

Abstract
Twenty-one cats with hypertrophic cardiomyopathy were enrolled in this study to determine if the administration of benazepril (0.5 mg/kg body weight [BW], PO, q24h) to cats with subclinical hypertrophic cardiomyopathy improves cardiac diastolic function and reverses left ventricular hypertrophy when compared with diltiazem controlled delivery (CD) (10 mg/kg BW, PO, q24h). Cats were evaluated at day 0 and after 3 and 6 months of therapy.
In the benazepril group (n = 11), the diastolic transmitral flow of the E and A waves ratio (E/A ratio) increased significantly between 0 and 6 months (P = 0.009) and the thickness of the left ventricular free wall in systole (LVFWs) decreased significantly between 0 and 3 months (P = 0.04). In the diltiazem CD group (n = 5), none of the parameters varied significantly throughout the study. There was no difference between the benazepril and the diltiazem CD group throughout the study. Therefore, the variations observed for the E/A ratio and the LVFWs may have been incidental. Further studies will be needed to establish the role of benazepril in subclinical hypertrophic cardiomyopathy in cat.
Résumé
Le bénazépril et la cardiomyopathie hypertrophique subclinique chez le chat: une étude prospective. Vingt et un chats asymptomatiques ayant obtenu un diagnostic échographique de cardiomyopathie hypertrophique ont participé à cette étude prospective. Les objectifs de l’étude étaient de démontrer que l’administration de bénazépril (0,5 mg/kg, per os (PO), q 24h) peut réduire l’hypertrophie ventriculaire et induire une amélioration de la fonction diastolique, et ce, d’une façon plus convaincante que le diltiazem CD (10 mg/kg, PO, q 24h). Tous les chats ont été évalués à T0, T3 et T6 mois.
Pour le groupe bénazépril (n = 11), le ratio du flot transmitral diastolique de l’onde E et A (ratio E/A) a significativement augmenté entre T0–T6 mois (P = 0,009) et l’épaisseur de la paroi postérieure du ventricule gauche en systole a diminué significativement entre T0–T3 mois (P = 0,04). Pour le groupe diltiazem CD (n = 5), aucun des paramètres évalués n’a varié significativement durant l’étude. Aucune différence significative n’a été observée lorsque les deux groupes étaient comparés. Ainsi les changements observés pour le ratio E/A et l’épaisseur de la paroi postérieure du ventricule gauche en systole constituent possiblement des trouvailles non significatives d’un point de vue clinique. Des études ultérieures seront nécessaires afin de déterminer le rôle du bénazépril lors de CMH subclinique féline.
(Traduit par les auteurs)
Introduction
Hypertrophic cardiomyopathy (HCM) is a primary myocardial disease leading to concentric, symmetric, or asymmetric hypertrophy with alteration of diastolic function of the left ventricle. It is the most common cardiac disease diagnosed in cats, but humans and other species are also affected (1–3). In humans, HCM has been identified as a familial disease since 1958 (4). In approximately 50% of cases of familial HCM, an autosomal dominant sarcomeric gene mutation has been demonstrated, while in the other 50%, spontaneous mutations have been identified (5–8). The etiology of HCM in cats remains unknown, although inheritance through an autosomal dominant trait with 100% penetrance has recently been proposed in a colony of Maine coon cats (9–11).
In cats, clinical presentation varies from the incidental finding of a heart murmur, a gallop sound, or arrhythmias in an otherwise asymptomatic cat, to an acute onset of congestive heart failure, arterial thromboembolism, or sudden death (2). Treatment of asymptomatic cats remains controversial, since the administration of either calcium channel blockers, β-adrenergic blockers, or angiotensin-converting enzyme inhibitors (ACEI) has no proven effect on disease progression or survival (12). A calcium channel blocker (diltiazem), a β-adrenergic blocker (atenolol), or both, are commonly used, based on their theoretical ability to improve ventricular diastolic function, to control sinus tachycardia, or to reduce obstructive outflow tract pressure gradients when present (12).
Biochemical and genetic evidence for the existence of a cardiac renin-angiotensin system in different species has been accumulating since the 1980s (13–16). Angiotensin II has been shown to induce myocyte hypertrophy and fibroblast hyperplasia (2,17–24). Interestingly, the administration of ACEI can reverse cardiac hypertrophy in a pressure overload model, such as subaortic stenosis and systemic hypertension (25). However, when hypertrophy was due to HCM, the administration of ACEI failed to show regression of the hypertrophy in humans (19,26). This may suggest that the molecular basis of hypertrophy in HCM is different from hypertrophy in a pressure overload model (27). The utilization of ACEI in HCM remains interesting, as the angiotensin-converting enzyme (ACE) gene may modify the phenotypic expression of the HCM. It is now known that in presence of HCM, human patients expressing D/D genotype for the ACE gene show an increased level of serum ACE, have an increased risk of sudden death, and present an increased severity of hypertrophy (28–31). Intracoronary administration of enalapril maleate (an ACEI) to humans with HCM improved diastolic function, which suggested that blocking the tissue renin-angiotensin system may be beneficial in HCM, but when the captopril (another ACEI) was administered sublingually, the improvement in diastolic function was no longer seen (20). This knowledge has generated interest in ACEI as a potential therapeutic tool to prevent or reduce myocardial fibrosis, and thus, perhaps, reduced risk of arrhythmias and sudden death, and to reduce the progression of diastolic dysfunction in feline HCM (23,32).
The objective of this prospective study was to 1) determine if the administration of benazepril (an ACEI) to cats with subclinical HCM improves cardiac diastolic function and reverses cardiac hypertrophy and 2) to compare those results to results in cats with subclinical HCM receiving diltiazem controlled delivery (CD).
Materials and methods
Animals
Forty asymptomatic cats were referred to the Small Animal Veterinary Teaching Hospital, University of Montreal from February 2001 to March 2003 for evaluation of a heart murmur or a gallop sound. Twenty-one cats with subclinical HCM, conf irmed echocardiographically, (interventricular septum in diastole, left ventricular free wall in diastole > 6mm, or both) were enrolled in this double-blind, randomized, prospective study (33). None of the cats enrolled had other forms of heart disease, intracavitary thrombi, a history of thromboembolism, congestive heart failure, or previous administration of cardiac medications. The cats were otherwise considered healthy, based on normal results on physical examination.
The animals were cared for according to the principles outlined in the National Institute of Health guide for the care and use of laboratory animals. Written consent was obtained from the owner prior to enrolment.
Protocol
The cats were randomly assigned to either group 1, which were to be treated with benazepril (Fortekor; Novartis Animal Health Canada, Mississauga, Ontario), approximately 0.5 mg/kg body weight (BW), PO, q24h, or group 2, which were to be treated with diltiazem CD (Apo-diltiazem CD; Apotex, Weston, Ontario), approximately 10 mg/kg BW, PO, q24h (34–36). The pharmacist was responsible for the group distribution, so the single clinician in charge of all 21 cats, the clinician performing the echocardiography, the radiologist, and the owner were blinded to the treatment administered. Cats were assigned by the pharmacist to each group alternately, independent of sex.
At the initial visit (day 0), baseline data obtained included results from a complete physical examination (including heart rate determined by auscultation by the clinician in charge), a complete blood (cell) count (CBC) (Cell-Dyn 3500; Abbott Laboratories, Mississauga, Ontario), a serum chemical profile (urea nitrogen, creatinine, alanine aminotransferase, aspartate aminotransferase, total bilirubin, albumin, globulin, glucose, sodium, potassium, chloride, calcium, phosphorous) (Synchron CX-5; Beckman Coulter, Fullerton, California, USA), a total thyroxine level (if the cat was > 6 y old) (DPC Coat-a-count Gamma-Counter C-12; Diagnostic Products, Los Angeles, California, USA), an indirect measurement of systolic arterial blood pressure (SABP) (Ultrasonic Doppler Flow Detector 811; Parks Medical Electronics, Aloha, Oregon, USA), thoracic radiographs (lateral and ventrodorsal) (Gigantos 1012MP; Siemens, Pointe-Claire, Quebec), an electrocardiogram (ECG) (Auto Cardiner FCP 4101AN; Fukuda Denshi, Tokyo, Japan), and an echocardiography (HDI 5000; Philips Medical Systems, Bothell, Washington, USA). Any cat showing arrhythmias resulting in altered systemic perfusion (pale mucous membrane, prolonged capillary refill time, weak pulse) or signs of another underlying systemic disease, including hyperthyroidism, hypertension (SABP > 180 mmHg), was excluded and placed under conventional therapy. Cats in the study were reevaluated after 3 and 6 mo of therapy; each time, a physical examination, an SABP, an ECG, and an echocardiographic examination were carried out. A renal profile (urea nitrogen, creatinine, albumin, globulin, glucose, sodium, potassium, chloride, calcium, phosphorous) was also obtained at the 6-month visit.
Echocardiography
Standard measures —
Standard 2-dimensional (2D), Doppler, and M-mode echocardiographs were obtained by a single investigator according to standard recommendations (37). The interventricular septum in diastole (IVSd), the interventricular septum in systole (IVSs), the left ventricular internal dimension in diastole (LVIDd), the left ventricular internal dimension in systole (LVIDs), the left ventricular free wall in diastole (LVFWd), the left ventricular free wall in systole (LVFWs), and the fractional shortening were acquired on a cardiac long-axis view by using M-mode echocardiography. The 2D circumference of the left atrium was measured on a cardiac long-axis view at end-systole. The left atrium (LA) to aorta (Ao) ratio was acquired on a cardiac long-axis view by using M-mode echocardiography. Presence of valvular regurgitation was identified by using colorflow Doppler imaging (CDI). Systolic cranial motion of the mitral valve was assessed by using M-mode in a right parasternal long-axis view of the left ventricular outflow tract (LVOT) at the level of the mitral valve. The existence of a pressure gradient in the LVOT was evaluated by using pulsed-wave (PW) and continuous-wave (CW) Doppler at the left apical window; the gradient was considered significant whenever outflow tract velocity exceeded 2.0 m/s (38). Butorphanol (Torbugesic; Ayerst Veterinary Laboratories [Wyeth-Ayerst Canada], Guelph, Ontario), 0.4 mg/kg BW, was administered, IV, prior to each echocardiographic examination in an attempt to decrease stress-induced tachycardia.
Diastolic function evaluation —
The isovolumetric relaxation time (IVRT) and the diastolic transmitral flow profiles (E/A ratio) were measured according to the standard protocols (38–40). These measures were done using a simultaneous ECG display during the echocardiogram.
Left ventricular mass assessment
The left ventricular mass (VM) was calculated by using the area length method, a model validated in the dog and considered acceptable for clinical use in the cat by some authors (41–46).
 |
The areas A1 and A2 were determined echocardiographically by measuring, respectively, the outer and inner surface of a cross-section of the left ventricle. Measurements were taken from a right parasternal short axis view at end-diastole and at the level of the chordea tendinae (Figure 1) (41–46). The volume of the ellipsoid 1 and 2 were determined with the following geometric formulas:
Figure 1.
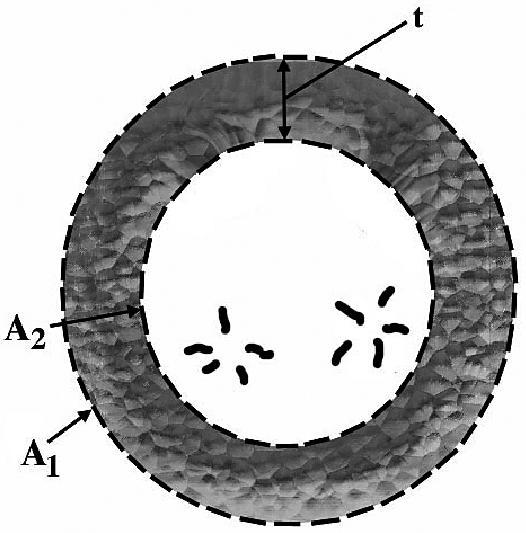
Schematic illustration of a left ventricular cross section (short axis) at the level of the chordea tendinae. A1: area of the left ventricular epicardial cross section; A2: Area of the left ventricular endocardial cross section; t: Thickness of the myocardial cross section.
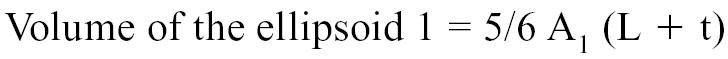 |
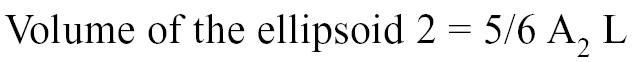 |
where
A1 = Area of the left ventricular epicardial cross-section,
A2 = Area of the left ventricular endocardial cross-section,
L = Long axis of the left ventricle, and
t = Thickness of the myocardial cross-section.
The long axis of the left ventricle (L) was derived from the right parasternal longitudinal view. Measurements were taken at end-diastole by recording the length of the left ventricle from the apparent endocardial apex to the closed mitral annulus (41–46). The thickness of the myocardial cross-section was calculated with the formula:
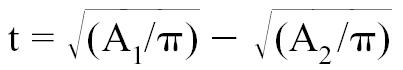 |
Myocardial density being 1.05 g/cm3, the left ventricular mass could therefore be determined with the following formula:
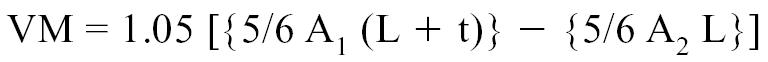 |
Values for left ventricular mass were obtained on initial assessment and at the 3- and 6-month visits.
Statistical analysis
Results are expressed as the median and range. A Wilcoxon signed-rank test was used to determine if there were variations within groups at the 3- and at 6-month visits compared with the baseline values. Values at baseline were compared between groups by using a Wilcoxon rank-sum test. Changes from baseline at the 3- and 6-month visits were compared between groups by using a Wilcoxon rank-sum test. A Fisher’s exact test was used to determine if the proportion of cats positive for systolic cranial motion of the mitral valve varied between groups, when considered as binary data (present or absent). The significance level was set at P < 0.05 based on a two-tailed hypothesis. All statistical analyses were carried out with proprietary statistical software programs (Number Cruncher Statistical System 2001; NCSS Statistical Software, Kaysville, Utah, USA).
Results
Group 1 (n = 11) cats received 0.60 ± 0.06 mg/kg BW, PO, q24h of benazepril, for 6 mo and group 2 (n = 5) cats received 13.3 ± 1.2 mg/kg BW, PO, q24h of diltiazem CD for 6 mo. All the cats in group 1 completed the 6 mo study. In group 2, 5 of the 10 cats enrolled initially were excluded between the initial and the 3-month visit due to detection of a systemic disease (diabetes mellitus [n = 1], hepatic lipidosis [n = 1]), and lack of owner compliance (n = 3). The discontinuation of the drug by the owner was not due to undesirable drug effects. These cats were not included in the statistical analyses.
Signalments of the selected cats
Table 1 shows the signalments for cats of group 1 and 2. Body weight was significantly greater in group 1 than in group 2.
Table 1.
Signalments for cats in group 1 and group 2 upon initial visit
| Group 1 (n = 11) | Group 2 (n = 5) | |
|---|---|---|
| Sex | ||
| Female | 2 (18%) | 1 (20%) |
| Male | 9 (82%) | 4 (80%) |
| Age (years) | 6.1 ± 3.3 | 6.2 ± 4.2 |
| Body weight (kg) | 6.6 ± 1.5a | 4.9 ± 0.7a |
| Breed | 1 Maine coon cat
10 domestic cats |
5 domestic cats |
Data are expressed as mean ± standard deviation (s)
Statistically significant (P < 0.05)
Minimum database
Results from the physical examination were unremarkable in all cats, with the exception of a systolic murmur of variable intensity, heard best at the left cardiac apex (n = 6) or at the sternal border (n = 9). One cat had an intermittent systolic murmur, best heard at the sternal border when the cat was tachycardic.
Electrocardiography
One cat (Maine coon) in group 1 had a cardiac left axis deviation at baseline that persisted throughout the study (mean electrical axis −90°). At the 6-month visit, 2 cats in group 1 had an irregular heart rhythm at auscultation and pulse deficits. The ECG revealed occasional ventricular premature complexes in 1 cat.
Thoracic radiographs
In group 1, the thoracic radiographs revealed a normal thorax (n = 1); mild cardiomegaly (n = 6), accompanied by a valentine-shaped heart (n = 2); and moderate cardiomegaly accompanied with a valentine-shaped heart (n = 4). Pulmonary abnormalities suggesting subclinical airway diseases were observed in 2 cats. Although congestive heart failure could not be excluded, it was felt to be unlikely given the pulmonary interstitial lesion was minor, the pulmonary vessels were normal, and the cat was asymptomatic. In group 2, the thoracic radiographs revealed a normal thorax (n = 1) and mild cardiomegaly (n = 4). The lung fields in all cats were normal.
Echocardiography
Standard measurements —
The mitral valve appeared normal and did not show evidence of primary mitral disease in any of the cats. Secondary mitral valve regurgitation was identified in 13 of the 16 (81%) cats at baseline and was qualitatively characterized as mild (n = 9), moderate (n = 3), and moderate to severe (n = 1), based on CDI. Three cats had no detectable mitral valve regurgitation on CDI.
Tables 2 and 3 show the echocardiographic data for groups 1 and 2, respectively. In group 1, the thickness of the LVFWs decreased significantly between 0 and 3 mo (P < 0.05), but at 6 mo, the thickness was not statistically different from the thickness observed at baseline. None of the other parameters evaluated varied significantly between baseline and the 3- and 6-month visits in both groups. None of the parameters evaluated varied significantly when the groups were compared.
Table 2.
Echocardiographic and physiologic data for group 1
| Parameters | Day 0 | 3 months | 6 months |
|---|---|---|---|
| HR (beat/min) | 196 (160;216) | 200 (160;264) | 204 (150;260) |
| SABP (mmHg) | 143 (108;180) | 140 (102;178) | 140 (120;180) |
| LA circum (cm) | 5.25 (3.97;8.39) | 5.18 (4.39;8.96) | 4.97 (4.11;10.51) |
| LA/Ao | 1.4 (0.9;2.2) | 1.3 (1;2.7) | 1.5 (0.9;2.4) |
| IVSd (mm) | 7.4 (5;8.5) | 7.0 (5;10) | 7.0 (5;9) |
| IVSs (mm) | 9.7 (8.1;11.4) | 9.7 (7;12) | 9.0 (8;12) |
| LVIDd (mm) | 12.8 (8.6;14.1) | 13.0 (9;17) | 12.0 (8;17) |
| LVIDs (mm) | 5.4 (2;8.1) | 6.0 (3;8) | 5.0 (2;8) |
| LVFWd (mm) | 6.1 (5;8.1) | 6.0 (3.9;10) | 6.0 (4;9) |
| LVFWs (mm) | 9.2 (8;11.7) | 9.0b (7.4;11) | 10.0 (7;11) |
| FS (%) | 55 (37;82) | 59 (47;77) | 59 (36;82) |
| IVRT (ms) | 55 (37;145) | 50 (40;75) | 50 (30;75) |
| E wave (cm/s) | 54 (21;66) | 55 (38;69) | 55 (49;78) |
| A wave (cm/s) | 67 (54;104) | 63b (45;81) | 73 (42;96) |
| E/A ratio | 0.6 (0.4;1) | 0.9a (0.6;1.1) | 0.7a (0.6;1.4) |
| Long axis (cm) | 2.21 (1.87;3.37) | 2.54 (1.92;3.07) | 2.51 (1.8;3.18) |
| LV mass (g) | 13.28 (8.4;25.51) | 14.46 (9.26;28.93) | 13.34 (9.4;25.65) |
| PG LVOT (mmHg) | 4.0 (4;29) | 5.8 (4;100) | 7.8 (4;100) |
Day 0, 3-, and 6-month values are expressed as median (minimum;maximum)
HR — heart rate; SABP — systolic arterial blood pressure; LA circum — left atrium circumference; LA/Ao — left atrium/aorta ratio; IVSd — interventricular septum in diastole; IVSs — interventricular septum in systole; LVIDd — left ventricular internal dimension in diastole; LVIDs — left ventricular internal dimension in systole; LVFWd — left ventricular free wall in diastole; LVFWs — left ventricular free wall in systole; FS — fractional shortening; IVRT — isovolumic relaxation time; LV mass — left ventricular mass; PG LVOT — pressure gradient in the left ventricular outflow tract
Statistically different from baseline data (P < 0.01)
Statistically different from baseline data (P < 0.05)
Table 3.
Echocardiographic and physiologic data for group 2
| Parameters | Day 0 | 3 months | 6 months |
|---|---|---|---|
| HR (beat/min) | 200 (108;212) | 180 (160;240) | 226 (168;246) |
| SABP (mmHg) | 121 (110;122) | 138.5 (97;152) | 124 (120;127) |
| LA circum (cm) | 5.59 (5.25;5.93) | 5.36 (4.77;8.68) | 5.65 (5.53;5.98) |
| LA/Ao | 1.6 (1.4;2.1) | 1.5 (1.3;2.8) | 1.4 (1.3;1.5) |
| IVSd (mm) | 7.8 (6.2;8) | 6.3 (6;9) | 6.0 (4;8) |
| IVSs (mm) | 10.7 (7.6;13) | 10.0 (6.7;12.3) | 9.0 (7;11) |
| LVIDd (mm) | 12.0 (7.6;15.1) | 12.5 (8;16.4) | 12.0 (9;15) |
| LVIDs (mm) | 5.4 (3;9.9) | 6.3 (5;9.2) | 5.5 (4;6) |
| LVFWd (mm) | 8.1 (5.8;10) | 8.3 (7;10) | 7.5 (5;10) |
| LVFWs (mm) | 10.0 (9;13) | 11.0 (8;12.1) | 10.0 (8;13) |
| FS (%) | 57.5 (34;75) | 44.0 (38;62) | 56.5 (45;67) |
| IVRT (ms) | 55.0 (30;100) | 55.0 (35;60) | 54.0 (40;80) |
| E wave (cm/s) | 64.5 (55;90) | 56.5 (48;82) | 75.5 (54;95) |
| A wave (cm/s) | 75.0 (48;82) | 73.0 (39;96) | 75.0 (57;93) |
| E/A ratio | 1.0c (0.8;1.2) | 0.7 (0.6;1.6) | 0.9 (7;13) |
| Long axis (cm) | 2.38 (1.48;2.97) | 2.40 (2.05;3.06) | 2.61 (2.5;2.71) |
| LV mass (g) | 16.16 (8.83;25.7) | 17.09 (11.7;27.85) | 16.63(12.7;20.09) |
| PG LVOT (mmHg) | 13.0 (4;46.2) | 16.0 (4;60) | 16.7 (4;46) |
Day 0, 3-, and 6-month values are expressed as median (minimum;maximum)
HR — heart rate; SABP — systolic arterial blood pressure; LA circum — left atrium circumference; LA/Ao — left atrium/aorta ratio; IVSd — interventricular septum in diastole; IVSs — interventricular septum in systole; LVIDd — left ventricular internal dimension in diastole; LVIDs — left ventricular internal dimension in systole; LVFWd — left ventricular free wall in diastole; LVFWs — left ventricular free wall in systole; FS — fractional shortening; IVRT — isovolumic relaxation time; LV mass — left ventricular mass; PG LVOT — pressure gradient in the left ventricular outflow tract
Statistically different from group 1 at baseline (P < 0.05)
Pressure gradient in the LVOT —
The values for the pressure gradient in the LVOT did not vary significantly between baseline and the 3- and 6-month visits in both groups. The pressure gradient in the LVOT did not vary significantly when the groups were compared.
Systolic cranial motion of the mitral valve —
At baseline, 2 cats in group 1 and 1 cat in group 2 displayed a systolic cranial motion (SAM) of the mitral valve. In group 1, SAM of the mitral valve was observed at 0, 3, and 6 mo in 1 cat, at baseline only in 1 cat, and at 3 and 6 mo but not at baseline in 1 cat. This last cat had no identif iable pressure gradient at baseline but had a 100 mmHg gradient 3 and 6 mo. When the groups were compared, the proportion of cats positive for systolic cranial motion of the mitral valve did not vary significantly throughout the study.
Diastolic function —
The IVRT did not vary significantly between baseline and the 3- and 6-month visits in both groups. The IVRT did not vary significantly when the groups were compared.
In group 1, the E/A ratio increased between baseline and 3 mo and remained stable thereafter (P < 0.01). The E wave amplitude did not vary significantly between baseline and the 3- and 6-month visits. Therefore, the variation in the E/A ratio was thought to result from a significant decrease in A wave amplitude between baseline and 3 mo (P < 0.05). The A wave amplitude obtained at 6 mo was not statistically different from that at baseline evaluation. At baseline, the E/A ratio was significantly higher in group 2 compared with group 1 (P < 0.05). This difference was no longer observed at the 3- and 6-month visits.
Left ventricular mass —
The left ventricular mass and the long axis length of the left ventricle were not statistically different between baseline evaluation and the 3- and 6-month visits in both groups. The left ventricular mass did not vary significantly when the groups were compared.
Power analysis
In this study, the null hypothesis was that the difference between T0 and T6-month, for a given parameter, was similar when comparing group 1 with group 2. The IVRT and the E/A ratio were used to evaluate the diastolic function. The probability of rejecting a false null hypothesis for the IVRT and the E/A ratio was, respectively, 24% and 17%. Which means, that if IVRT decreased significantly between T0 and T6-month in group 1 when compared with group 2, there was only a 24% chance of detecting this difference. The IVSd and the LVFWd were the parameters used to support the diagnosis of ventricular wall hypertrophy. In this study, the probability of rejecting a false null hypothesis for the IVSd and the LVFWd was 22% and 6%, respectively.
Discussion
Since the 1980s, biochemical and genetic evidence for the existence of a cardiac renin-angiotensin system in different species has been accumulating (13–16). Angiotensin II has been shown to induce myocyte hypertrophy and fibroblast hyperplasia, 2 phenomena known to be involved in the progression of the HCM (2,17–24,38). Diastolic dysfunction is another factor that characterized HCM (38). In HCM, the intracoronary administration of enalapril maleate improved diastolic function, which suggests that blocking tissue renin-angiotensin system may be beneficial in HCM (20). However, when the captopril was administered sublingually, the improvement in diastolic function was no longer seen (20). The affinity for the cardiac tissue may be different for those 2 ACEI, but more probably, the systemic effect of the ACEI may hide the beneficial effect observed after local ACEI administration.
Interestingly, the administration of ACEI had been proven to reverse cardiac hypertrophy in a pressure overload model such as sub-aortic stenosis and systemic hypertension (25). In HCM, the hypertrophy is due to a myocardial disease and not to pressure overload, unless an obstructive form of HCM contributes secondarily to the hypertrophy. In humans, when hypertrophy was due to HCM, the administration of ACEI failed to show regression of the hypertrophy (19,26). This suggests that the molecular basis of hypertrophy in HCM is different from hypertrophy in pressure overload models (27). Nevertheless, the administration of ACEI remains interesting in HCM, as angiotensin-converting enzyme (ACE) gene may modify the phenotypic expression of the HCM. It is now known that in presence of HCM, patients expressing D/D genotype for ACE gene show an increased level of serum ACE, have an increased risk of sudden death, and present an increased severity of hypertrophy (28–31).
This knowledge has generated interest in ACEI as a potential therapeutic tool to prevent or reduce myocardial fibrosis, perhaps reduce the risk of arrhythmias and sudden death, and reduce the progression of diastolic dys-function in feline HCM (23, 32).
At baseline, 3 females (14%) and 18 males (86%) were diagnosed with HCM, which is similar to the sex ratios reported elsewhere (47–49). All patients enrolled in the study were domestic cats, except for 1 Maine coon cat. Kittleson et al (9–11,50) suggested that in the Maine coon cat, HCM was transmitted as an autosomal dominant trait. An identical mode of transmission for HCM has also been suspected in a family of American shorthair and in a family of mixed-breed cats (51,52). The body weight was significantly greater in group 1 when compared with group 2 at baseline. This difference was not expected to influence the statistical analyses, since each cat was its own control and since body weight is only weakly correlated to echocardiographic parameters in this species (53).
As 0.5 mg/kg BW, PO, q24h of benazepril has been proven to inhibit the plasma renin-angiotensin system and possibly the cellular renin-angiotensin system, all of the cats in group 1 received at least this dose (34,35). Diltiazem CD, a sustained-release form of diltiazem, was used as a control drug. The dose of 10 mg/kg BW, PO, q24h was used, as it is known to produce serum levels that are considered therapeutic (36).
All the cats in our study had a systolic heart murmur and none had a gallop sound. However, it is possible to have HCM without the presence of a heart murmur or a gallop sound (49). In HCM, the systolic heart murmur is suspected to be due to mitral valve regurgitation, resulting from SAM of the mitral valve, from distortion of the mitral valve apparatus related to hypertrophy, or both (2,54). Since SAM of the mitral valve was identified in only 3 cats at baseline, the mitral valve regurgitation in the remaining 10 cats was suspected to be due to distortion of the mitral valve apparatus related to hypertrophy. Three cats in this study had no detectable mitral valve regurgitation with CDI. The 1st cat had a mild pressure gradient in the LVOT (19 mmHg), which could have participated in generating the systolic heart murmur (50). The 2nd cat had an intermittent systolic heart murmur, which was present only with tachycardia. Therefore, it is possible that butorphanol administration had prevented tachycardia by reducing stress induced by manipulation in such a way that subtle abnormalities became unrecognizable once the cat was sedated. The origin of the heart murmur could not be identified in a 3rd cat, and butorphanol administration was again suspected to have prevented detection of subtle turbulence of the flow in the LVOT.
Mild to moderate radiographic cardiomegaly was observed in 14 cats and probably reflected the cardiac hypertrophy, atrial dilation, or both. The valentine-shaped heart (n = 6) was suspected to be due to enlarged atria in 2 cats where the LA/Ao ratio was > 2. The LA/Ao ratio was within normal limits for the remaining 4 cats and the valentine-shaped heart could have been due to hypertrophy of the base of the left ventricle.
Electrocardiographic abnormalities were rarely identified. A left axis deviation was noted in the Maine coon cat (group 1) and persisted throughout the study. A bundle branch block was suspected. Unfortunately, precordial leads were not recorded at any time to clarify the exact origin of the block. At the 6-month revaluation, arrhythmias, not reported previously, were found in 2 cats (group 1). The ECG revealed occasional single ventricular premature complexes in 1 cat. The 2nd cat had a normal ECG and arrhythmias were not identified at that time. It is probable that intermittent supraventricular or ventricular premature complexes were occurring; a Holter examination was not done since the cat was subclinical (38).
In group 1, normal variation or measurement error was likely to account for the signif icant decrease in thickness of the LVFWs at 3 mo, as changes in loading conditions were not reported. The fact that the thickness of the LVFWs at 6 mo was not different from the value obtained at baseline also support this hypothesis. In a retrospective study, Rush et al (32) reported a signif icant decrease in IVSd, IVSs, and LVFWd when a group of HCM cats receiving a polytherapy that included enalapril was compared with another group of patients receiving a polytherapy that excluded enalapril. In that study, 15 of the 19 cats were initially in congestive heart failure and only 1 remained in congestive heart failure at revaluation 3 to 6 mo later. Therefore, it was not clear whether the improvement in the LV and IVS thickness were due to a reduction of hypertrophy, a manifestation of the natural evolution of the disease, or changes in the cardiac loading condition (32). In another prospective study, Amberger et al (23) reported a significant decrease in LVFWd when a group of cats receiving both diltiazem and benazepril were compared with a group of cats receiving diltiazem only.
The pressure gradient in the left ventricular outflow tract usually results from SAM of the mitral valve or prominent hypertrophy of the base of the septum. In both situations, an obstructive narrowing of the outflow tract is responsible for the acceleration of blood flow and the pressure gradient (38,49,54). Oyama et al (55) showed that the administration of benazepril to cats with the dynamic obstructive form of HCM did not cause a significant elevation of the pressure gradient in the LVOT (55).
In group 1, the pressure gradient in the LVOT worsened in 4 of the 11 cats. The 1st cat had no detectable pressure gradient, SAM, and mitral valve regurgitation at baseline, but it had a pressure gradient of 100 mmHg, SAM, and a mild mitral valve regurgitation at 3 and 6 mo. Theoretically, benazepril could cause the development of SAM, a pressure gradient in the LVOT through a decrease in afterload, or both (32). In this cat, the thickness of the basal portion of the septum remained stable throughout the study, and the systolic blood pressure (130 mmHg at baseline) was 165 mmHg at 6 mo. The pressure gradient in this cat was suspected to result from the recently acquired SAM. In the other 3 cats, the pressure gradient in the LVOT at baseline was 0, 16, and 19 mmHg and reached 17, 25, and 34 mmHg, respectively, at 6 mo. All 3 cats had experienced a thickening of the IVSs at 6 mo, when compared with baseline values. Therefore, the worsening of the pressure gradient in the LVOT was possibly due to the progression of the septal hypertrophy. These cats appeared calm and anxiety was not thought to be a major factor to explain the variation observed in the pressure gradient.
In group 1, 1 cat had SAM of the mitral valve and a mild pressure gradient in the LVOT (25 mmHg) at base-line. However, the SAM of the mitral valve was not detected at 3 and 6 mo and the pressure gradient in the LVOT decreased to 7 mmHg at 6 mo. This cat’s systolic blood pressure remained stable throughout the study (120 mmHg initially to 130 mmHg at 6 mo). It was hypothesized that the normalization of the pressure gradient may have been due to a decrease in IVSs thickness (from 8.0 to 7.0 mm), which could alleviate the pressure gradient and the SAM of the mitral valve. Butorphanol administration could also have caused the pressure gradient in the LVOT to decrease, as it may prevent stress induced tachycardia and anxiety.
In 1990, Spirito et al (56) evaluated the relationship between hypertrophy and diastolic function in HCM and concluded that these 2 parameters were independent. Therefore, sole consideration of ventricular thickness may not be an adequate predictor of the natural course of HCM in cats. The time constant of left ventricular pressure fall (Tau) is the preferred method used to assess relaxation. The determination of Tau is invasive and its clinical use is therefore limited (40). Schober et al (57) found that a linear and moderately strong correlation (r = 0.78) exists between IVRT and Tau in normal cats. In our study, the IVRT was evaluated as an indicator of the left ventricular diastolic function. In group 1, a trend towards a shortening of the IVRT (P = 0.06) was observed at 6 mo. The loading conditions at 6 mo were not significantly different from those at baseline, since the LVIDd, LVIDs, the long axis of the left ventricle, and the heart rate at 6 mo were not statistically different from those at baseline. Therefore, the trend towards a shortening of the IVRT could suggest an improvement in diastolic function in group 1. However, as no significant difference was found when comparing both groups, the trend toward a shortening of the IVRT in group 1 may have been incidental and not significant from a clinical standpoint.
In HCM, the early transmitral flow wave (E wave) may decrease due to impaired relaxation and the late inflow signal (A wave) may increase due to increased contribution of the atrial contraction to ventricular filling (39). At baseline, the E/A ratio in group 2 was greater than in group 1, which could suggest a more normal diastolic function in group 2. However, the IVRT was equivalent for both groups, therefore, the E/A ratio was possibly influenced by other factors such as the heart rate. In group 1, an increase in the E/A ratio was observed between baseline and 3 mo, and baseline and 6 mo. The increase in the E/A ratio between baseline and 3 mo was attributed to a significant decrease in the A wave, since the E wave was not statistically different from baseline. The decrease in the A wave could be attributed to technical variations in the measurements or presence of a restrictive lesion. At 6 mo, the elevation of the E/A ratio compared with that at baseline resulted from a trend towards elevation of the E wave (P = 0.06) which may represent an actual improvement in ventricular relaxation and compliance (39). However, this statistical finding may not reflect a real clinical improvement as the E/A ratio was still < 1, suggesting an ongoing impaired relaxation. Butorphanol may also have altered indices of the transmitral fillings.
Pseudonormalization is a phenomenon whereby, with time, the IVRT and the E/A ratio normalize even though diastolic function continues to deteriorate (38,39). This phenomenon is possibly explained by the fact that left ventricular stiffness increases to an extent where blood flow through the mitral valve annulus decreases. The compensatory rise in left atrial pressure eventually masks the impaired relaxation pattern and the mitral valve flow profile may appear normal (38,39). In this case, Doppler evaluation of the pulmonary flow can sometimes help to differentiate normal from abnormal diastolic function. High velocity reversal of flow lasting for the entire atrial contraction period is observed if left ventricular diastolic pressure increases abnormally during atrial contraction, regardless of atrial flow profile (38,39). Technically, it was not possible to assess the flow through the pulmonary vein in this study. Velocity of flow propagation by color M-mode echography and tissue Doppler imaging can also help to differentiate a normal from a pseudonormal left ventricular filling pattern (58). However, it appeared unlikely that the improvement of the E/A ratio, as a marker of the diastolic function, was related to pseudonormalization, given the size of the left atria and the fact that the LA/Ao ratio remained stable throughout the study.
Coleman et al (59) reported that the area length model was sensitive enough to detect variations in LV mass over time in a volume overload model. To the author’s knowledge, such a study using HCM as a model has not been reported. The area length method used here has been validated in dogs and was considered as a potentially valid tool for left ventricular mass determination in cats (41–43,45,46). Since evaluating left ventricular thickness may not be adequate to assess progressive disease or subtle changes, such as focal form of HCM, the left ventricular mass was determined in an attempt to establish if this parameter could be a useful clinical tool. In this study, each cat was its own control and variations in LV mass were studied through the 6-month duration of the study. The LV mass did not vary significantly within and between groups throughout the study. It is possible that reduction of LV hypertrophy did not occur, but lack of sensitivity of the mathematical model was likely. Recently, MacDonald et al (60) reported that cardiac magnetic resonance imaging when compared with echocardiographs was more accurate to quantify left ventricular mass in domestic cats.
No statistical difference was found for any parameters in group 2, most probably due to the small number of cats in this group. In group 1, interesting variations or trends regarding IVRT and E/A ratio were observed, but when group 1 is compared with group 2 for the differences obtained between baseline and 3 mo and baseline and 6 mo, no statistical difference was observed for any of the parameters.
Sixteen of the 21 cats enrolled completed the 6-month study. None of the 5 cats excluded was removed due to cardiac causes. Of the 16 cats that completed the study, 14 remained asymptomatic at the writing of this article. Two cats died within 5 mo after completion of the study. Those 2 cats maintained a good body condition throughout the study period. The 1st cat (Maine coon) was found dead at home, 5 mo after completion of the study, while he was still receiving benazepril. The owner did not authorize a postmortem examination. However, sudden death from lethal arrhythmias or massive thromboembolism was suspected, since no clinical signs had been identified previously by the owner. The 2nd cat died of congestive heart failure after being anesthetized (isoflurane) for a minor procedure. The owner did not authorize a postmortem examination. At the time, the cat was still receiving benazepril and a decrease in LA circumference (8.4 cm to 7.2 cm), in LA/Ao ratio (2.2 to 2.1), and in IVRT (55 ms to 30 ms) was suggestive of an improvement in diastolic function.
One of the major limitations of this study was the small number of patients recruited. As shown by the power analysis, if IVRT, E/A ratio, IVSd, and LVFWd decreased significantly between T0 and T6 mo in group 1 when compared with group 2, there was, respectively, a 24%, 17%, 22%, and 6% chance to detect this difference. This shows that the absence of statistical difference between groups observed in this study may be due to a lack of power (type II error). A larger number of patients in each group would have increased the power of the statistical analysis and our chances to reject a false null hypothesis.
Butophanol was administered in order to reduce tachycardia that could result from catecholamine release due to the stress and the anxiety induced by the manipulations. The administration of butorphanol may have influenced the result of our study, as it may alter the transmitral filling indices and decrease the detection of otherwise present dynamic LVOT pressure gradient. In an attempt to minimize the influence of the butorphanol administration on our results, each cat acted as its own control. Also, to minimize the variation in the level of sedation, each cat received the same dose (on a mg/kg BW basis) of butorphanol prior to each echocardiographic examination.
Because of the controversy surrounding treatment strategies for subclinical HCM, having a control group under no treatment would have helped to determine if the changes observed were real or reflected measurement variations. However, due to ethical concerns, it was decided not to leave any cat diagnosed with HCM without treatment.
It is known that benazepril has a good affinity for cardiac tissue in rats (61). However further studies are needed to confirm that the chosen dosage of benazepril is adequate to inhibit the feline cardiac renin-angiotensin system. Also taking into consideration that little is known about the natural course of the disease, a 6-month study period may not have been long enough to evaluate the long-term effects of benazepril.
Finally, from what is known about the pathophysiology of the HCM and the central role that angiotensin II may play in the development of cardiac hypertrophy, benazepril appeared to be an interesting drug. We demonstrated that administration of benazepril, 0.5 mg/kg BW, PO, q24h, to cats with subclinical HCM might improve diastolic function. However, the variations observed may have been incidental, as no difference was found between the benazepril and the diltiazem CD group. Multicenter studies will be needed to gather a sufficient number of cases to establish the role, if any, of benazepril in subclinical HCM.
Acknowledgments
The authors thank Mr. Maxim Moreau for statistical analysis assistance and Dr. Luc Breton for the interpretation of the thoracic radiographs. CVJ
Footnotes
This work was supported by a grant from Novartis Animal Health Canada Inc. and from “Le Fond du Centenaire” of the Faculté de Médecine Vétérinaire, Université de Montréal.
References
- 1.Liu S-K, Roberts WC, Maron BJ. Comparison of morphologic findings in spontaneously occurring hypertrophic cardiomyopathy in humans, cats and dogs. Am J Cardiol. 1993;72:944–951. doi: 10.1016/0002-9149(93)91112-u. [DOI] [PubMed] [Google Scholar]
- 2.Kittleson MD, Kienle RD. Small Animal Cardiovascular Medicine. St-Louis: Mosby, 1998:347–361.
- 3.Maron BJ, Spirito P. Implications of the left ventricular remodeling in hypertrophic cardiomyopathy. Am J Cardiol. 1998;81:1339–1344. doi: 10.1016/s0002-9149(98)00164-7. [DOI] [PubMed] [Google Scholar]
- 4.Maron BJ, Mulvihill JJ. The genetics of hypertrophic cardiomyopathy. Ann Intern Med. 1986;105:610. doi: 10.7326/0003-4819-105-4-610. [DOI] [PubMed] [Google Scholar]
- 5.Wynne J, Braunwald E. The cardiomyopathies and myocarditides. In: Breaunwald E, Zipes DG, Libby P, eds. Heart Disease: A Textbook of Cardiovascular Medicine, 6th ed. Philadelphia: WB Saunders, 2001:1751–1806.
- 6.Seidman CE, Seidman JG. Mutations in cardiac myosin heavy chain genes cause familial hypertrophic cardiomyopathy. Mol Biol Med. 1991;8:159–166. [PubMed] [Google Scholar]
- 7.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 8.Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol. 2002;17:242–252. doi: 10.1097/01.HCO.0000013803.40803.6A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kittleson MD, Kittleson JA, Mekhamer Y. Development and progression of inherited hypertrophic cardiomyopathy in Maine Coon cats [abstract] J Vet Intern Med. 1996;10:165. [Google Scholar]
- 10.Kittleson MD, Meurs KM, Kittleson JA, et al. Heritable characteristics, phenotypic expression, and natural history of hypertrophic cardiomyopathy in Maine Coon cats. J Vet Intern Med. 1998;12:198. [Google Scholar]
- 11.Kittleson MD, Meurs KM, Munro MJ, et al. Familial hypertrophic cardiomyopathy in Maine Coon cats: an animal model of human disease. Circulation. 1999;99:3172–3180. doi: 10.1161/01.cir.99.24.3172. [DOI] [PubMed] [Google Scholar]
- 12.Petrie JP. Feline cardiomyopathy: update and review [abstract]. Proc 21st Am Coll Vet Intern Med Forum 2003:148.
- 13.Dzau VJ. Cardiac renin-angiotensin system. Am J Med. 1988;84:22–26. doi: 10.1016/0002-9343(88)90201-x. [DOI] [PubMed] [Google Scholar]
- 14.Manwen J, Wilhelm MJ, Lang RE, et al. Endogenous tissue reninangiotensin systems. Am J Med. 1988;84:28–36. doi: 10.1016/0002-9343(88)90202-1. [DOI] [PubMed] [Google Scholar]
- 15.Schunkert H, Jackson B, Shih Tang S, et al. Distribution and functional significance of cardiac angiotensin-converting enzyme in hypertrophied rat hearts. Circulation. 1993;87:1328–1339. doi: 10.1161/01.cir.87.4.1328. [DOI] [PubMed] [Google Scholar]
- 16.Endo-Mochizuki Y, Mochizuki N, Sawa H, et al. Expression of renin and angiotensin-converting enzyme in human hearts. Heart Vessels. 1995;10:285–293. doi: 10.1007/BF02911386. [DOI] [PubMed] [Google Scholar]
- 17.Brilla CG, Maisch B. Regulation of the structural remodeling of the myocardium: from hypertrophy to heart failure. Eur Heart J. 1994;15:45–52. doi: 10.1093/eurheartj/15.suppl_d.45. [DOI] [PubMed] [Google Scholar]
- 18.Booz GW, Baker KM. Protein kinase C in angiotensin II signaling in neonatal rat cardiac fibroblasts. Role in the mitogenic response. Ann N Y Acad Sci. 1995;752:158–167. doi: 10.1111/j.1749-6632.1995.tb17419.x. [DOI] [PubMed] [Google Scholar]
- 19.Takeda A, Takeda N. Different pathophysiology of cardiac hypertrophy in hypertension and hypertrophic cardiomyopathy. J Mol Cell Cardiol. 1997;29:2961–2965. doi: 10.1006/jmcc.1997.0531. [DOI] [PubMed] [Google Scholar]
- 20.Kyriakidis M, Triposkiadis F, Dernellis J, et al. Effects of cardiac versus circulatory angiotensin-converting enzyme inhibition on the left ventricular diastolic function and coronary blood flow in hypertrophic obstructive cardiomyopathy. Circulation. 1998;97:1342–1347. doi: 10.1161/01.cir.97.14.1342. [DOI] [PubMed] [Google Scholar]
- 21.Gray MO, Long CS, Kalinyak JE, et al. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 22.Ambrose J, Pribnow DG, Giraud GD, et al. Angiotensin type 1 receptor antagonism with irbesartan inhibits ventricular hypertrophy and improves diastolic function in the remodeling post- myocardial infarction ventricle. J Cardiovasc Pharmacol. 1999;33:433–439. doi: 10.1097/00005344-199903000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Amberger CN, Glardon O, Glaus T, et al. Effects of benazepril in the treatment of feline hypertrophic cardiomyopathy. Results of a prospective, open-label, multicenter clinical trial. J Vet Cardiol. 1999;1:19–26. doi: 10.1016/S1760-2734(06)70026-1. [DOI] [PubMed] [Google Scholar]
- 24.Booz GW, Dostal DE, Baker KM. Paracrine actions of cardiac fibroblasts on cardiomyocytes: implications for the cardiac renin-angiotensin system. Am J Cardiol. 1999;83:44H–47H. doi: 10.1016/s0002-9149(99)00257-x. [DOI] [PubMed] [Google Scholar]
- 25.Ke YS, Gao H, Yang T. Effect of combination of valsartan with benazepril on blood pressure and left ventricular hypertrophy in SHR. Acta Pharmacol Sin. 2000;21:1043–1047. [PubMed] [Google Scholar]
- 26.Hartmann A, Putz A, Hopf R. Effect of long-term ACE-inhibitor therapy in hypertrophic cardiomyopathy (HCM) [abstract] J Am Coll Cardiol. 1995;25:234A. [Google Scholar]
- 27.Marian AJ, Robert R. Recent advances in molecular genetics of hypertrophic cardiomyopathy. Circulation. 1995;92:1336–1347. doi: 10.1161/01.cir.92.5.1336. [DOI] [PubMed] [Google Scholar]
- 28.Marian A, Yu Q, Workman R, et al. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden death. Lancet. 1993;342:1085–1086. doi: 10.1016/0140-6736(93)92064-z. [DOI] [PubMed] [Google Scholar]
- 29.Lechin M, Quinones MA, Omran A, et al. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation. 1995;92:1808–1812. doi: 10.1161/01.cir.92.7.1808. [DOI] [PubMed] [Google Scholar]
- 30.Rigat, B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of the serum enzyme level. J Clin Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jan Danser AH, Schalekamp M, Bax WA, et al. Angiotensin-converting enzyme in human heart. Effect of the deletion/insertion polymorphism. Circulation. 1995;92:1387–1388. doi: 10.1161/01.cir.92.6.1387. [DOI] [PubMed] [Google Scholar]
- 32.Rush JE, Freeman LM, Brown DJ, et al. The use of enalapril in the treatment of feline hypertrophic cardiomyopathy. J Am Anim Hosp Assoc. 1998;34:38–41. doi: 10.5326/15473317-34-1-38. [DOI] [PubMed] [Google Scholar]
- 33.Fox PR, Lui SK, Maron BJ. Echocardiographic assessment of spontaneously occurring feline hypertrophic cardiomyopathy. An animal model of human disease. Circulation. 1995;92:2645–2651. doi: 10.1161/01.cir.92.9.2645. [DOI] [PubMed] [Google Scholar]
- 34.King JN, Humbert-Droz E, Maurer M. Pharmacokinetics of benazepril and inhibition of plasma ACE activity in cats [abstract]. Proc 14th Am Coll Vet Intern Med Forum 1996:745.
- 35.King JN, Humbert-Droz E, Maurer M. Plasma angiotensin-converting enzyme activity and pharmacokinetics of benazepril and benazeprilat in cats after single and repeated oral administration of benazepril HCl. J Vet Pharmacol Ther. 1999;22:360–367. doi: 10.1046/j.1365-2885.1999.00230.x. [DOI] [PubMed] [Google Scholar]
- 36.Johnson LM, Atkins CE, Keene BW, Bai SA. Pharmacokinetic and pharmacodynamic proprieties of conventional and CD-formulated diltiazem in cats. J Vet Intern Med. 1996;10:316–320. doi: 10.1111/j.1939-1676.1996.tb02069.x. [DOI] [PubMed] [Google Scholar]
- 37.Thomas WP, Gaber CE, Jacobs GJ, et al. Recommendations for standards in transthoracic two-dimensional echocardiography in the dog and cat. J Vet Intern Med. 1993;7:247–252. doi: 10.1111/j.1939-1676.1993.tb01015.x. [DOI] [PubMed] [Google Scholar]
- 38.Fox PR. Feline cardiomyopathies. In: Fox PR, Sisson DD, Moise NS, eds. Canine and Feline Cardiology, Principles and Clinical Practice. 2nd ed. Philadelphia: WB Saunders, 1999:621–641.
- 39.Bright JM, Herrtage ME, Schneider JF. Pulsed Doppler assessment of left ventricular diastolic function in normal and cardiomyopathic cats. J Am Anim Hosp Assoc. 1999;35:285–291. doi: 10.5326/15473317-35-4-285. [DOI] [PubMed] [Google Scholar]
- 40.Constable P, Muir W, Sisson DD. Clinical assessment of left ventricular relaxation. J Vet Intern Med. 1999;13:5–13. [PubMed] [Google Scholar]
- 41.Wyatt HL, Heng MK, Meerbaum S, et al. Cross-sectional echocardiography I. Analysis of mathematic models for quantifying mass of the left ventricle in dogs. Circulation. 1979;60:1104–1113. doi: 10.1161/01.cir.60.5.1104. [DOI] [PubMed] [Google Scholar]
- 42.Schiller NB, Skiôldebrand CG, Schiller EJ, et al. Canine left ventricular mass estimation by two-dimensional echocardiography. Circulation. 1983;68:210–216. doi: 10.1161/01.cir.68.1.210. [DOI] [PubMed] [Google Scholar]
- 43.Stack RS, Ramage JE, Bauman RP, et al. Validation of in vivo two-dimensional echocardiographic dimension measurements using myocardial mass estimates in dogs. Am Heart J. 1987;113:725–731. doi: 10.1016/0002-8703(87)90713-7. [DOI] [PubMed] [Google Scholar]
- 44.Schiller NB, Shah PM, Crawford M, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 45.Nyland T, Matton J. Veterinary Diagnostic Ultrasound. Philadelphia: WB Saunders, 1995:208–211.
- 46.Kittleson MD, Kienle RD. Echocardiography. In: Kittleson MD, Kienle RD, eds. Small Animal Cardiovascular Medicine. St-Louis: Mosby, 1998:95–115.
- 47.Harpster NK. Feline myocardial diseases. In: Bonagura JD, ed. Kirk’s Current Veterinary Therapy IX. Philadelphia: WB Saunders, 1986:380–398.
- 48.Atkins CE, Gallo AM, Kurzmann ID, et al. Risks factors, clinical signs and survival of idiopathic hypertrophic cardiomyopathy: 74 cases (1985–1989) J Am Vet Med Assoc. 1992;201:613–618. [PubMed] [Google Scholar]
- 49.Rodriguez DB, Harpster N. Feline hypertrophic cardiomyopathy: etiology, pathophysiology, and clinical features. Compend Contin Educ Pract Vet. 2002;24:364–372. [Google Scholar]
- 50.Kittleson MD. CVT Update: feline hypertrophic cardiomyopathy. In: Bonagura JD, ed. Kirk’s Current Veterinary Therapy XII. Philadelphia: WB Saunders, 1995:854–862.
- 51.Meurs KM, Kittleson MD, Towbin J, et al. Familial systolic anterior motion of the mitral valve and/or hypertrophic cardiomyopathy is apparently inherited as an autosomal dominant trait a family of American shorthair cats [abstract] J Vet Intern Med. 1997;11:138. [Google Scholar]
- 52.Kraus MS, Calvert CA, Jacobs GJ. Hypertrophic cardiomyopathy in a litter of f ive mixed-breed cats. J Am Anim Hosp Assoc. 1999;35:293–296. doi: 10.5326/15473317-35-4-293. [DOI] [PubMed] [Google Scholar]
- 53.Sisson DD, Knight DH, Helinski C, et al. Plasma taurine concentrations and M-mode echocardiographic measures in healthy cats and in cats with dilated cardiomyopathy. J Vet Intern Med. 1991;5:232–238. doi: 10.1111/j.1939-1676.1991.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 54.Abbott JA. Feline myocardial disease — I. Proc 16th ACVIM Forum 1998:110.
- 55.Oyama MA, Gidlewski J, Sisson DD. Effect of ACE-inhibition on the dynamic left ventricular obstruction in cats with hypertrophic cardiomyopathy [abstract]. Proc 21st Am Coll Vet Intern Med Forum 2003:84.
- 56.Spirito P, Maron BJ. Relation between extent of left ventricular hypertrophy and diastolic filling abnormalities in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1990;15:808–813. doi: 10.1016/0735-1097(90)90278-w. [DOI] [PubMed] [Google Scholar]
- 57.Schober KE, Luis Fuentes V, Bonagura JD. Comparison between invasive hemodynamic measurements and noninvasive assessment of left ventricular diastolic function by use of Doppler echocardiography in healthy anesthetized cats. Am J Vet Res. 2003;64:93–103. doi: 10.2460/ajvr.2003.64.93. [DOI] [PubMed] [Google Scholar]
- 58.Poerner TC, Goebel B, Unglaub P, et al. Detection of a pseudonormal mitral inflow pattern: an echocardiographic and tissue doppler study. Echocardiography. 2003;20:345–356. doi: 10.1046/j.1540-8175.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- 59.Coleman B, Cothran LN, Ison-Franklin EL, et al. Estimation of the left ventricular mass in conscious dogs. Am J Physiol. 1986;251:H1149–1157. doi: 10.1152/ajpheart.1986.251.6.H1149. [DOI] [PubMed] [Google Scholar]
- 60.MacDonald KA, Kittleson MD, Larson RF, Wisner ER. Cardiac magnetic resonance imaging more accurately quantifies LV mass than echocardiography in domestic cats [abstract] J Vet Intern Med. 2004;18:404. [Google Scholar]
- 61.Chen B, Perich R, Jackson B, et al. Benazepril: profile of a new ACE inhibitor. Proc R Soc Med. 1990;166:17–27. [Google Scholar]