrolysis by hCE1 toward Pyrethroid-like Fluorescent Substrates. The overall domain structure of hCE1 and general mechanism of biochemical assays are shown in Figure 2. hCE1 was chosen as the model for manual docking studies because the other crystal structure of a mammalian CE (e.g., rabbit liver CE, rCE) lacks important loops adjacent to the active site that may affect proper configuration of placed molecules. Cypermethrin or fenvalerate analogues were considered to enter the substrate-binding gorge from the direction opposite the catalytic site. After cleavage, the alcohol moiety (or cyanohydrin) exits the active site gorge, and then rearranges immediately into a fluorescent aldehyde under neutral to basic conditions. Production of the resulting fluorescent aldehyde was subsequently measured with a fluorescence spectrophotometer. At the substrate-binding site, there are two loops of interest that presumably affect the binding of the substrate and hCE1 (Figure 3). These loops are referred to as Ser loop (219-225), which contains the catalytic serine 221, and the oxyanion hole (140-144) containing the Gly 142-Gly 143 pocket for stabilization of the transition state. Both of two loops were considered to be rigid, a supposition supported by the low crystallographic thermal displacement parameters (B-factors) observed for these regions relative to the remainder of the structure. When the 1S trans, αR and 1R trans, αS cypermethrin analogues were manually docked into the active site, few structural differences were evident that explained the 2.5-fold preference in specific activity (Table 1). However, when 1S cis, αR was modeled over 1S trans, αR, a clear difference was observed (Figure 3a). Switching the chirality from trans to cis made the dichlorovinyl group protrude into the main chain of the oxyanion hole effectively blocking the active site Ser 221 from accessing the carbonyl carbon. Similar steric clashes were observed in 1R cis-isomers, but not in 1R trans-isomers. These steric clashes likely explain the >20-fold difference in specific activity between cis- and trans-isomers of cypermethrin analogues (Table 1). Similarly, for fenvalerate analogues, a reversal of chirality from 2R, αS into 2S, αR caused a severe steric clash by both isomers of fenvalerate analogues. The stereoselective hydrolysis of fenvalerate analogues by hCE1 resulted from a differential degree in the steric clash, although it was much smaller for 2R-isomers (e.g., 2R, αS) (Figure 3b left panel) than for 2S-isomers (e.g., 2S, αR) (Figure 3b, right panel). The fact that all fenvalerate analogues produced steric clashes also explains why the highest activities for fenvalerate analogues (2R-isomers) were similar to those of cis-isomers of cypermethrin analogues (Table 1), which also exhibited steric clashes.
Figure 2.
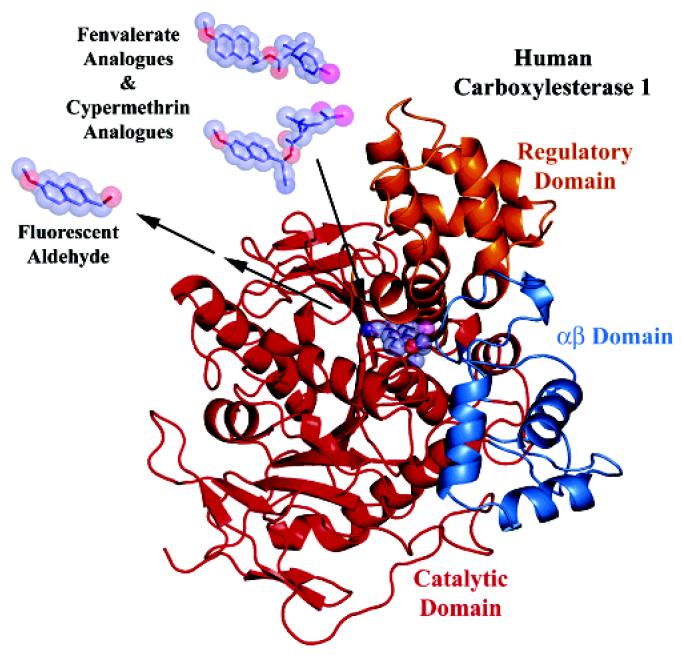
Overall domain structure of human carboxylesterase 1 (hCE1) and general mechanism of activity assays. Cypermethrin and fenvalerate structures are shown in stick models with transparent CPK surface. Arrows represent mechanistic steps resulting in fluorescent aldehyde which is measured spectrophotometrically. hCE1 domains are colored red (catalytic domain), blue (αβ domain), and gold (regulatory domain).
Figure 3.

Proposed binding of cypermethrin and fenvalerate analogues just prior to catalysis: comparison of stereochemistry and activity. Active site loops with terminal amino acids labeled. Loop 140-144 represents the oxyanion hole, while loop 219-225 represents the active site serine loop. Ser 221 is shown in stick model, labeled in gray. Ligands are shown in stick models with CPK transparent surfaces. (a) Cypermethrin analogues 1S cis αR shown in blue with 1S trans αR shown in green. (b) Fenvalerate analogues 2R, αS shown in yellow with 2S, αR shown in blue.
Discussion
In insects, mammals, and humans, major metabolic enzymes that detoxify pyrethroids in the first phase of drug metabolism are esterases and monooxygenases (21, 22). Although these enzymes distribute in multiple organs and tissues, the highest activities of these enzymes are found to be in mammalian livers (2). Indirect and direct evidence has indicated that pyrethroids are largely metabolized in mammalian livers (10, 11, 15, 23-26). Thus, further metabolic studies with hepatic carboxylesterases and other enzymes may address organ selective metabolism.
Pyrethroids and pyrethroid-like fluorescent substrates are stereoselectively hydrolyzed by hepatic CEs. Among ester-containing pyrethroids, fenvalerate is the best studied in terms of stereoselective hydrolysis by hepatic CEs. Experiments suggest that the hydrolysis of the four isomers of fenvalerate in mice (in vivo) follows the same pattern of stereoselectivity (i.e., 2R-isomers of fenvalerate are more rapidly hydrolyzed than its 2S-isomers) as that seen in in vitro studies with the mouse extracts (26). This stereoselective pattern was further supported by studies with purified mouse liver CE (e.g., BAC36707 (11)). Similarly, this stereoselective pattern is also generally predictable for fenvalerate analogues.
In this study, a similar pattern of stereoselective hydrolysis of four isomers of fenvalerate analogues was followed by purified hCE1, BAC 36707, NM_133960, and a mixture of isozymes such as CEs from rabbit liver. However, there is a moderate deviation from this stereoselective pattern for porcine liver CEs. The activity toward 2R, αS was the highest among the four isomers, whereas the activity toward 2R, αR was significantly lower that those toward 2S, αR and 2S, αS. These results were in agreement with a previous study with a racemic mixture of 2R, αR/S or 2S, αR/S, in which 2R-isomers of fenvalerate analogues were preferred to its 2S-isomers by human and murine liver microsomes but not porcine liver CEs (27). For cypermethrin, a complete spectrum of stereoselective hydrolysis of eight stereoisomers by hepatic CEs is not available. However, it is well-known that trans-isomers of cypermethrin and other pyrethroids with a cyclopropane ring (e.g., permethrin, deltamethrin) are generally more rapidly hydrolyzed by hepatic CEs than the corresponding cis-isomers (12, 23, 25). This general pattern is also supported by the fact that the trans-isomers of cypermethrin analogues were generally preferred over the cis-isomers (Table 1). Unlike the unique pattern of stereoselective hydrolysis of fenvalerate and its analogues by hepatic CEs, however, the detailed stereoselectivity of cypermethrin analogues varied greatly across the spectrum of CEs. For example, hCE1 preferred all trans-isomers of cypermethrin analogues to its cis-isomers, but 1R cis, αS was a better substrate for NM_133960 than two isomers with the 1R trans-configuration in the acid moiety. In addition, the chiral center(s) in the acid moiety for cypermethrin analogues influenced hydrolytic rates more dramatically than that in the alcohol moiety (cyanohydrin) for all examined hepatic CEs, similar to fenvalerate results.
The origin of these general patterns of stereoselective hydrolysis of pyrethroid-like fluorescent substrates byhCE1 was examined through structural modeling and produced a proposed mechanism (Figures 2 and 3). As a result of sharing the same acid moiety between pyrethroids and pyrethroid fluorescent substrates, this proposed mechanism probably also fits for hydrolysis of pyrethroids by hCE1. Steric clashes between the specific isoforms and the active sites of hCE1 appear to explain why trans-isomers of other pyrethroids (e.g., permethrin, deltamethrin) with a cyclopropane ring are metabolized faster in hCE1 or humans than their corresponding cis isomer. For example, levels of trans-isomers in human serum were below the limits of detection within 25 h after oral ingestion of an emulsifiable concentrate formulation of permethrin, whereas cis-permethrin was still present at detectable levels 10 days after exposure (28). A similar pattern of stereoselective hydrolysis of pyrethroids and their fluorescent analogues by mammalian and human hepatic CEs (14) suggests that the interaction between pyrethroid-like fluorescent substrates and the substrate-binding site of hCE1 may be a general phenomenon for hepatic CEs and all other carboxylesterases because all carboxylesterases share similar three-dimensional structure (1). However, stereoselectivity is inherently difficult to characterize because it strongly relies on the three-dimensional structures involved, and these can vary considerably due to intramolecular motions and intermolecular interactions. For example, there was >10-fold difference in selective activities between 1R trans- and 1S trans-isomers of cypermethrin analogues by both recombinant murine liver CEs. Similarly, under the best configuration (1S trans) in the acid moiety of cypermethrin analogues, the activity of the isomer (1S trans, αS) with S-cyanohydrin was higher than that (1S trans, αR) with R-cyanohydrin. Two possible structural explanations for these observations are that (1) the substrate-binding sites in the mouse CEs, in an undetermined way, sterically restrict 1R trans-isomers (but not 1S trans-isomers) from aligning properly for a nucleophilic attack by the catalytic serine; or (2) the orientation of 1S trans-isomers in the substrate-binding sites facilitates the hydrogen-bond formation between the catalytic histidine and the oxygen (C-O-C) of the ester bond, but the orientation of 1R trans-isomers does not. The latter has been used as an explanation for why lipases from Candida rugosa and C. antarctica exhibit a strong preference for the R-ester with a secondary alcohol to its S-ester (29, 30). A missing hydrogen bond between the catalytic histidine and the oxygen of S-esters has been suggested to account for these particular observations of stereoselectivity. Additional structural analyses will be required to establish what the detailed nature of such stereoselectivities is.
The structure-guided explanation of stereoselective hydrolysis of pyrethroid-like substrates by CEs presented in this paper provides information that may be useful in the development of improved inhibitors for pyrethroidhydrolyzing esterases and the development of novel insecticides. The aim of designing efficacious inhibitors for pyrethroid-hydrolyzing esterases is to control the propagation of agricultural pests and disease vectors (e.g., mosquitoes), in which an enhanced esterase-mediated metabolism is a major metabolic mechanism of pyrethroid resistance (31, 32), while having little effect on mammals. Thus, inhibition of esterase-mediated hydrolysis in resistant pests will potentially restore the insecticidal efficacy of pyrethroids on previously resistant insects. Although carbamates and organophosphates are excellent inhibitors of esterases, and they have been used as synergists of ester-containing pyrethroids in some countries, they are not selective, and some are toxic to mammals and nontarget organisms. Inhibitors with structures similar to pyrethroids may be more selective than general carbamates and organophosphates, and they may offer less risk of toxicity. On the basis of the specific activity (Table 1) and structural examination, compounds with a three-dimensional configuration matching (1S trans, αS) appear to the best structure for pyrethroid-selective inhibitors. In contrast, efforts to develop new ester-containing pyrethroids with a cyclopropane ring should avoid 1S trans-configuration, and those directed further developments of ester-containing compounds with a noncyclopropane ring should avoid a 2R-configuration since ester-containing compounds with 1S trans- or 2R-configuration are easily hydrolyzed, and thus, detoxified by mammalian carboxylesterases.
Acknowledgments
This work was supported in part by NIEHS Grant R37 ES02710, NIEHS Superfund Grant P42 ES04699, NIEHS Center for Environmental Health Sciences Grant P30 ES 05707, and NIH grant AI58267 (B.D.H.) and NIH Grant CA98468 (M.R.R.).
References
- (1).Satoh T, Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Annu. Rev. Pharmacol. Toxicol. 1998;38:257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- (2).Hosokawa M, Maki T, Satoh T. Characterization of molecular species of liver microsomal carboxylesterases of several animal species and humans. Arch. Biochem. Biophys. 1990;277:245–253. doi: 10.1016/0003-9861(90)90572-g. [DOI] [PubMed] [Google Scholar]
- (3).Ollis DL, Chea E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Vershueren KHG, Goldman A. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- (4).Elliott M, Janes NF. Synthetic pyrethroidssa new class of insecticide. Chem. Soc. Rev. 1978;7:473–505. [Google Scholar]
- (5).Class TJ, Kintrup J. Pyrethroids as household insecticides-analysis, indoor exposure and persistence. Effects of pyrethroid insecticides on subjects engaged in packaging pyrethroids. Fresenius’. J. Anal. Chem. 1991;340:446–453. [Google Scholar]
- (6).He F, Sun J, Han K, Wu Y, Yao P. Effects of pyrethroid insecticides on subjects engaged in packaging pyrethroids. Br. J. Ind. Med. 1988;45:548–551. doi: 10.1136/oem.45.8.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Go V, Garey J, Wolff MS, Pogo GT. Estrogenic potential of certain pyrethroid compounds in the MCF-7 human breast carcinoma cell line. Environ. Health Perspect. 1999;107:173–177. doi: 10.1289/ehp.99107173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mani U, Islam F, Prasad AK, Kumar P, Suresh Kumar V, Maji BK, Dutta K. Steroidogenic alterations in testes and sera of rats exposed to formulated fenvalerate by inhalation. Hum. Exp. Toxicol. 2002;21:593–597. doi: 10.1191/0960327102ht298oa. [DOI] [PubMed] [Google Scholar]
- (9).Repetto RC. In: Repetto R, Baliga SS, editors. WRI (World Resources Institute), National Center for Food & Agricultural Policy; Washington, DC: 1996. [Google Scholar]
- (10).Extension Toxicology Network . Pyrethroids. Pesticide Information Profiles. (accessed Oct 20, 2004) [Google Scholar]
- (11).Cantalamessa F. Acute toxicity of two pyrethroids, permethrin and cypermethrin, in neonatal and adult rats. Arch. Toxicol. 1993;67:510–513. doi: 10.1007/BF01969923. [DOI] [PubMed] [Google Scholar]
- (12).Stok JE, Huang H, Jones PD, Wheelock CE, Morisseau C, Hammock BD. Identification, expression and purification of a pyrethroid-hydrolyzing carboxylesterase from mouse liver microsomes. J. Biol. Chem. 2004;279:29863–29869. doi: 10.1074/jbc.M403673200. [DOI] [PubMed] [Google Scholar]
- (13).Huang H, Stok JE, Stoutamire DW, Shirley JG, Hammock BD. Development of optically pure pyrethroid-like fluorescent substrates for carboxylesterases. Chem. Res. Toxicol. 2005;18:516–527. doi: 10.1021/tx049773h. [DOI] [PubMed] [Google Scholar]
- (14).Shan G, Hammock BD. Development of sensitive esterase assays based on R-cyano-containing esters. Anal. Biochem. 2001;299:54–62. doi: 10.1006/abio.2001.5388. [DOI] [PubMed] [Google Scholar]
- (15).Soderlund DM, Abdel-Aal YAI, Helmuth DW. Selective inhibition of separate esterases in rat and mouse liver microsomes hydrolyzing malathion, trans-permethrin and cis-permethrin. Pestic. Biochem. Physiol. 1982;17:162–169. [Google Scholar]
- (16).Bencharit S, Morton CL, Howard-Williams EL, Danks MK, Potter PM, Redinbo MR. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat. Struct. Biol. 2002;9:337–342. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- (17).Bencharit S, Morton CL, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous drug-processing enzyme. Nat. Struct. Biol. 2003;10:349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- (18).Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- (19).Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr., Sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- (20).DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. (accessed Sept 25, 2004) [Google Scholar]
- (21).Abernathy CO, Ueda K, Engel JL, Gaughan LC, Casida JE. Substrate-specificity and toxicological significance of pyrethroids-hydrolzying esterases of mouse liver microsomes. Pestic. Biochem. Physiol. 1973;3:300–311. [Google Scholar]
- (22).Soderlund DM. Metabolic consideration in pyrethroid design. Xenobiotica. 1992;22:1185–1194. doi: 10.3109/00498259209051872. [DOI] [PubMed] [Google Scholar]
- (23).Miyamoto J. Degradation, metabolism and toxicity of synthetic pyrethroids. Environ. Health Perspect. 1976;14:15–28. doi: 10.1289/ehp.761415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Casida JE, Gaughan LC, Ruzo LO. Comparative metabolism of pyrethroids derived from 3-phenoxybenzyl and R-cyano-3-phenoxybenzyl alcohols. In: Geissbuehler H, editor. Advance in Pesticide Science. Vol. 2. Pergamon; New York: 1979. p. 182. [Google Scholar]
- (25).Casida JE, Ruzo LO. Metabolic chemistry of pyrethroid insecticides. Pestic. Sci. 1980;11:257–269. [Google Scholar]
- (26).Takamatsu Y, Kaneko H, Abiko J, Yoshitake A, Miyamoto J. In vivo and in vitro stereoselective hydrolysis of four isomers of fenvalerate. J. Pestic. Sci. 1987;12:397–404. [Google Scholar]
- (27).Wheelock CE, Wheelock AM, Zhang R, Stok JE, Morisseau C, LeValley SE, Green CE, Hammock BD. Evaluation of R-cyanoesters as fluorescent substrates for examining interindividual variation in general and pyrethroid-selective esterases in human liver microsomes. Anal. Biochem. 2003;315:208–222. doi: 10.1016/s0003-2697(03)00002-2. [DOI] [PubMed] [Google Scholar]
- (28).Gotoh Y, Kawakami M, Matsumoto N, Okada Y. Permethrin emulsion ingestion: clinical manifestations and clearance of isomers. Clin. Toxicol. 1998;36:57–61. doi: 10.3109/15563659809162587. [DOI] [PubMed] [Google Scholar]
- (29).Cygler M, Grochulski P, Kazlaukas RJ, Schrag JD, Bouthillier FB, Rubin B, Serrequi AN, Gupta AK. A structural basis for the chiral preferences of lipases. J. Am. Chem. Soc. 1994;116:3180–3186. [Google Scholar]
- (30).Uppenberg J, Öhrner N, Norin M, Hult K, Kleywegt G, Patkar S, Waagen V, Anthosen T, Jones TA. Crystallographic and molecular-modeling studies of lipase B from Candida antarctica reveal a stereospecificity pocket for secondary alcohols. Biochemistry. 1995;34:16838–16851. doi: 10.1021/bi00051a035. [DOI] [PubMed] [Google Scholar]
- (31).Oppenoorth FJ. Biochemistry and genetic of insecticide resistance. In: Kerkut GA, Gilbert LI, editors. Comprehensive Insect Physiology, Biochemistry and Pharmacology. Vol. 12. Pergamon Press; Oxford, U.K: 1985. pp. 731–773. [Google Scholar]
- (32).Grant DF, Bender DM, Hammock BD. Quantitative kinetic assays for glutathione S-transferase and general esterase in individual mosquitoes using an EIA reader. Insect Biochem. 1989;19:741–751. [Google Scholar]