

Abstract
Soluble epoxide hydrolase (sEH) inhibitors have been demonstrated to have cardiovascular protective actions. This hydrolase enzyme converts fatty acid epoxides to their corresponding diols, and this conversion can alter the biologic activity of these metabolites. We hypothesized that 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), a sEH inhibitor, would protect stroke-prone spontaneously hypertensive rats from cerebral ischemia. AUDA was administered to 6-week-old male rats for 6 weeks, during which blood pressure was measured by telemetry. Cerebral ischemia was induced by middle cerebral artery occlusion, the size of the cerebral infarct was assessed after 6 hours of ischemia, and the results were expressed as a percentage of the hemisphere infarcted (%HI). Vascular structure and function were assessed using a pressurized arteriograph. Plasma levels of AUDA at the end of the treatment period averaged 5.0 ± 0.4 ng/mL, and the urinary excretion rate was 99 ± 21 ng/d. AUDA-treated rats had significantly smaller cerebral infarcts than control rats (36 ± 4% vs 53 ± 4% HI, treated versus control, P < 0.05, n = 6). This difference occurred independently of changes in blood pressure. AUDA treatment increased the passive compliance of the cerebral vessels but had no effect on vascular structure. The results of this study provide novel evidence suggesting that the sEH inhibitor AUDA is a possible therapeutic agent for ischemic stroke.
Keywords: hypertension, ischemia, epoxyeicosatrienoic acids, soluble expoxide hydrolase
Eighty-eight percent of strokes occurring in the population are ischemic in nature, and hypertension exacerbates the damage caused by ischemic stroke. When cerebral ischemia is induced by middle cerebral artery (MCA) occlusion, the volume of the resultant infarct is greater in stroke-prone spontaneously hypertensive rats (SHRSP) than in Wistar Kyoto (WKY) rats.1 This may be because of differences in cerebral blood vessel structure and function between the 2 strains. In WKY rats, when an occlusion occurs, the collateral vessels around it dilate to facilitate perfusion of the brain to prevent a large infarct. This increase in collateral blood flow does not occur in SHRSP. SHRSP vessels have smaller vessel diameters before an ischemic insult and an impaired ability to dilate in response to ischemia.2 Therefore, when an occlusion occurs, there is insufficient tissue perfusion, and a large cerebral infarct is produced. Accordingly, treatment of SHRSP with compounds that alter the vasculature to improve blood flow should also reduce cerebral infract size.
Soluble epoxide hydrolase (sEH) inhibitors have been demonstrated to reduce blood pressure in hypertension, decrease hypertension-induced renal damage, attenuate vascular smooth muscle cell proliferation, and decrease tissue injury associated with lipopolysaccharide-induced systemic inflammation.3-6 In rats and humans, sEH is involved in the metabolism of arachidonic acid and linoleic acid epoxides to their corresponding diols.7-9 Likewise, the arachidonic acid epoxides, epoxyeicosatrienoic acids (EETs), are excellent therapeutic candidates for hypertensive ischemic cerebrovascular disease.10-14 EETs dilate cerebral blood vessels and are metabolized to dihydroxyeicosatrienoic acids (DHETs) by sEH, and DHETs have significantly less activity than EETs.10,11,13 The development of potent and stable urea-based inhibitors of sEH has facilitated long-term in vivo studies.15,16 These sEH inhibitors are competitive, tight-binding inhibitors with namomolar KI values that efficiently reduce epoxide hydrolysis in several in vivo and in vitro models.15,16 Recent modifications to the urea-based inhibitors have resulted in potent sEH inhibitors with favorable solubility properties that allow for oral administration.16 2-(3-Adamantan-1-yl-ureido)-dodecanoic acid (AUDA) is an N′-carboxylic acid substitution that increases water solubility without an appreciable reduction in the potency of sEH inhibition.16 We hypothesized that the sEH inhibitor, AUDA, would reduce the size of the cerebral infarct produced after induction of ischemia in SHRSP. We further hypothesized that AUDA would have this effect by altering the structure of the cerebral blood vessels or increasing the vasodilator capacity of these vessels.
MATERIALS AND METHODS
Animals
Male SHRSP obtained from the colony at the Medical College of Georgia were maintained on a 12-hour light-dark cycle and allowed access to food and water ad libitum. These studies complied with the protocols for animal use outlined by the American Physiological Society and were approved by the institutional animal care and use committee. AUDA (25 mg/L) was dissolved in a drinking water solution containing βcyclodextrin (0.5 g/L) and ethanol (0.05%) that was sonicated for 1 hour. Rats from 6 to 12 weeks of age were treated with AUDA or vehicle. Vascular structure and the effects of cerebral ischemia were assessed in 12-week-old rats.
Measurement of Oxylipid and AUDA Levels
The levels of the arachidonic acid metabolites, EETs and DHETs, and the linoleic acid metabolites, epoxyoctadecanoic acid (EPOME) and dihydroxyoctadecanoic acid (DHOME), were measured in the plasma and urine as described previously.17 Rats were housed in metabolic cages that separated urine from food and feces, and urine was collected for a 24-hour period. At the end of the experimental period, a plasma sample was also obtained. AUDA and 4-(3-adamantan-1-yl-ureido)-butanoic acid (AUBA), an inactive metabolite of AUDA, were measured as described previously using high-performance liquid chromatography with tandem mass spectrometry.15
Measurement of Blood Pressure
Telemetry transmitters (Data Sciences, St. Paul, MN) were implanted, and mean arterial pressure (MAP) and heart rate data were collected as described previously.18 Baseline measurements were obtained for 5 days before beginning the treatment.
MCA Occlusion
At 12 weeks of age, after 6 weeks of treatment with AUDA or vehicle, cerebral ischemia was induced. Rats were anesthetized with sodium pentobarbital (50 mg/kg IP), and body temperature was maintained at 37°C during anesthesia. The MCA was permanently occluded by use of the intralumenal thread occlusion technique.19 Briefly, the common carotid artery was exposed by a midline incision, and the branches of the external carotid artery were cauterized. A 3-0 monofilament thread with a rounded tip was introduced into the external carotid artery in a retrograde fashion. This was advanced cranially into the internal carotid artery over a distance of exactly 19 mm, measured from the bifurcation of the common carotid artery. The thread was left in place, and the rats were allowed to recover. Because SHRSP have very large cerebral infarcts that can result in significant mortality, we used a 6-hour period of ischemia that causes significant ischemic damage. Postocclusion, the rats were anesthetized and decapitated, and the brains were carefully removed. The area of the infarction was quantified using 2,3,5-triphenyltetrazolium (TTC) staining as described previously.20 Blood flow to the region surrounding the MCA was measured using a laser Doppler flow probe placed 5 mm lateral and 1 mm posterior to the bregma to confirm MCA occlusion.
MCA Function and Structure
The structure and function of the MCA was assessed in 12-week-old rats after 6 weeks of treatment with AUDA or vehicle. MCAs were isolated and placed in cold physiological salt solution (PSS) (in mM: 141.9 NaCl, 4.7 KCl, 1.7 MgSO4, 0.5 EDTA, 2.8 CaCl2, 10.0 HEPES, 1.2 KH2PO4, and 5.0 glucose). The first branch-free segment of the MCA most proximal to the Circle of Willis was mounted on 2 glass micropipettes in an arteriograph (Living Systems Instrumentation, Burlington, VT). Vessels were bathed with warm oxygenated PSS. Videomicroscopy and a calibrated video dimension analyzer were used to measure lumen diameter and wall thickness. The intraluminal pressure was set at 75 mm Hg. Cumulative dose-response curves were generated for bradykinin and serotonin (10-9-10-5 M). Lumen diameter was measured after a 5-minute equilibration with each dose of agonist, and results were expressed as a percentage of the maximum dilation obtained with sodium nitroprusside (10-5 M). The percentage myogenic tone was calculated at intraluminal pressures of 75 and 125 mm Hg using the following formula: 1 - (active diameter/passive diameter) × 100.21 These 2 pressures were selected because they fall within the range of autoregulatory pressures for the MCA.
The MCAs were then bathed in calcium-free PSS containing 2 mM EGTA, and the intraluminal pressure was increased from 0 to 180 mm Hg in 20 mm Hg increments; measurements of lumen diameter and wall thickness were taken at each pressure after a 5-minute equilibration. The external diameter, wall/lumen ratio, and circumferential wall stress and strain were calculated using the method of Baumbach and Hadju.22 The β-coefficient, a measure of wall stiffness, was calculated from the exponential curve fit for each individual stress-strain curve.
Statistics
All results are represented as a mean ± SEM. Blood pressure and cerebral vascular data were analyzed using an ANOVA. The cerebral infarct size, β-coefficients, and oxylipid levels were compared using an unpaired t test. A P value ≤ 0.05 was considered significant.
RESULTS
MAP and heart rate were continuously monitored for the duration of the study using telemetry (Fig. 1). There was no difference in the MAP or heart rate between the SHRSP treated with AUDA and the control rats.
FIGURE 1.
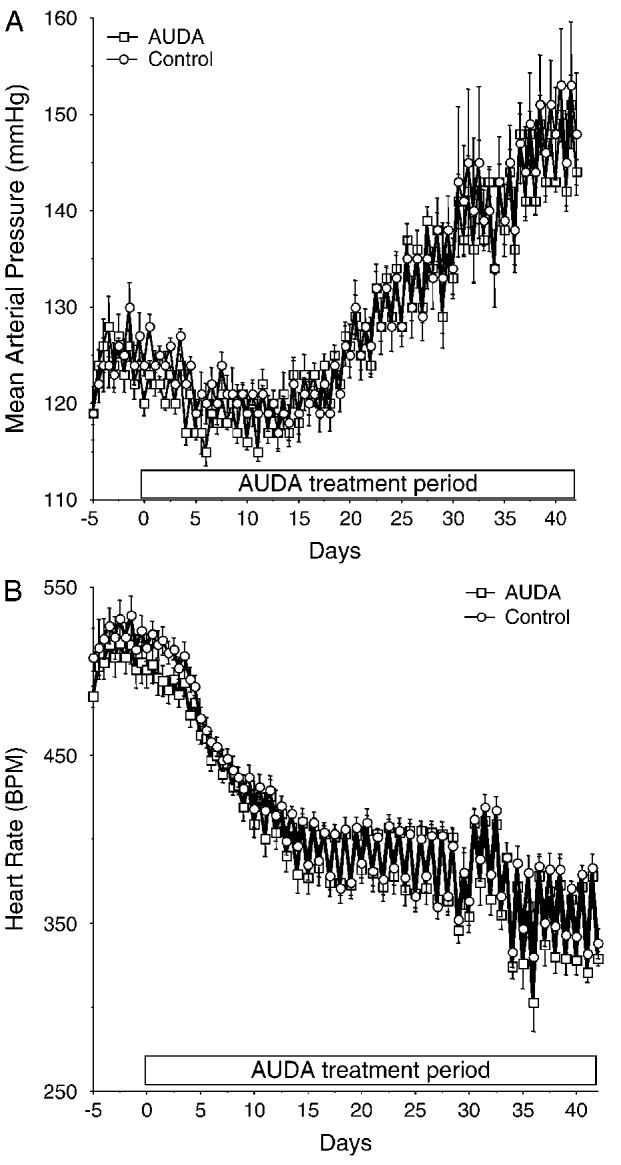
A, Mean arterial pressure measured by radiotelemetry in the AUDA-treated and control rats. The average 12-hour day and night pressures are shown for the 5 days before and for the duration of the treatment. B, Heart rate over the same time (n = 6 in each group).
To investigate the effect of sEH inhibition on the outcome of cerebral ischemia, the MCA was occluded, and infarct size measured in 12-week-old rats after 6 weeks of treatment with AUDA or vehicle. Treatment of SHRSP with AUDA caused a marked reduction in percentage of the hemisphere damaged by ischemia (36 ± 4% vs 53 ± 4% AUDA versus vehicle, P < 0.05, n = 6) (Fig. 2). The infarct was limited to the cortex and the basal ganglia. The percentage drop in cerebral blood flow at the time of occlusion, assessed by laser Doppler, was similar in both groups (68% for the AUDA treated and 69% for the control group).
FIGURE 2.
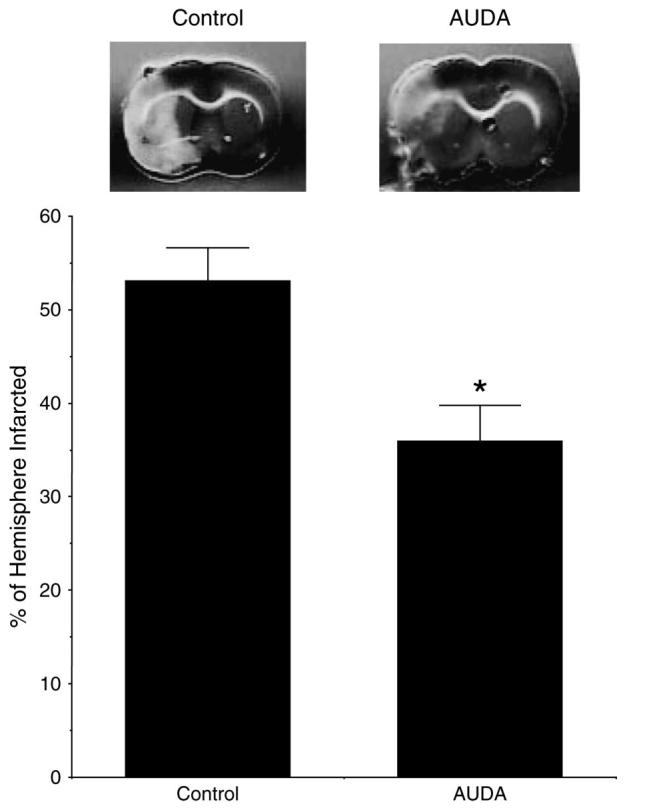
The upper panel shows representative cerebral infarcts for AUDA-treated and control rats; the gray area is viable tissue; and the white area is tissue damaged by ischemia. The lower panel shows the percentage of the hemisphere infarcted (n = 6 in each group *P < 0.05).
A pressurized arteriograph was used to study the function and the structure of the MCA after AUDA treatment. The responsiveness of the MCA to bradykinin and serotonin was evaluated. AUDA treatment did not affect the responsiveness of the MCA to either agonist; both the EC50 and the maximal response were similar between the groups (Fig. 3). Similarly, the treatment of the SHRSP with AUDA had no effect on the vessels’ ability to generate tone. The percentage tone at 75 mm Hg was 12.5 ± 3.1% for the AUDA-treated group and 9.2 ± 5.1% for the control group, and at 125 mm Hg the percentage tone was 18.6 ± 4.8 and 13.0 ± 4.8 for the AUDA-treated and control groups, respectively. The structure of the MCA was assessed under calcium-free conditions to eliminate the effects of myogenic tone. Lumen diameter and wall/lumen ratio of the MCA were similar in the rats treated with AUDA compared with control (Fig. 4). There was no difference in external vessel diameter at any intraluminal pressure between the 2 groups (at 120 mm Hg intraluminal pressure, 274 ± 14 μm control and 279 ± 7 μm AUDA). Wall thickness was also measured, and it did not differ between the groups (at 120 mm Hg intraluminal pressure, 38.1 ± 3.1 μm control and 38.8 ± 5.5 μm AUDA). When wall stress was plotted against wall strain, the curve produced for the vessels from AUDA-treated rats was shifted to the right when compared with the curve produced for the vessels from control rats (Fig. 5), indicating that the passive compliance was increased in the AUDA-treated vessels. The βcoefficient was calculated from the exponential curve fit for each individual stress-strain curve to produce a measure of vessel stiffness. The β-coefficient was greater for the vessels from the control rats, indicating that these vessels were stiffer than those from the AUDA-treated group.
FIGURE 3.
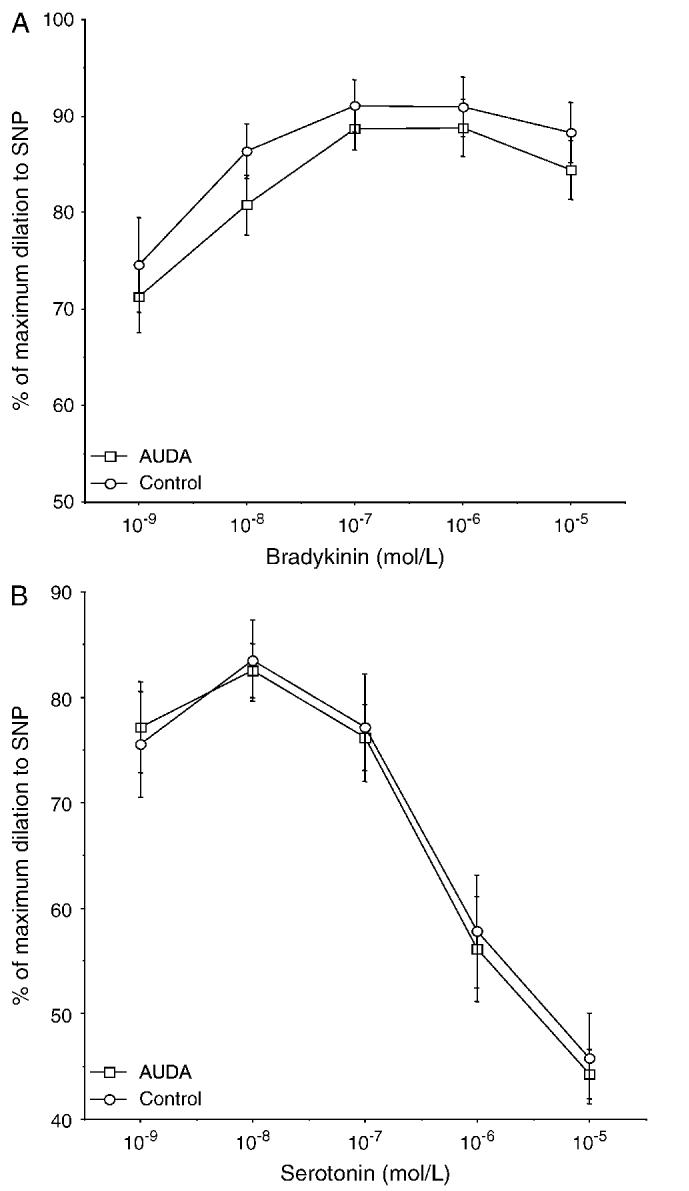
A, Relaxation of the MCA in response to bradykinin (10-9-10-5 M); the results are expressed as a percentage of the maximum dilation otained in response to SNP (10-5 M). B, Constriction of the MCA in response to serotonin (10-9-10-5 M); the results are expressed as a percentage of the maximum lumen diameter as described above (n = 10 for AUDA and 8 for control).
FIGURE 4.
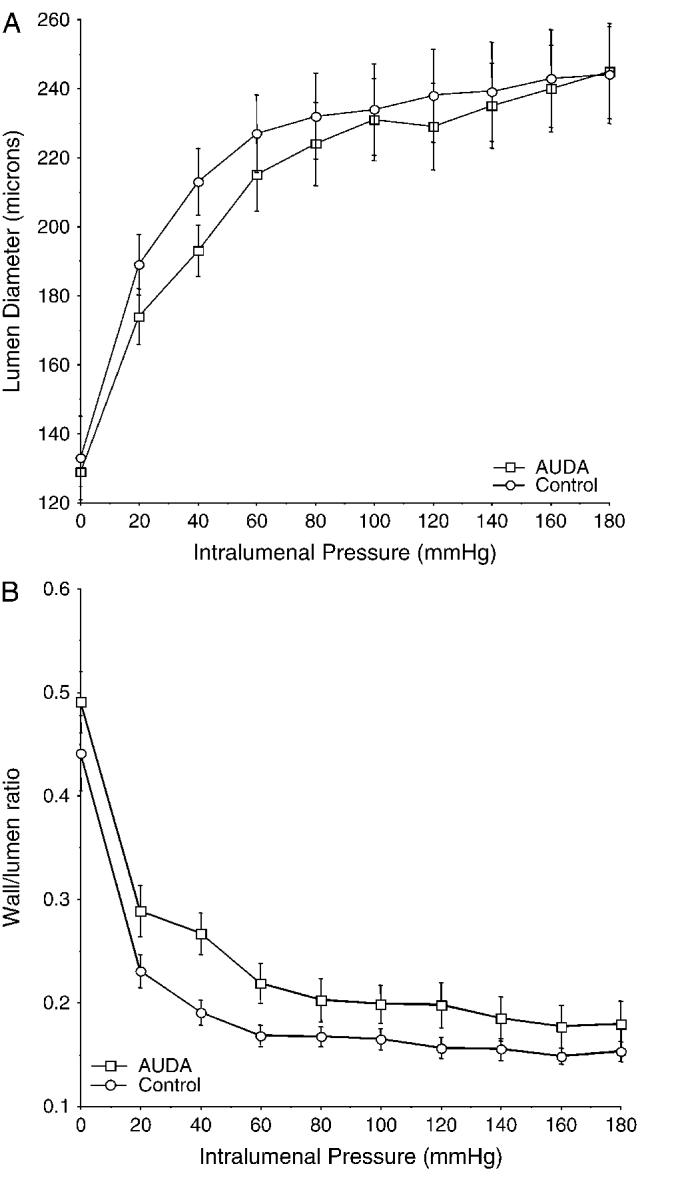
A, Lumen diameter in micrometerss of the MCA over a range of intraluminal pressures (0-180 mm Hg) for the AUDA-treated and the control rats. B, Wall/lumen ratio (n = 10 for AUDA and 8 for control).
FIGURE 5.
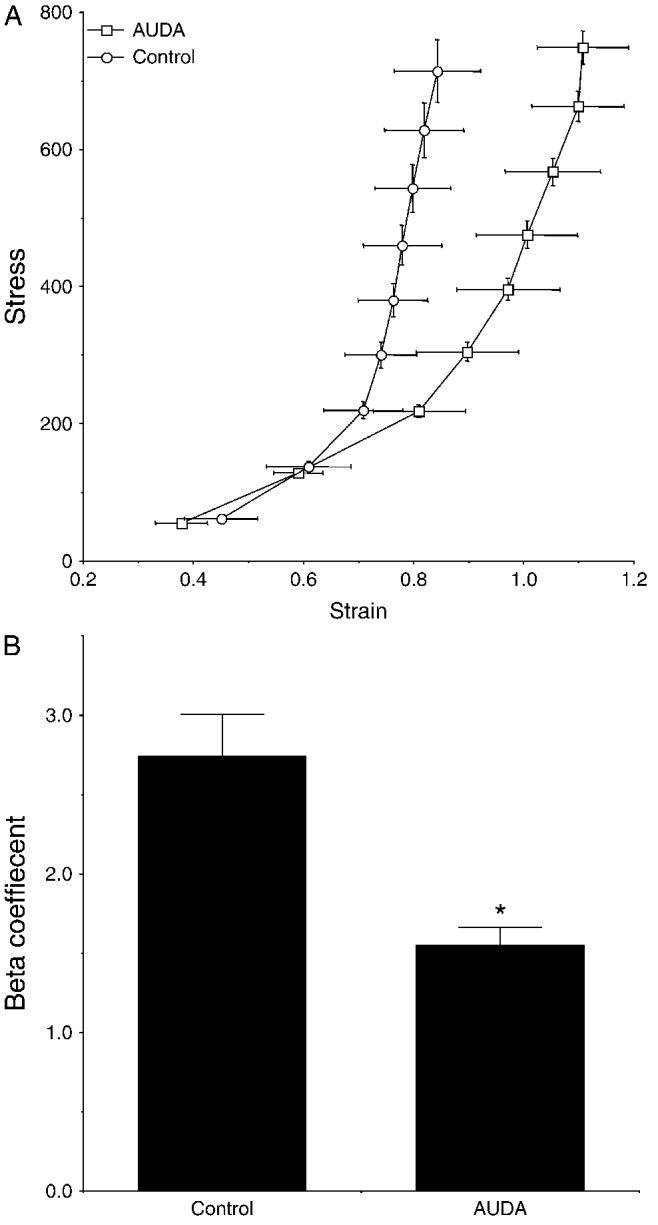
A, Average stress-strain curves generated for the MCAs from AUDA-treated and control SHRSP (n = 8). B, β-Coefficients calculated from the individual stress-strain curves for the MCAs from AUDA-treated and control SHRSP.
Analysis of urinary and plasma levels of AUDA and AUBA and epoxide metabolites was carried out at the end of the treatment period. The concentrations of AUDA in the plasma and urine were 5.0 ± 0.4 ng/mL and 99.3 ± 21.1 ng/d, respectively, and confirm that AUDA reached the systemic circulation. AUBA is a terminal β-oxidation metabolite lacking biologic activity. AUBA levels were measured to determine the importance of the β-oxidation pathway in the degradation of AUDA. AUBA levels were found at significant levels in the plasma, and it is the only AUDA metabolite detected at high levels in the urine. The levels of AUBA in the plasma and the urine were 7.2 ± 0.6 ng/mL and 2617 ± 1008 ng/d (plasma n = 5, urine n = 7). We also observed small measurable amounts of AUDA (0.4 ± 0.1 nmol/L) in the cerebral spinal fluid. The levels of the arachidonic and linoleic acid metabolites in the urine and plasma are shown in Table 1. AUDA treatment had little effect on the plasma levels of the fatty acid metabolites. AUDA did result in changes in the urinary epoxides and diols of arachidonic and linoleic acid. The urinary EPOME:DHOME ratio was increased by 32% in the AUDA-treated rats, and the EET:DHET ratio was increased by 75%. The change in the urinary epoxide to diol ratio results from a decrease in diol levels with maintained epoxide levels.
TABLE 1.
.Plasma and Urine CYP450 Metabolites in AUDA-Treated Rats
| Oxylipid Metabolites | Control Plasma | AUDA Plasma | Control Urine | AUDA Urine |
|---|---|---|---|---|
| EPOMEs (pmol/mL) | 292.5 ± 26.2 | 290.7 ± 14.4 | 2568 ± 625 | 2090 ± 131 |
| DHOMEs (pmol/mL) | 56.7 ± 5.7 | 52.9 ± 6.9 | 1,170 ± 33 | 722 ± 52 |
| EPOME:DHOME ratio | 5.2 | 5.5 | 2.2 | 2.9 |
| EETs (pmol/mL) | 95.0 ± 20.7 | 72.9 ± 21.1 | 25.7 ± 1.12 | 28.7 ± 3.91 |
| DHETs (pmol/mL) | 30.0 ± 1.8 | 23.9 ± 2.1 | 30.8 ± 9.1 | 20.9 ± 0.8 |
| EET:DHET ratio | 3.2 | 3.1 | 0.8 | 1.4 |
| 12,13-EPOME | 89 ± 2% | 91 ± 1% | 75 ± 1% | 76 ± 1% |
| 9,10-EPOME | 11 ± 2% | 9 ± 1% | 25 ± 1% | 24 ± 1% |
| 12,13-DHOME | 58 ± 3% | 46 ± 1% | 74 ± 2% | 72 ± 1% |
| 9,10,-DHOME | 42 ± 3% | 54 ± 1% | 26 ± 2% | 28 ± 1% |
| 14,15-EET | 39 ± 7% | 40 ± 4% | 57 ± 1% | 55 ± 2% |
| 11,12-EET | 14 ± 1% | 16 ± 1% | 43 ± 1% | 45 ± 3% |
| 8,9-EET | 24 ± 8% | 17 ± 6% | 0 ± 0% | 0 ± 0% |
| 5,6-EET † | 24 ± 1% | 27 ± 2% | ND | ND |
| 14,15-DHET | 8 ± 1% | 10 ± 1% | 14 ± 2% | 12 ± 1% |
| 11,12-DHET | 7 ± 2% | 8 ± 1% | 0 ± 0% | 0 ± 0% |
| 8,9-DHET | 6 ± 1% | 6 ± 1% | 0 ± 0% | 0 ± 0% |
| 5,6-DHET | 79 ± 3% | 76 ± 1% | 86 ± 2% | 88 ± 1% |
Values are mean ± SE.
5,6-EET recoveries with the implemented procedure are roughly 25%. Loss of this compound is apparently through internal cyclization to the 5,6-delta lactone, not hydrolysis to the 5,6-DHET (29). “%” indicates the percentage of specific EPOMEs, DHOMEs, EETs, and DHETs in total EPOMEs, DHOMEs, EETs, and DHETs respectively.
Abbreviations: SHRSP, stroke-prone spontaneously hypertensive rats; EET, epoxyeicosatrienoic acid; DHET, dihydroxyeicosatrienoic acids; AUDA, adamantyl dodecanoic acid; EPOME, epoxyoctadecanoic acid; DHOME, dihydroxyoctadecanoic acid.
DISCUSSION
Recent evidence suggests that administering sEH inhibitors during disease states will have cardiovascular protective actions.3-6,23 In the current study we demonstrate that the sEH inhibitor, AUDA, reduces the damage caused by MCA occlusion in SHRSP. Interestingly, the cerebral protection provided by sEH inhibition in the SHRSP was independent of any effect on blood pressure.
As expected, blood pressure increased and heart rate decreased in the SHRSP over the duration of the study. The absence of an effect of AUDA on blood pressure was intriguing because previous studies suggest that epoxide hydrolase is an important modulator of blood pressure in angiotensin II-dependent hypertension3,23 and SHR.4 On the other hand, other studies using SHR showed that there is variability in the blood pressure response to sEH inhibition that is dependent on the substrain of SHR used and that a polymorphism in the sEH gene exists. Activity of the sEH enzyme is greater in rats carrying the variant allele; however, inheritance of this allele does not correlate with blood pressure in the F2 progeny of a cross between this SHR substrain and WKY rats.24 It has not yet been determined if the SHRSP used in the current study also express the variant allele of the sEH gene. Another possibility is the low dose of the sEH inhibitor used in the current study. We have previously reported that the sEH inhibitor 1-cyclohexyl-3-dodecylurea (CDU) reached plasma levels of 23 ng/mL and lowered blood pressure in chronic angiotensin II-induced hypertension.3 Likewise, AUDA at a plasma concentration of 15 ng/mL lowers blood pressure in angiotensin II-induced hypertension.25 These concentrations are considerably higher than the circulating sEH inhibitor levels attained in the current study. On the other hand, decreased urinary diol levels and increased epoxide to diol ratios suggest that the sEH enzyme is being inhibited. In any case, it would appear from our studies that despite not lowering blood pressure, AUDA had beneficial cerebral effects in the SHRSP, suggesting that these beneficial effects occur via a blood pressure-independent mechanism.
One possible explanation for the cerebral protective actions of AUDA is that long-term sEH inhibition reduces the structural alterations in cerebral blood vessels that are normally observed in hypertensive rats. Nevertheless, we did not observe changes in the diameter or wall thickness of the MCAs from the rats treated for 6 weeks with AUDA. Vascular smooth muscle cell proliferation in cultured human aortic cells is reduced by sEH inhibition.5 Under these conditions, any effects of flow, pressure, or circulating factors are lost. Although we did not measure smooth muscle cell proliferation directly, we did not observe changes in the structure or contractile function of the MCA, two smooth muscle cell-driven events. Interestingly, we did observe a change in the passive compliance or stiffness of the blood vessels between the 2 groups. AUDA treatment causes the vessels from the SHRSP to be less stiff or more compliant. The mechanism for this change is not clear; normally changes in compliance result from alterations in collagen and elastin synthesis or deposition in the vessels. In the absence of marked changes in vascular structure and function, it seems likely that this is the case. Interestingly, we have previously reported decreased kidney collagen levels in sEH inhibitor-treated angiotensin-hypertensive rats.3 A similar change in the cerebral vasculature collagen levels could be occurring in the SHRSP. Thus, it is possible that an increase in compliance in the vessels around the ischemic blockade could allow for enhanced perfusion of the area and thus a smaller area of neuronal damage.
Current evidence in the literature also suggests that a cerebral vascular or direct neuroprotective action could explain the decreased neuronal damage in the SHRSP. Arachidonic acid epoxides are effective dilators of the cerebral vasculature.10,11 They are also produced in the brain by astrocytes26,27 and are present in cerebrospinal fluid in micromolar concentrations.14 In addition, astrocytes are unique cells that play an important role in regulating neuronal function.28,29 Because small but measurable amounts of AUDA were present in the cerebral spinal fluid, it is possible that sEH inhibition could be acting directly on the astrocytes. This cell type has been proposed to form the communication between neurons and the cerebral microvasculature, and EETs may be the signaling molecules responsible for matching blood flow to neuronal metabolic activity.14 When glutamate is infused into the subdural space, a reactive hyperemia occurs that is blunted when the production of EETs is pharmacologically inhibited.11,30 Interestingly, a recent study demonstrated enhanced postischemic recovery in mouse hearts that overexpressed the CYP2J2 epoxygenase enzyme in cardiomyocytes.31 This protection of the heart involved the mitochondrial ATP-sensitive K+ channel.31 It is therefore possible that greater availability of cerebral EETs during sEH inhibition will increase cerebral blood flow and therefore reduce ischemic damage. On the other hand, Sellers et al32 recently reported that intracerebroventricular delivery of AUDA actually increased blood pressure in the SHR. Thus, it is more likely that the protection from ischemic damage resulted from the changes in the cerebral vasculature in AUDA treated animals.
Although greater availability of cerebral EETs during AUDA treatment could explain the protection of the cerebral vasculature from ischemic damage, the lack of an increase in plasma or urinary EET levels would suggest that other mechanisms could contribute to the vascular protective effect of the sEH inhibitor. Other consequences that have been attributed to sEH inhibition include increased cellular incorporation and retention of EETs into endothelial phospholipids.33 Likewise, endothelium-dependent relaxation is enhanced by sEH inhibition;33 however, the present study failed to demonstrate an enhanced dilation of cerebral vessels in response to bradykinin. Inhibitors of sEH also enhance the flux of EETs into alternative β-oxidation and chain elongation metabolic pathways.34,35 The possible contribution of incorporation of EETs into endothelial phospholipids or metabolism of EETs by other metabolic pathways to the protection of the cerebral vasculature in AUDA-treated SHRSP remains unknown. Last, the N′-carboxylic acid substitution that increases water solubility of sEH inhibitors has also been demonstrated to confer peroxisome proliferator-activated receptor α (PPAR-α) activity to AUDA. AUDA at the high doses of 3 and 10 μM produced 1.5- and 3-fold increases in PPAR-α activation.36 Interestingly, the PPAR-α activator fenofibrate has been demonstrated to reduce the susceptibility to stroke in apolipoprotein E-deficient mice as well as decrease the cerebral infarct size in wild-type mice.37 Thus, AUDA could be providing protection from ischemic damage by mechanisms other than increasing circulating EET bioavailability in the SHRSP.
CONCLUSIONS
These studies add to the growing list of cardiovascular benefits imparted by sEH inhibitors. The present study provides novel and exciting data demonstrating the possible therapeutic use for sEH inhibitors as preventative therapies to provide neuronal protection from cerebral ischemia. We show that AUDA treatment reduces cerebral infarct size in the absence of changes in blood pressure. We propose that AUDA has this effect by enhancing the matching of blood flow to metabolic needs of the brain or protecting the neurons directly from ischemic damage. Thus, it may be possible to use sEH inhibitor treatment in patients at risk for ischemic stroke.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Ann Schreihofer and Constantine Zaharis for technical assistance.
Footnotes
This work was supported by National Institutes of Health grants HL-59699, DK-38226, HL-074167 (J.D.I.), R37 ES02710, P42 ES04699 (B.D.H.), and HL077385 (A.M.D.). Support also came from an American Heart Association Established Investigator Award (J.D.I.) and a Scientist Development Grant 0130364N (A.M.D.).
REFERENCES
- 1.Coyle P, Jokelainen PT. Differential outcome to middle cerebral artery occlusion in spontaneously hypertensive rats (SHRSP) and Wistar-Kyoto (WKY) rats. Stroke. 1983;14:605–611. doi: 10.1161/01.str.14.4.605. [DOI] [PubMed] [Google Scholar]
- 2.Coyle P, Heistad DD. Blood flow through cerebral and collateral vessels one month after middle cerebral artery occlusion. Stroke. 1987;18:407–411. doi: 10.1161/01.str.18.2.407. [DOI] [PubMed] [Google Scholar]
- 3.Zhao X, Yamamoto T, Newman JW, et al. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:12442–12445. [PubMed] [Google Scholar]
- 4.Yu Z, Xu F, Huse LM, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 5.Davis BB, Thompson DA, Howard LL, et al. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA. 2002;99:2222–2227. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmelzer KR, Kubala L, Newman JW, et al. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 8.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 9.Spector AA, Fang X, Snyder GD, et al. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 10.Ellis EF, Police RJ, Yancey L, et al. Dilation of cerebral arterioles by cytochrome P-450 metabolites of arachidonic acid. Am J Physiol. 1990;259:H1171–H1177. doi: 10.1152/ajpheart.1990.259.4.H1171. [DOI] [PubMed] [Google Scholar]
- 11.Leffler CW, Fedinec AL. Newborn piglet cerebral microvascular responses to epoxyeicosatrienoic acids. Am J Physiol. 1997;273:H333–H338. doi: 10.1152/ajpheart.1997.273.1.H333. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Harder DR. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic Acid. Stroke. 2002;33:2957–2964. doi: 10.1161/01.str.0000037787.07479.9a. [DOI] [PubMed] [Google Scholar]
- 13.Gebremedhin D, Ma YH, Falck JR, et al. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263:H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 14.Harder DR, Zhang C, Gebremedhin D. Astrocytes function in matching blood flow to metabolic activity. News Physiol Sci. 2002;17:27–31. doi: 10.1152/physiologyonline.2002.17.1.27. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe T, Morisseau C, Newman JW, et al. In vitro metabolism of the mammalian soluble epoxide hydrolase inhibitor, 1-cyclohexyl-3-dodecylurea. Drug Metab Dispos. 2003;31:846–853. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
- 16.Kim IH, Morisseau C, Watanabe T, et al. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem. 2004;47:2110–2122. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 17.Newman JW, Watanabe T, Hammock BD. The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J Lipid Res. 2002;43:1563–1578. doi: 10.1194/jlr.d200018-jlr200. [DOI] [PubMed] [Google Scholar]
- 18.Sasser JM, Pollock JS, Pollock DM. Renal endothelin in chronic angiotensin II hypertension. Am J Physiol. 2002;283:R243–R248. doi: 10.1152/ajpregu.00086.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zea Longa E, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 20.Dorrance AM, Osborn HL, Grekin R, et al. Spironolactone reduces cerebral infarct size and epidermal growth factor receptor mRNA in stroke-prone rats. Am J Physiol. 2001;281:R944–R950. doi: 10.1152/ajpregu.2001.281.3.R944. [DOI] [PubMed] [Google Scholar]
- 21.Baumbach GL, Hajdu MA. Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension. 1993;21:816–826. doi: 10.1161/01.hyp.21.6.816. [DOI] [PubMed] [Google Scholar]
- 22.Cipolla MJ, Curry AB. Middle cerebral artery function after stroke: the threshold duration of reperfusion for myogenic activity. Stroke. 2002;33:2094–2099. doi: 10.1161/01.str.0000020712.84444.8d. [DOI] [PubMed] [Google Scholar]
- 23.Imig JD, Zhao X, Capdevila JH, et al. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 24.Fornage M, Hinojos CA, Nurowska BW, et al. Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension. 2002;40:485–490. doi: 10.1161/01.hyp.0000032278.75806.68. [DOI] [PubMed] [Google Scholar]
- 25.Imig JD, Zhao X, Pollock DM, et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2003;42:395. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alkayed NJ, Narayanan J, Gebremedhin D, et al. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- 27.Murphy S, Pearce B, Jeremy J, et al. Astrocytes as eicosanoid-producing cells. Glia. 1988;1:241–245. doi: 10.1002/glia.440010402. [DOI] [PubMed] [Google Scholar]
- 28.Temburni MK, Jacob MH. New functions for glia in the brain. Proc Natl Acad Sci USA. 2001;98:3631–3632. doi: 10.1073/pnas.081073198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- 30.Alkayed NJ, Birks EK, Narayanan J, et al. Role of P-450 arachidonic acid epoxygenase in the response of cerebral blood flow to glutamate in rats. Stroke. 1997;28:1066–1072. doi: 10.1161/01.str.28.5.1066. [DOI] [PubMed] [Google Scholar]
- 31.Seubert J, Yang B, Bradbury JA, et al. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 32.Sellers KW, Sun C, Diez-Freire C, et al. Novel mechanism of brain epoxide hydrolase-mediated blood pressure regulation in the spontaneously hypertensive rat. FASEB J. 2005;19:626–628. doi: 10.1096/fj.04-3128fje. [DOI] [PubMed] [Google Scholar]
- 33.Weintraub NL, Fang X, Kaduce TL, et al. Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am J Physiol. 1999;277:H2098–H2108. doi: 10.1152/ajpheart.1999.277.5.H2098. [DOI] [PubMed] [Google Scholar]
- 34.Fang X, Kaduce TL, Weintraub NL, et al. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276:14867–14874. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 35.Fang X, Weintraub NL, McCaw RB, et al. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am J Physiol. 2004;287:H2412–H2420. doi: 10.1152/ajpheart.00527.2004. [DOI] [PubMed] [Google Scholar]
- 36.Fang X, Hu S, Watanabe T, et al. Activation of peroxisome proliferator-activated receptor α by substituted urea-derived soluble epoxide hydrolase inhibitors. J Pharmacol Exp Ther. 2005;314:260–270. doi: 10.1124/jpet.105.085605. [DOI] [PubMed] [Google Scholar]
- 37.Deplanque D, Gele P, Petrault O, et al. Peroxisome proliferator-activated receptor-α activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]