

Abstract
The mechanisms of the rearrangements and stereoinversion of azametallacyclobutenes generated via [2+2] cycloaddition of allenes and imidozirconium complexes have been studied. Metallacycles derived from allenes bearing β-hydrogen atoms racemize at room temperature by reversible β-hydride elimination, a process which is also responsible for their eventual conversion to monoazadiene complexes. Metallacycles derived from diarylallenes racemize by reversible thermal bond homolysis at 95 °C; racemization of these metallacycles is also catalyzed by mild oxidants.
Introduction
The hydroamination of alkynes and allenes is a powerful method for transforming unsaturated hydrocarbons into substituted amines or imines.1–5 A variety of titanium and zirconium complexes have been shown to efficiently catalyze this transformation.6–14 Prior work in this laboratory established that a key step in zirconium-catalyzed hydroamination reactions is a [2+2] cycloaddition of the unsaturated organic substrate with a zirconium imido complex (LnZr=NR) to generate an azamet-allacyclobutane.13,14 Understanding the factors that govern the formation and rearrangement of these metallacycles is important in controlling the regio- and enantioselectivity of hydroamination reactions and in the development of new reactions based on this mechanism.
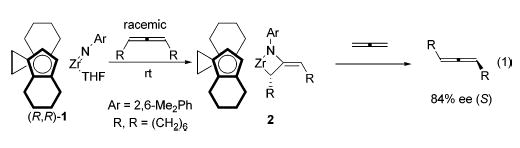
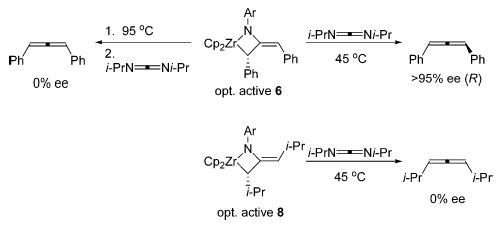
Recently, we reported an unusual selective stereoinversion of allenes promoted by enantioenriched ethylenebis(tetrahy-droindenyl) (ebthi) imidozirconium complexes.15 In that report, enantioenriched ebthi imidozirconium complex 1 reacted with 1 equiv of racemic 1,2-cyclononadiene to generate only a single diastereomer of the corresponding metallacycle (eq 1). The 1,2-cyclononadiene could be released from the metal center by treatment of the metallacycle with parent allene (C3H4) and was found to be considerably enantioenriched (84% ee). More recently, we communicated preliminary results of a study of the mechanism of this remarkable stereoinversion.16 In the present report, we describe a complete synthetic and mechanistic investigation of the formation, racemization, and rearrangements of allene metallacycles derived from ebthi imido complex 1 and achiral zirconocene imido complex 3 (Scheme 1). Consideration of both new and previously proposed mechanisms by which the multifold rearrangements of allene-derived metallacycles proceed follows the presentation of the data.
Scheme 1.
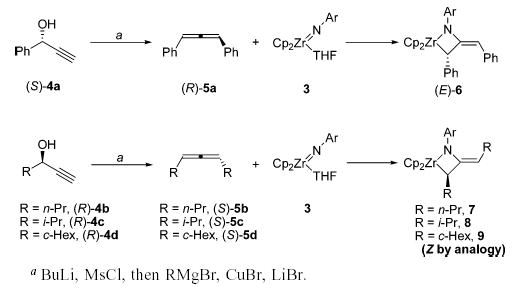
Synthesis and Reactions of Enantiopure Allenes with Zirconocene Imido Complex 3a
Results
Cycloaddition.
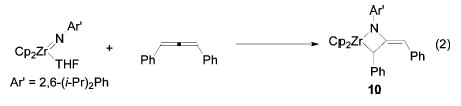
To ascertain the stereospecificity of the cycloaddition step, as well as to study the subsequent metallacycle isomerizations in a simpler system, the reactions of enantioenriched 1,3-disubstituted allenes with achiral zir-conocene imido complex 3 were examined. Enantioenriched allenes 5a–d were prepared by stereoselective SN2′ displacement of the appropriate propargylic alcohols 4a–d (Scheme 1).17 As a control, heating a solution of enantiopure allene (R)-5a at 95 °C for 24 h resulted in no loss of enantioenrichment. Enantiopure 1,3-disubstituted allenes underwent [2+2] cycloaddition with achiral zirconocene imido complex 3 to generate deep blue or purple azametallacyclobutanes 6–9. Metallacycles 6–9 possess two types of stereoisomerism: the configuration of the alpha stereocenter (R or S), and the orientation of the exocyclic double bond (E or Z).
E/Z Isomerization.
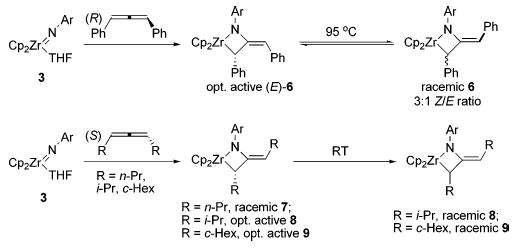
The configuration of the exocyclic double bond in the initially formed metallacycles was strongly dependent on the allene substituents. Diphenylmetallacycle 6 was initially formed exclusively as the E isomer, as determined by NOESY spectroscopy. After being heated at 95 °C for 24 h, however, (E)-6 was transformed to an equilibrium mixture of both isomers, favoring (Z)-6 3:1 (Scheme 2). The E and Z isomers of this complex were consistently distinguishable by the 1H NMR chemical shift of the vinyl proton of the exocyclic olefin. This resonance appears at 5.72 ppm for the E isomer, but is shifted downfield to 6.38 ppm for the Z isomer due to deshielding of this proton from the α-phenyl group. X-ray crystallographic analysis of the closely related metallacycle 10 confirmed the structure of the kinetically formed metallacycle and its assignment as the E isomer. An ORTEP diagram of 10 and relevant metric parameters are given in Figure 1. The X-ray crystal structure of diphenylmetallacycle 10 reveals that the four-membered ring is substantially puckered (angle between the planes = 33° ). The nitrogen atom is planar (sum of bond angles = 359.5° ), but this plane is canted by 29° relative to the Zr1–N1–C12 plane. This lessens the overlap between the filled p orbital on N and the exocyclic double bond, but it allows partial overlap between this filled p orbital and an empty in-plane acceptor orbital on Zr.
Scheme 2.
E/Z Isomerization and Racemization of Metallacycles
Figure 1.
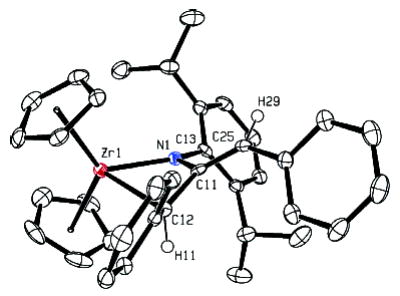
X-ray crystal structure of diphenylallene metallacycle 10. 50% probability boundaries are indicated. Bond distances: Zr1–N1 2.081 Å, Zr1–C12 2.348 Å, N1–C11 1.430 Å, C11–C12 1.503 Å, C11–C25 1.357 Å. Bond angles: N1–Zr1–C12 64.1°, Zr1–N1–C11 95.8°, N1–C11–C12 107.2°.
In contrast, treatment of imido complex 3 with dialkylallenes 5b,c initially formed metallacycles 7 and 8 with Z exocyclic double bond geometries. These assignments were also determined by NOESY spectroscopy. Due to the complexity of the NMR spectrum of dicyclohexylmetallacycle 9, the configuration of the double bond could not be definitively determined, but is presumed to be Z by analogy. Instead of affording the corresponding E isomers, heating the dialkylmetallacycles provided an unexpected rearrangement product, the formation of which will be discussed in detail below.
Inversion (Racemization).
To determine if the initial [2+2] cycloaddition was concerted, the CD spectra of a dilute hexane solution of metallacycles 6–9 were measured. A solution of isolated diphenylmetallacycle 6 displayed a strong CD signal with maxima at 234, 257, and 294 nm, demonstrating that this metallacycle is optically active. Although the maxima at 234 and 257 nm coincide closely with the spectrum of free (R)-diphenylallene, the CD signal at 294 nm is new, and NMR spectroscopy shows no free allene is present. Heating this solution at 45 ° C for 18 h resulted in no change in the CD spectrum, indicating no racemization under these conditions. However, after heating at 95 ° C for 24 h, the CD spectrum indicated that the metallacycle had lost all of its optical activity. It was established by 1H NMR and UV spectroscopy that metallacycle decomposition does not occur under these conditions.
Solutions of diisopropyl- and dicyclohexylmetallacycles 8 and 9 generated by treatment of a suspension of imido complex 3 with an excess of enantioenriched allene displayed CD signals at approximately 300 and 480 nm (Figure 2). The free dialkylallenes display no CD signal at wavelengths greater than 240 nm. In contrast to the high temperatures required for the racemization of diphenylmetallacycle 6, solutions of these metallacycles lost their optical activity over the course of 18 h at room temperature. Even more dramatically, treatment of imido complex 3 with di-n-propylallene resulted in a solution of metallacycle 7 that was optically inactive after only 15 min at room temperature.
Figure 2.
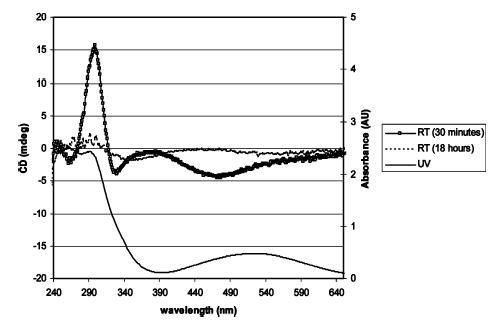
Representative CD and UV spectra from the racemization of metallacycle 8.
Interestingly, most disubstituted allenes could be released from the metal center by treatment of the metallacycles with either allene (C3H4) or diisopropylcarbodiimide (DIC) under mild heating. The enantiopurity of the allenes recovered in this fashion was measured to confirm the results of the CD experiments above (Scheme 3). As expected, treatment of optically active diphenylmetallacycle 6 with DIC at 45 ° C generated 1,3-diphenylallene of unchanged enantiopurity (>95% ee). Also, after heating 6 at 95 ° C for 24 h to effect its racemization, the diphenylallene recovered upon treatment with DIC was racemic. Furthermore, 1,3-diisopropylallene was recovered from reaction of metallacycle 8 with DIC, but only after several hours at 45 ° C, conditions that were sufficient to fully racemize 8, and the recovered allene in this case was racemic.
Scheme 3.
Enantiopurity of Recovered Allenes
The racemization of diisopropylmetallacycle 8 was monitored by CD spectroscopy at 25, 35, and 45 ° C. This transformation displayed clean first-order kinetics at all three temperatures, and the first-order rate constants are given in Table 1. The rate of racemization of 8 was independent of the concentration of excess free allene. Activation parameters of ΔH‡ = 19 ± 1 kcal/mol and ΔS‡ = −13 ± 3 cal/mol·K were obtained from an Eyring plot of the data in Table 1.
Table 1.
Racemization Kinetics for Metallacycle 8
| temp (° C) | kH (s −1)a | kD (s −1) | kH/kD |
|---|---|---|---|
| 25 | 1.48 × 10−4 | ND | NA |
| 35 | 4.25 × 10−4 | 1.49 × 10−4 | 2.8 |
| 45 | 1.16 × 10−3 | 4.08 × 10−4 | 2.8 |
Two runs for the first entry gave kobs = 1.46 and 1.51 × 10−5 s−1, so the error was estimated to be ±5%, and errors in activation parameters and isotope effect were determined on the basis of this value.
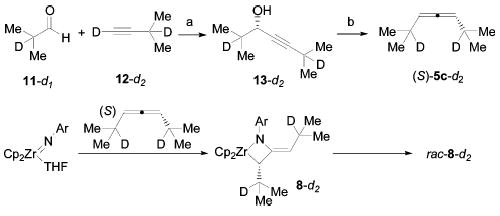
To provide information about the racemization mechanism, enantiopure deuterium-labeled allene 5c-d2 was synthesized via the route depicted in Scheme 4. The synthesis was accomplished by the enantioselective coupling of appropriately labeled aldehyde (11-d1) and alkyne (12-d2) partners by the method of Carreira,18–20 followed by stereospecific rearrangement of the propargylic alcohol (13-d2) to the enantioenriched allene by the method of Myers.21 Deuteration of the N-methylephedrine ligand in the alkyne addition reaction was required to avoid loss of label in the aldehyde coupling partner. The racemization of the deuterated metallacycle 8-d2 was also monitored by CD spectroscopy at 35 and 45 ° C, and the first-order rate constants are given in Table 1. A normal primary isotope effect of kH/kD =2.8 ± 0.3 was observed at both temperatures.
Scheme 4.
Synthesis and Racemization of Deuterated Diisopropylmetallacycle
Monoazadiene Complex Formation.
Dialkylmetallacycles 7–9 rearranged slowly to new isomeric complexes upon mild heating (45 ° C) in the absence of trapping agent (Scheme 5). The rate of this rearrangement was always slower than that of racemization and depended on the size of the alkyl substituents, being fastest with the smallest substituents (n-Pr > (CH2)6 > i-Pr ≈ c-Hex). These new complexes were identified as the monoazadiene (MAD) complexes 14–16 by 2D NMR spectroscopy (NOESY, TOCSY, HMQC). Cyclononadiene-derived metallacycle 17 also rearranged at 45 ° C, but in this case a 3:1 mixture of two MAD complex isomers 18a,b was observed. Attempted recovery of di-n-propylallene from metallacycle 7 by treatment with diisopropylcarbodiimide resulted in quantitative formation of MAD complex 14, indicating that rearrangement to 14 is faster than retrocyclization to form free allene.
Scheme 5.
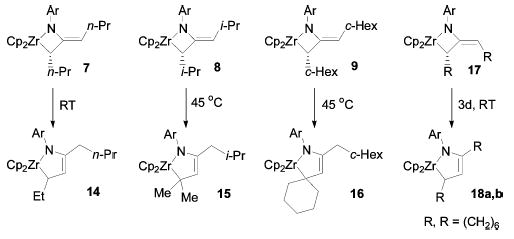
MAD Complex Formation
To obtain more information about the MAD complexes and their structures, complex 18a was crystallized from THF/Et2O and an X-ray crystal structure was determined. An ORTEP diagram of the structure is given in Figure 3. The MAD moiety is strongly bent with respect to the Zr–N–C plane, but Zr–C11 and Zr–C12 bond distances of 2.65 and 2.54 Å, respectively, reveal that 18 is best described as a metallacyclopentene, rather than as an η4-MAD complex. This observation has been made in other reports from the literature.22–30 The strain of the bridgehead double bond is evident in the reduced torsion angle about that bond (157° ). When crystals of 18a were redissolved in C6D6, the same 3:1 mixture of 18a and 18b was observed by 1H NMR spectroscopy. NOESY spectroscopy established that the crystal structure corresponds to the major isomer 18a and that the minor isomer 18b is likely to be that depicted in Figure 3. Interestingly, the proton on C13 resonates at 3.0 ppm in 18a, but at −0.4 ppm in 18b, a large difference that has also been observed previously.22 This difference presumably reflects a large shielding of this proton in 18b from the adjacent cyclopentadienyl ring which is lacking in 18a.
Figure 3.
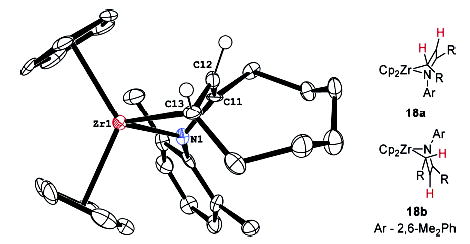
X-ray structure of MAD complex 18a. 50% probability boundaries are indicated. Bond distances: Zr1–N1 2.160 Å, Zr1–C13 2.254 Å, Zr1–C11 2.652 Å, Zr1–C12 2.497 Å, N1–C11 1.385 Å, C11–C12 1.386 Å, C12–C13 1.441 Å. Bond angles: N1–Zr1–C13 83.7° .
Matched and Mismatched Metallacycles.
Metallacycles derived from enantiopure ebthi imido complexes were synthesized to simultaneously monitor the rates of epimerization and E/Z isomerization by NMR spectroscopy in the same system. Treatment of imido complex (R,R)-1 with (R)-diphenylallene gave exclusively metallacycle diastereomer 19 (Scheme 6). This is the diphenylallene enantiomer that reacts more rapidly with the (R,R)-imido complex, that is, the “matched” metallacycle. In contrast, the reaction of (S)-diphenylallene with imido complex (R,R)-1 yielded an equilibrium mixture of starting imido complex (R,R)-1 and “mismatched” metallacycle 20, again as a single diastereomer (Keq ≈ 1). Removal of the released THF under vacuum drove the cycloaddition to completion and allowed for the isolation of the pure compound. Both diastereomers displayed proton resonances at approximately 5.81 ppm, which suggest an E geometry of the exocyclic olefin, and the configuration of both metallacycles was further confirmed by NOESY spectroscopy. Treatment of matched metallacycle 19 with diisopropylcarbodiimide generated (R)-diphenylallene with no erosion of enantiopurity (>95% ee). Likewise, cleavage of mismatched metallacycle 20 with carbodiimide formed (S)-diphenylallene (>95% ee), indicating that no racemization occurred during the cycloaddition/cycloreversion sequence for either antipode of the allene.
Scheme 6.
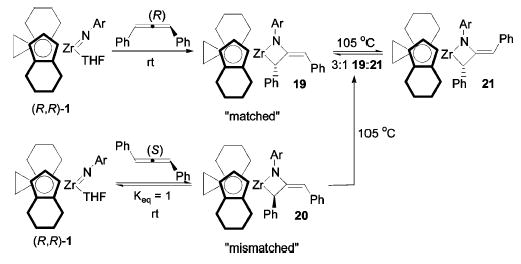
Formation of Matched and Mismatched Metallacycles and Establishment of the Equilibrium Mixture
Matched metallacycle 19 did not rearrange at temperatures lower than 95 ° C. However, after 24 h at 105 ° C, an equilibrium mixture of the starting material and its Z isomer 21 was formed, favoring the original metallacycle 3:1 (Scheme 6). The same equilibrium mixture of 19 and 21 was also obtained upon heating mismatched metallacycle 20 at 105 ° C. Under milder conditions, 20 rearranged at 45 ° C to give a mixture of all four possible metallacycle diastereomers. Initially, first-order decay of 20 to give a 2.5:2.5:1 mixture of 21, 22, and 19 was observed. Newly formed metallacycle 22 then decayed more slowly to give a mixture of 19 and 21. Metallacycles 19 and 21 do not interconvert appreciably at this temperature, but the same 3:1 equilibrium ratio of 19 and 21 was observed upon heating to 105 ° C. The concentrations of all four metallacycles were measured as a function of time at 45, 55, and 65 ° C. These data were fitted to the pathways illustrated in Scheme 7 with the aid of the Gepasi computer modeling program.31–33 The RMS error of each fit was <0.9%, and the estimated standard deviation for most rate constants was <2%.34 When a rate constant (k5) for the conversion of 22 to 21 was included in the modeling, its optimum value was essentially zero, and leaving this parameter out of the modeling had an insignificant effect on the total error. Approximate activation parameters for each rate constant were obtained from Eyring plots and are listed in Table 2.
Scheme 7.
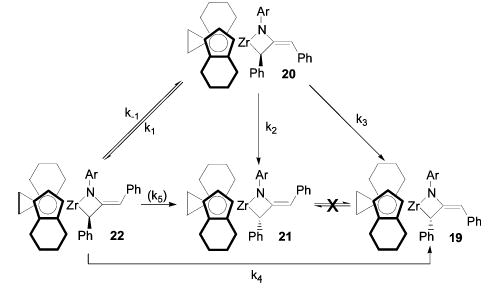
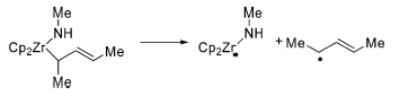
Rearrangement of Mismatched Metallacycle 20 at 45–65 ° C
Table 2.
First-Order Rate Constants and Eyring Parameters for Rearrangement of Metallacycle 20
| rate constanta | @ 45 ° C (s−1) | @ 55 ° C (s−1) | @ 65 ° C (s−1) | ΔH‡ (kcal/mol) | ΔS‡ (eu) |
|---|---|---|---|---|---|
| k1 | 4.08 × 10−5 | 1.51 × 10−4 | 5.72 × 10−4 | 27.5 | +8 |
| k−1 | 4.28 × 10−5 | 1.62 × 10−4 | 6.17 × 10−4 | 27.8 | +9 |
| k2 | 1.54 × 10−5 | 5.52 × 10−5 | 2.07 × 10−4 | 27.1 | +4 |
| k3 | 6.52 × 10−6 | 2.68 × 10−5 | 1.00 × 10−4 | 28.5 | +7 |
| k4 | 3.00 × 10−6 | 1.06 × 10−5 | 4.20 × 10−5 | 27.5 | +3 |
The error in isolated rate constants is estimated to be ±5%, and the error in activation parameters is estimated to be ±1 kcal/mol and ±3 eu.
Catalytic Isomerization.
Attempted functionalization of diphenylmetallacycle 6 by treatment with benzophenone did not result in insertion of the carbonyl moiety into the Zr–C bond; rather, the 1:3 equilibrium mixture of E and Z isomers was formed within 30 min at room temperature (Scheme 8). This E/Z isomerization was slower in the presence of 4,4′ -dimeth-ylbenzophenone, and faster in the presence of 4,4′ -dichloroben-zophenone. Catalytic quantities of these reagents were sufficient to promote the isomerization, and the presence or absence of light had no effect on its rate. Pivaldehyde and di-tert-butyl ketone failed to promote the isomerization, but di-p-tolyl disulfide, and ferricenium and cobaltocenium salts, also catalyzed the E/Z isomerization of the metallacycle, suggesting that these isomerizations proceed by an electron-transfer mechanism. CD spectroscopy revealed that racemization of the metallacycle took place concomitantly.
Scheme 8.
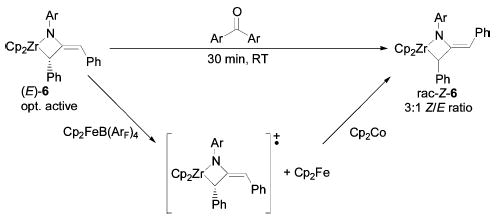
Catalytic Racemization and Isomerization of Metallacycle 6
Treatment of the metallacycle 6 with a full equivalent of ferricenium tetrakis(pentafluorophenyl)borate in chlorobenzene-d5 resulted in the formation of a red solution that displayed no sharp resonances by NMR or EPR spectroscopy, except for that corresponding to the quantitative formation of ferrocene (δ 4.05 ppm). Immediate addition of 1 equiv of cobaltocene to this mixture regenerated a blue solution of the metallacycle as an equilibrium mixture of E and Z isomers. However, if the red solution is allowed to stand at room temperature for 2 h, addition of cobaltocene fails to generate any recognizable products.
Insertions.
To further investigate possible electron-transfer reactions, the metallacycle 6 was treated with known radical traps. For example, metallacycle 6 reacted with Me3SnH within 18 h at room temperature to give the product of formal insertion into the Zr–C bond 23 (Scheme 9). The 1H NMR spectrum of 23 displayed broad resonances due to hindered rotation about the N–C bond. The two protons on the benzylic methylene group are diastereotopic at room temperature (δ 4.1 and 2.57 ppm), but their signals coalesce at high temperatures. The structure of 23 was confirmed by X-ray crystallographic analysis; an ORTEP diagram is displayed in Figure 4. In contrast, the poorer radical trap Et3SiH failed to react with metallacycle 6, even at 105 ° C. Treatment of metallacycle 6 with bis(4-trifluoromethylphenyl)disulfide at room temperature gave insertion product 24, which displayed similar hindered rotation.
Scheme 9.
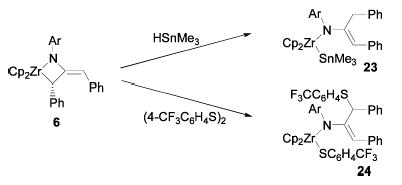
Insertion Reactions of Diphenylmetallacycle 6
Figure 4.
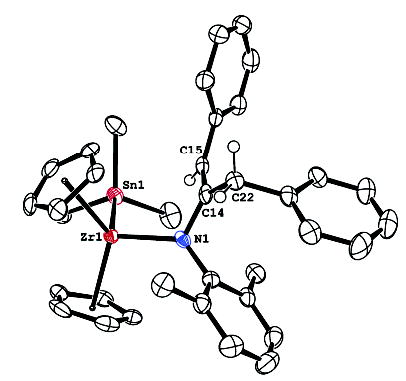
X-ray crystal structure of complex 23. 50% probability boundaries are indicated. Bond distances: Zr1–Sn1 2.969 Å, Zr1–N1 2.133 Å, Zr1–C14 3.00 Å, Zr1–C15 3.28 Å, N1–C14 1.416 Å, C14–C15 1.347 Å, C14–C22 1.525 Å. Bond angles: N1–Zr1–Sn1 107.9° , Zr1–N1–C14 113.9° .
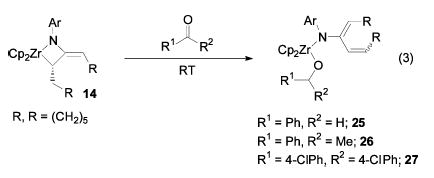
Instead of the expected insertion product, treatment of cyclononadiene metallacycle 14 with benzaldehyde for 1 h at room temperature gave benzyloxide product 25 (eq 3). This product presumably results from β-hydride elimination followed by hydride insertion. Acetophenone and 4,4′ -dichlorobenzophe-none yielded similar insertion products.
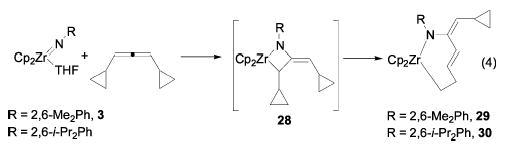
Dicyclopropylallene.
In an additional effort to detect radical intermediates, zirconocene imido complex 3 was treated with dicyclopropylallene. This did not yield the (presumably initially formed) metallacycle 28; instead, a new metallacycle was formed quantitatively in 15 min at room temperature (eq 4). This species was identified as the seven-membered ring met-allacycle 29 by 2D NMR spectroscopy. The unusual trans double bond in a seven-membered ring was confirmed by the large coupling constant (J = 15 Hz) and by X-ray crystallographic analysis of related complex 30. An ORTEP diagram and relevant metric parameters are given in Figure 5. The double bond is not coordinated to the Zr, as shown by the Zr–C13 and Zr–C14 distances of 2.88 and 3.06 Å, and is somewhat strained, as shown by the C12–C13–C14–C15 torsion angle of 155° (rather than 180° ) and the Zr1–C11 bond distance of 2.41 Å.
Figure 5.
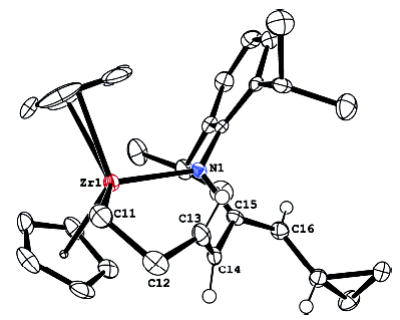
X-ray structure of dicyclopropylallene metallacycle 30. 50% probability boundaries are indicated. Bond distances: Zr1–N1 2.235 Å, Zr1–C11 2.410 Å, C13–C14 1.327 Å, C15–C16 1.333 Å. Bond angles: N1–Zr1–C11 117.9° , Zr1–C11–C12 108.1° .
Discussion
The results presented above provide insight into the mechanisms of the isomerization and racemization of allene-derived metallacycles, and a discussion of these mechanisms follows. First, it is important to establish whether the racemization of allenes occurs during the initial cycloaddition step or is due to a subsequent isomerization event. After answering this question, potential mechanisms for E/Z isomerization and racemization will be presented, and the kinetic and thermodynamic selectivities in the cycloaddition process and subsequent isomerizations will be discussed.
Stereospecificity of Cycloaddition.
In our initial report discussing the selective stereoinversion of cyclononadiene,15 we proposed that this stereoinversion could be the result of a stepwise [2+2] cycloaddition mechanism which proceeds through an achiral diradical or zwitterionic intermediate 31 (path a, Scheme 10). If this were the case, reaction of enantiopure allenes with achiral imido complexes should yield racemic products because intermediate 31 is achiral. However, the initially formed zirconocene metallacycles (with the exception of di-n-propylmetallacycle 7) in this study exhibited strong CD signals and were therefore optically active. Furthermore, the structures of the cycloadducts of (R)- and (S)-diphenylallene with enantiopure ebthi imido complex (R,R)-1, 19 and 20, respectively, reveal that the original stereochemistry of the allene is retained through the cycloaddition/cycloreversion process. Therefore, the [2+2] cycloaddition process is almost certainly concerted and stereospecific for all allenes, initially generating enantiopure metallacycles that subsequently racemize by another mechanism (path b). This racemization is too rapid to observe in the case of the di-n-propylmetallacycle 7, but is slow enough for the other metallacycles to allow observation of optical activity.
Scheme 10.
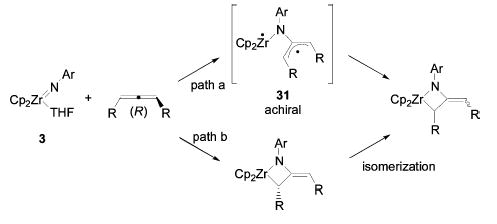
Stepwise versus Concerted Cycloaddition Processes
Potential Metallacycle Rearrangement Mechanisms.
After establishing that the cycloaddition is concerted and stereospecific, we began investigating the nature and mechanism of the subsequent isomerizations of the allene-derived metallacycles. Two different types of stereoisomerization were observed: E/Z isomerization and epimerization of the alpha stereocenter. Isomerization of either the alpha stereocenter or the exocyclic double bond results in net inversion of the stereochemistry of the allene fragment of the metallacycle and thus could be responsible for the overall allene stereoinversion. Several mechanistic possibilities for the various isomerization processes are depicted in Scheme 11. Although diradical (or zwitterionic) species 31 is not an intermediate in the initial cycloaddition process, simple reversible fragmentation of the Zr–C bond of the metallacycle to generate intermediate 31 can explain both the racemization and the E/Z isomerization processes (eq 5). Alternatively, coordination of the exocyclic double bond to the Zr center to generate η4-azatrimethylenemethane intermediate (or transition state) 32,35–37 followed by collapse to the opposite terminus, can also explain both types of isomerization processes (eq 6). Reversible β-hydride elimination, with transient formation of achiral Zr hydride 33, would result in racemization of the metallacycle, but not E/Z isomerization, and is only available to dialkylmetallacycles (eq 7). Net E/Z isomerization can be accomplished by simple rotation around the double bond (eq 8) or by retrocyclization followed by cycloaddition to the opposite face of the allene (eq 9), but neither pathway allows for racemization of the metallacycle. These potential mechanisms will serve as a guide for the following discussion of the experimental results.
Scheme 11.
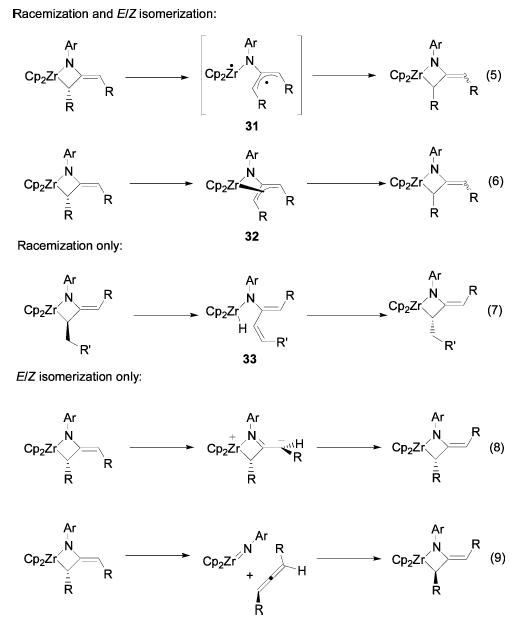
Possible Isomerization and Racemization Mechanisms
Kinetic and Thermodynamic Olefin Geometries.
On the basis of a simple analysis of the steric factors in the [2+2] cycloaddition, it was anticipated that the substituent on the exocyclic double bond would prefer to be oriented away from the bulky imido substituent and, therefore, that the E isomers of the metallacycles would be favored (Figure 6). This model holds for the kinetic cycloadducts of diphenylallene with both zirconocene and ebthi imido complexes. In fact, even in the reaction of imido complex (S,S)-1 with (R)-diphenylallene, where an unfavorable interaction between the bulk of the ebthi ligand and the α-phenyl group could be avoided by forming the Z isomer, only the E isomer was observed. This preference also explains the observed rate difference in the formation of the matched and mismatched isomers.
Figure 6.
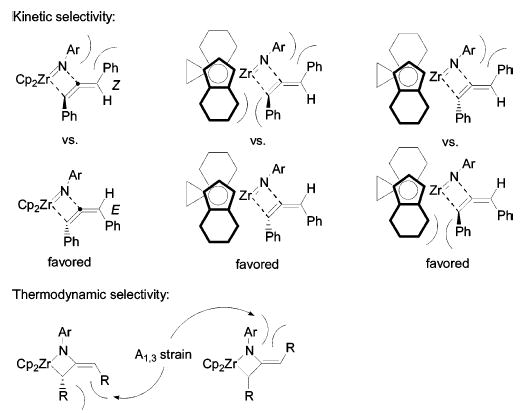
Analysis of the kinetic and thermodynamic selectivities leading to E and Z isomers.
In contrast to the strong kinetic preference for formation of E metallacycles from diphenylallene, the energies of the E and Z isomers are fairly similar. In fact, for the zirconocene metallacycle 6, the Z isomer is slightly more stable than the E isomer (Keq = 3 at 95 ° C). In the ebthi series, the equilibrium between 20 and 22 also favors the Z isomer (k1/k−1 = Keq = 6 at 45 ° C), but in the epimeric pair (19 and 21), the E isomer is more stable (Keq = 3 at 105 ° C). However, in all cases, the difference in energy between the isomers is small (ΔG° < 1.2 kcal/mol). There appears to be a subtle balance between two types of allylic strain involving orientation of the exocyclic phenyl group toward either the α-phenyl substituent or the imido substituent (Figure 6). Although the imido substituent is larger, the X-ray crystal structure of metallacycle 10 reveals that, due to puckering of the metallacycle, the α-phenyl substituent is closer to being coplanar with the exocyclic phenyl group (26° vs 39° , Figure 1), thereby increasing the magnitude of steric interaction for this substituent, and compensating for its smaller size.
For the dialkylmetallacycles 7–9, only the Z isomer was ever observed, so it is unclear whether the stereochemistry about the exocyclic double bond is due to kinetic or thermodynamic preference. DFT calculations indicate that the Z isomer is thermodynamically more stable than the E isomer for all dialkylmetallacycles. We cannot say, however, whether the Z isomer is formed upon initial cycloaddition or if the E isomer is kinetically preferred and a rapid E/Z isomerization follows. If such a rapid E/Z isomerization is occurring, it must do so without racemization of the metallacycle, as this process is substantially slower (t1/2 > 1 h) than the hypothetical E/Z isomerization (t1/2 < 5 min).
Monoazadiene Complex Formation.
All metallacycles with available hydrogen atoms beta to the metal center eventually rearrange to monoazadiene (MAD) complexes. The rate of this rearrangement is affected by the size of the substituents (n-Pr > (CH2)6 > i-Pr ≈ c-Hex). The failure to isolate free allene upon treatment of di-n-propylmetallacycle 7 with diisopropyl-carbodiimide indicates that rearrangement of 7 to the MAD complex is faster than retrocycloadditon. A mechanism consistent with these observations is depicted in Scheme 12 (path a). Initial β-hydride elimination to generate zirconium hydride complex 34 is followed by intramolecular hydrozirconation of the enamide double bond to give η2-imine complex 35. A [1,3]-shift of the zirconium center then generates the MAD complex.38 An alternative mechanism initiated by dissociation of the allene followed by intermolecular C–H activation does not fit the observation that metallacycle 7 forms MAD complex 14 even in the presence of DIC (path b).
Scheme 12.
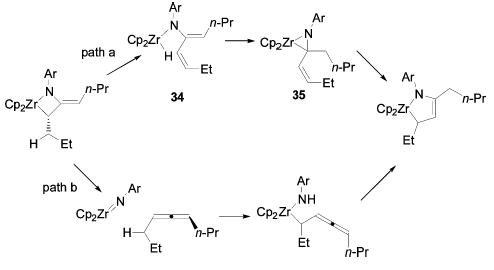
Mechanisms for MAD Complex Formation
Racemization.
The rate of racemization of metallacycles formed from 1,3-dialkylallenes parallels the rate of MAD complex formation (n-Pr > i-Pr ≈ c-Hex ≫ Ph). The initial β-hydride elimination step in the proposed mechanism for MAD complex formation suggested a possible route for racemization of dialkyl-substituted metallacycles and an explanation for their relative rates. Reversible β-hydride elimination would result in loss of optical activity via achiral intermediate 33 (Scheme 11, eq 7). To test this hypothesis, the racemization rate of enantiopure deuterated metallacycle 8-d2 was determined. This process exhibited a normal primary isotope effect (kH/kD = 2.8), similar in magnitude to that of reported β-hydride eliminations.39–41 The formation of the alkoxide complexes 25–27 upon treatment of cyclononadiene metallacycle 14 with carbonyl compounds (eq 3) provides additional evidence for the intermediacy of Zr hydride 33. Insertion of the ketone or aldehyde into the transient Zr hydride complex is evidently much faster than insertion into the Zr–C bond of the metallacycle. These data strongly indicate that the racemization of dialkylmetalla-cycles proceeds by a reversible β-hydride elimination mechanism (eq 7).
Rapid reversible β-hydride elimination explains the relative ease with which dialkylmetallacycles racemize, but such a mechanism is not available to diarylmetallacycles, which lack available beta hydrogen atoms. An alternative mechanism is therefore required to explain the racemization of these complexes. The E/Z isomerization and racemization of the diaryl-metallacycles occur simultaneously in both the zirconocene system and the ebthi system, so mechanisms that explain the E/Z isomerization but do not allow for racemization (eqs 8 and 9) are unlikely. On the basis of preliminary DFT calculations, we proposed a [1,3]-shift mechanism involving coordination of the exocyclic olefin to the zirconium to form an η4-azatrim-ethylenemethane complex 32 as either an intermediate or a transition state (Scheme 11, eq 6). Collapse of this species to the other terminus of the allyl system would result in racemization of the metallacycle. Analogous [1,3]-shifts proceeding through alternative azatrimethylenemethane isomers can also explain the E/Z isomerization (Figure 7). Each [1,3]-shift results in either inversion of the alpha stereocenter or E/Z isomerization, but not both. However, as discussed below, additional evidence has led us to postulate a different mechanism for the diaryl-metallcycle isomerization.
Figure 7.
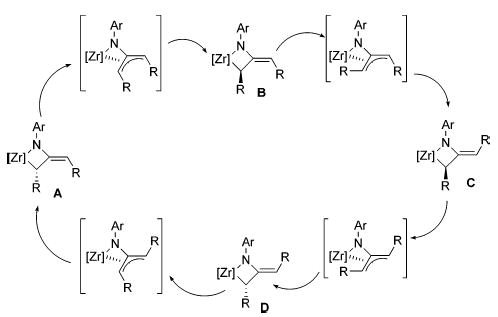
[1,3]-Shift mechanism for E/Z isomerization and racemization.
Matched and Mismatched Rearrangements.
To try to observe individual [1,3]-shifts, matched and mismatched met-allacycles 20 and 21 were synthesized by treatment of enantiopure ebthi imido complex (R,R)-1 with (R)- and (S)-diphenylallene, respectively. These isomers correspond to structures A and B in Figure 7. The rearrangement of the mismatched metallacycle 20 was observed to form all possible isomers of this complex through a complex series of reactions. Kinetic modeling revealed that this rearrangement followed the kinetic scheme depicted in Scheme 7. These rearrangements correspond to the conversions of B to C (k1), B to D (k2), B to A (k3), C to B (k−1), and C to A (k4) in Figure 7. The observed direct rearrangements of 20(B) to 21(D) and 22(C) to 19(A) (k2 and k4) are inconsistent with any of the simple [1,3]-shifts depicted in Figure 7.
Oxidative Isomerization.
Mild oxidants (benzophenone, disulfides, cobaltocenium, ferricenium) were found to catalyze the E/Z isomerization and racemization of diphenylmetallacycle 6. This process could be reproduced in a stepwise fashion by treatment of the metallacycle with ferricinium cation, followed by reduction with cobaltocene. The proposed mechanism for this transformation involves removal of an electron from the metallacycle, resulting in fragmentation of the Zr–C bond to generate a structure (36) containing a Zr cation and a highly stabilized diphenylallyl radical (eq 10). Ample precedents for this type of reactivity exist: bond fragmentation induced by one-electron oxidation of zirconium alkyl complexes has been described by Jordan et al.,42 and reversible one-electron oxidation of a zirconaazacyclobutene has been reported by Norton et al.43 These precedents suggest that oxidation could occur directly from the Zr–C bond or from the enamide π system. In either case, rapid fragmentation of the Zr–C bond should take place to form intermediate 36. Because this intermediate has lost the original stereochemical information of the allene, collapse to regenerate the metallacycle upon reduction occurs to give an equilibrium mixture of all possible isomers.
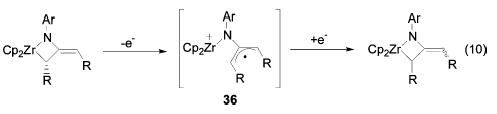
Intermediate 36 in the oxidative catalysis pathway suggested an alternative mechanism for the uncatalyzed racemization and E/Z isomerization processes. Thermal homolysis of the Zr–C bond in 6 would result in the formation of a structure (31) containing the same stabilized diphenylallyl radical and a Zr-(III) center (Scheme 11, eq 5). Radical recombination would regenerate the metallacycle, but because the original stereochemistry is lost in the diradical intermediate, an equilibrium mixture of the four isomers would result. Although none of these constitutes conclusive proof, several observations support this mechanism, rather than the [1,3]-shift mechanism for the racemization of diarylmetallacycles. First, the rearrangements of mismatched metallacycle 20 are not consistent with the predictions of the [1,3]-shift mechanism; that is, the direct rearrangements of 20 to 21 and 22 to 19 (k2 and k4) should not occur via a simple [1,3]-shift (see above). Second, the entropies of activation for these rearrangements are moderately positive (ΔS‡ = 3–9 eu), which is more consistent with a mechanism in which a bond is breaking, than with one in which a concerted [1,3]-shift is taking place.44–46 Third, the Zr–C bond length in diphenylmetallacycle 10 is 0.1 Å longer47 than the Zr–C bond in Cp2ZrMe2 (BDE = 67 kcal/mol),48,49 indicating that it may also be substantially weaker. Fourth, preliminary DFT calculations indicate that allylic Zr–C bonds have bond dissociation energies similar to the enthalpies of activation measured for the rearrangements (calc. BDE = 29 kcal/mol).50 Fifth, diphe-nylmetallacycle 6 undergoes insertion reactions with stannanes and disulfides that are reminiscent of radical trapping reactions (Scheme 9). Finally, the rearrangement product of the dicyclo-propylallene metallacycle 29 is formally the result of a radical ring-opening process. All of these observations taken together argue forcibly for reversible bond homolysis (Scheme 11, eq 5) as the mechanism of racemization and E/Z isomerization of diarylmetallacycles.
Conclusion
The stereoinversion of allenes promoted by imidozirconium complexes has been thoroughly investigated. The [2+2] cycloaddition of allenes and imido complexes is concerted and stereospecific, leading to the formation of metallacycles in which the original configuration of the allene has been retained. These metallacycles undergo racemization and E/Z isomerization processes, as well as subsequent rearrangement to monoazadiene (MAD) complexes. Reversible β-hydride elimination is the initial step in the racemization of dialkylmetallacycles and their rearrangements to MAD complexes. Diarylmetallacycles undergo racemization and E/Z isomerization processes via a reversible homolysis of the unusually weak Zr–C bond. The racemization and E/Z isomerization of diarylmetallacycles is also catalyzed by weak oxidants, proceeding through a radical cation intermediate. We hope to use the mechanistic insights disclosed here in the development of new catalytic and stoichiometric transformations of allenes mediated by imidozirconium complexes, especially potentially asymmetric versions.
Experimental Section
General Procedures.
Unless otherwise noted, reactions and manipulations were performed at ambient temperature in an inert atmosphere (N2) glovebox, or using standard Schlenk and high vacuum line techniques. Glassware was dried overnight at 150 ° C or flame dried under vacuum immediately prior to use. All NMR spectra were obtained at ambient temperature using Bruker AMX-300, AMX-400, or DRX-500 spectrometers. 1H NMR chemical shifts (δ) are reported in parts per million (ppm) downfield of TMS and are referenced relative to residual protiated solvent. 13C NMR chemical shifts (δ) are reported in ppm relative to the carbon resonance of the deuterated solvent. In cases where assignment of 13C resonances was ambiguous, standard DEPT 45, 90, and/or 135 pulse sequences or 13C–1H HMQC experiments were used. Infrared (IR) spectra were recorded as a Nujol mull or a thin film between NaCl plates. Elemental analyses were performed at the University of California, Berkeley Microanalytical facility on a Perkin-Elmer 2400 Series II CHNO/S Analyzer. Optical rotations were measured on a Perkin-Elmer model 241 polarimeter at room temperature.
Materials.
Unless otherwise noted, reagents were purchased from commercial suppliers and were used without further purification. Pentane, hexanes, benzene, and toluene (Fisher) were passed through a column of activated alumina (type A2, size 12 × 32, Purifry Co.) under nitrogen pressure and sparged with N2 prior to use. Diethyl ether and tetrahydrofuran (Fisher) were distilled from sodium/benzophenone ketyl under N2 prior to use. Deuterated solvents (Cambridge Isotope Laboratories) were purified by vacuum-transfer from the appropriate drying agent (Na/Ph2CO or CaH2) prior to use. Imido complexes 1 and 3 were prepared according to literature procedures.15,51 (rac)-1,2-Dicyclopropylallene was a generous gift of Dr. Armin DeMeijere.
General Procedures for Circular Dichroism Experiments.
All circular dichroism spectra were recorded at room temperature on a Jasco J-810 spectropolarimeter. In experiments to determine the change in optical activity with time and/or temperature, benzene or hexanes solutions of metallacycles were stored and/or heated in glass ampules to which a Kontes vacuum stopcock had been sealed. For metallacycle 6, an analytical solution of the metallacycle in hexanes was prepared in the glovebox. The solution was transferred to a 1 cm quartz glass cuvette, to which a Kontes vacuum stopcock had been sealed. For metallacycles 7 and 8, the imido complex 3 (2 mg) was stirred in hexanes (20 mL) for 30 min to allow it to dissolve, and then an excess (5–10 equiv) of the appropriate enantioenriched allene (5b or 5c) was added. The solution took on a pale purple color within 5 min at room temperature. The metallacycle solutions were filtered and transferred to an airtight cuvette, as described above. Initial spectra were taken within 30 min of mixing the reagents.
CD Kinetics of Racemization of Metallacycle 8.
Solutions of metallacycles 8 and 8-d2 were prepared as described above, and the CD signal at 300 nm was recorded as a function of time at 25, 35, and 45 ° C. Clean first-order kinetics were observed in all cases, and the observed rate constants were obtained from a plot of log(CD) versus time.
(E)-Cp2ZrN(2,6-Me2C6H3)C(=CHPh)C(H)Ph ((E)-6).
To a solution of 3 (0.097, 0.24 mmol) in benzene (3 mL) was added a solution of (R)-5a (0.45 g, 0.24 mmol). After 10 min at room temperature, the resulting deep blue solution was frozen (−30 ° C). The solvent was lyophilized, yielding a purple solid. Crystallization by diffusion of pentane into a concentrated THF solution at −30 ° C afforded pure (E)-6 as a brown powder (0.082 mg, 58%) (approximately 1 equiv of THF cocrystallized with (E)-6 and could not be removed, even after extended exposure of the crushed crystalline material to dynamic vacuum, as determined by 1H NMR spectroscopy). 1H NMR (C6D6, 500 MHz): δ 6.79–7.13 (m, 13H, ArH), 5.77 (s, 5H, C5H5), 5.72 (br d, J = 2 Hz, =CH), 5.63 (s, 5H, C5H5), 4.48 (br d, J = 2 Hz, 1H, ZrCH), 3.56 (br s, 4H, ((CH2CH2)2O), 2.22 (s, 3H, ArCH3), 2.14 (s, 3H, ArCH3), 1.40 (br s, 4H, ((CH2CH2)2O) ppm. 13C NMR (C6D6, 125 MHz): δ 151.4 (NC=C), 149.9 (Caryl), 140.6 (Caryl), 136.9 (Caryl), 132.8 (Caryl), 132.5 (Caryl), 130.7 (Caryl), 129.1 (Caryl), 129.0 (Caryl), 128.2 (Caryl), 127.7 (Caryl), 126.8 (Caryl), 123.9 (Caryl), 123.1 (Caryl), 121.4 (Caryl), 115.2 (C5H5), 113.6 (C5H5), 106.2 (=C(H)Ph), 68.2 ((CH2CH2)2O), 53.7 (ZrC), 26.1 ((CH2CH2)2O), 22.3 (ArCH3), 20.5 (ArCH3) ppm. FT-IR (Nujol): 3106 (w), 1590 (m), 1543 (s), 1376 (m), 1263 (m), 1213 (m), 1147 (m), 1067 (w), 1015 (m), 912 (m), 797 (s), 762 (m), 701 (m), 637 (m) cm−1. Anal. Calcd for ZrNC33H31(C4H8O): C, 73.46; H, 6.50; N, 2.32. Found: C, 73.25; H, 6.77; N, 2.69. Reaction of 3 with a slight excess of (R)-1,3-diphenylallene, followed by removal of solvent and trituration with pentane, afforded a quantitative yield of spectroscopically pure (S,E)-6.
(Z)-Cp2ZrN(2,6-Me2C6H3)C(=CHPh)C(H)Ph ((Z)-6).
A glass ampule was charged with a solution of (E)-6 (0.120 g, 0.23 mmol) in toluene (3 mL). The ampule was sealed (Kontes Teflon vacuum stopcock) and heated to 95 ° C for 32 h. Upon cooling to room temperature, the toluene solution was layered with pentane (5 mL) and stored at −30 ° C. After 2 d, blue blocky crystals were isolated and solvent was removed under reduced pressure to afford pure (Z)-6 as a blue solid (0.046 g, 33%) (exactly 0.6 equiv of toluene cocrystallized with (Z)-6 and could not be removed, even after extended exposure of the crushed crystalline material to dynamic vacuum, as determined by 1H NMR spectroscopy). 1H NMR (C6D6, 500 MHz): δ 7.30–7.36 (m, 4H, ArH), 6.93–7.01 (m, 8H, ArH), 6.93 (m, 1H, ArH), 6.68 (m, 1H, ArH), 6.63 (m, 1H, ArH), 6.38 (br d, J = 2 Hz, 1H, =CH), 5.79 (s, 5H, C5H5), 5.72 (s, 5H, C5H5), 4.25 (br d, J = 2 Hz, 1H, ZrCH), 2.18 (s, 3H, ArCH3), 2.10 (s, 1.8H, CH3C6H5), 1.87 (s, 3H, ArCH3) ppm. 13 C NMR (C6D6, 125 MHz): δ 150.5 (NC=C), 149.3 (Caryl), 139.7 (Caryl), 138.2 (Caryl), 132.6 (Caryl), 131.1 (Caryl), 129.7 (Caryl), 128.9 (Caryl), 128.7 (Caryl), 128.0 (Caryl), 127.8 (Caryl), 127.8 (Caryl), 127.6 (Caryl), 127.5 (Caryl), 126.0 (Caryl), 124.2 (Caryl), 122.1 (Caryl), 121.1 (Caryl), 115.2 (Caryl), 114.1 (C5H5), 113.5 (C5H5), 108.7 (=CH), 59.6 (ZrC), 23.6 (ArCH3), 21.8 (CH3C6H5), 20.8 (ArCH3) ppm. FT-IR (Nujol): 3057 (w), 1590 (m), 1545 (w), 1377 (m), 1292 (w), 1259 (m), 1232 (m), 1199 (w), 1115 (w), 1013 (m), 1069 (w), 1013 (m), 929 (w), 877 (w), 804 (s), 763 (m), 736 (m), 694 (m) cm−1. Anal. Calcd for ZrNC33H31(C7H8)0.6: C, 75.97; H, 6.14; N, 2.39. Found: C, 75.71; H, 6.22; N, 2.30.
(Z)-Cp2ZrN(2,6-Me2C6H3)C(=C(H)C3H7)CH(C3H7) (7).
Although it is initially formed cleanly, azazirconacyclobutane 7 rearranges slowly (t1/2 ≈ 10 h) to a new product at room temperature; therefore, it could not be isolated in analytically pure form. Rather, 7 was generated in situ and characterized by 1H and 13C NMR spectroscopy only. To a solution of 3 (0.016 g, 0.04 mmol) in C6D6 (0.5 mL) was added a solution of (S)-5b (0.007 g, 0.06 mmol) in C6D6 (0.5 mL). The resulting deep purple reaction mixture was transferred to an NMR tube, which was flame-sealed under static vacuum. 1H NMR (C6D6, 500 MHz): δ 5.93 (s, 5H, C5H5), 5.83 (s, 5H, C5H5), 5.02 (dt, J = 7 Hz, 2.5 Hz, 1H, =CH), 2.78 (m, 1H, ZrCH), 2.58 (m, 1H, ZrCHCH2), 2.34 (s, 3H, ArCH3), 2.06 (s, 3H, ArCH3), 2.05 (m, 1H, ZrCHCH2), 1.79 (m, 2H, =CHCH2), 1.37 (m, 4H, =CHCH2CH2, ZrCHCH2CH2), 1.18 (t, J = 7 Hz, 3H, ZrCH(CH2)2CH3), 0.83 (t, J = 7 Hz, =CH(CH2)2CH3) ppm. 13C NMR (C6D6, 125 MHz): δ 152.9 (NC=C), 131.3 (C6H3), 130.4 (C6H3), 129.2 (C6H3), 128.8 (C6H3), 120.5 (C6H3), 114.1 (=CH), 111.7 (C5H5), 110.8 (C5H5), 58.5 (ZrC), 36.1 (CH2), 31.4 (CH2), 25.7 (CH2), 23.8 (CH2), 23.3 (ArCH3), 20.4 (ArCH3), 15.5 (CH3), 14.8 (CH3) ppm. One of the aryl resonances in the 13C spectrum could not be resolved.
(Z)-Cp2ZrN(2,6-Me2C6H3)C(=C(H)CH(CH3)2)CH(CH(CH3)2) (8).
To a solution of 3 (0.095 g, 0.23 mmol) in benzene (5 mL) was added a solution of (S)-5c (0.034 g, 0.28 mmol) in benzene (1 mL). The resulting deep purple solution was stirred for 6 h at room temperature. The solvent was removed under reduced pressure, leaving a viscous purple material. This was taken up in pentane (10 mL) and filtered. Concentration of this solution afforded analytically pure 8 as a purple solid (0.090 g, 84%). 1H NMR (C6D6, 500 MHz): δ 7.04 (d, J = 7 Hz, 1H, m-ArH), 6.98 (d, J = 7 Hz, 1H, m-ArH), 6.78 (t, J = 7 Hz, 1H, p-ArH), 5.80 (s, 5H, C5H5), 5.67 (s, 5H, C5H5), 4.68 (dd, J = 10 Hz, 2.5 Hz, 1H, =CH), 2.34 (dd, J = 9.5 Hz, 2 Hz, ZrCH), 2.22 (s, 3H, ArCH3), 2.13 (m, 2H, CH(CH3)2), 1.94 (s, 3H, ArCH3), 1.29 (d, J = 6 Hz, 3H, CH(CH3)2), 0.97 (d, J = 6 Hz, 3H, CH(CH3)2), 0.78 (d, J = 6 Hz, 3H, CH(CH3)2), 0.66 (d, J = 6 Hz, 3H, CH(CH3)2) ppm. 13C NMR (C6D6, 125 MHz): δ 153.6 (NC=C), 130.6 (C6H3), 129.6 (C6H3), 129.5 (C6H3), 128.8 (C6H3), 124.2 (=CH), 120.7 (C6H3), 67.6 (ZrC), 30.6 (CH(CH3)2), 28.3 (CH(CH3)2), 26.9 (CH(CH3)2), 26.7 (CH-(CH3)2), 24.9 (CH(CH3)2), 22.9 (ArCH3), 22.6 (CH(CH3)2), 20.5 (ArCH3) ppm. FT-IR (Nujol): 3064 (w), 1590 (m), 1278 (s), 1236 (m), 1175 (w), 1109 (m), 1013 (m), 984 (w), 914 (w), 795 (s) cm−1. Anal. Calcd for ZrNC27H35: C, 69.77; H, 7.59; N, 3.01. Found: C, 69.40; H, 7.68; N, 3.33.
(Z)-Cp2ZrN(2,6-Me2C6H3)C(=C(H)C6H11)CH(C6H11) (9).
To a solution of 3 (0.107 g, 0.25 mmol) in benzene (4 mL) was added a solution of (S)-5d (0.052 g, 0.26 mmol) in benzene (1 mL). After 9.5 h at room temperature, the solvent was removed under vacuum, leaving a dark purple solid. Precipitation from a concentrated ether solution at −30 ° C afforded spectroscopically pure 9 (0.063 g, 45%). Several attempts to crystallize 9 to analytical purity were unsuccessful. 1H NMR (C6D6, 500 MHz): δ 7.05 (d, J = 7 Hz, 1H, m-ArH), 7.00 (d, J = 7 Hz, 1H, m-ArH), 6.81 (t, J = 7 Hz, 1H, p-ArH), 5.86 (s, 5H, C5H5), 5.73 (s, 5H, C5H5), 4.84 (dd, J = 9 Hz, 2 Hz, 1H, =CH), 2.50 (br d, 1H, CH(CH2)5), 2.47 (dd, J = 10 Hz, 2 Hz, 1H, ZrCH), 2.28 (s, 3H, ArCH3), 2.06 (s, 3H, ArCH3), 1.70–1.94 (m, 6H, C6H11), 0.770–1.47 (m, 15H, C6H11) ppm. 13C NMR (C6D6, 125 MHz): δ 154.0 (NC=C), 130.5 (C6H3), 129.9 (C6H3), 129.7 (C6H3), 129.0 (C6H3), 122.7 (=CH), 120.8 (C6H3), 111.8 (C5H5), 109.5 (C5H5), 66.2 (ZrC), 40.8 (C6H11), 38.8 (C6H11), 38.4 (C6H11), 37.7 (C6H11), 35.3 (C6H11), 33.3 (C6H11), 28.3 (C6H11), 28.1 (C6H11), 27.6 (C6H11), 27.5 (C6H11), 27.0 (C6H11), 27.0 (C6H11), 22.9 (ArCH3), 20.6 (ArCH3) ppm. FT-IR (Nujol): 3108 (w), 1590 (w), 1376 (m), 1283 (m), 1268 (m), 1235 (m), 1104 (w), 1012 (m), 800 (s), 759 (m), 718 (w) cm−1. MS (EI) m/z = 543 (M+). HRMS (EI): calcd, m/z = 543.2439; found, m/z = 543.2443.
(E)-Cp2ZrN(2,6-i-Pr2C6H3)C(=CHPh)C(H)Ph (10).
To a solution of Cp2Zr=N(2,6-i-Pr2C6H3)(THF)52 (0.082 g, 0.18 mmol) in toluene/pentane (1 mL/9 mL) was added a solution of 5a (0.033 g, 0.18 mmol) in pentane (2 mL). After 20 min at room temperature, the resulting deep blue solution was concentrated to dryness. Crystallization of the residue by diffusion of pentane into a concentrated toluene solution at −30 ° C afforded pure 10 as blue blocky crystals (0.062 g, 60%). One-sixth of an equivalent of toluene remained trapped in the crystals of 10, even after extended exposure to high vacuum, as determined by 1H NMR spectroscopy. 1H NMR (C6D6, 500 MHz): δ 6.92–7.15 (m, 9H, ArH), 6.77–6.82 (m, 4H, ArH), 5.87 (s, 5H, C5H5), 5.69 (s, 5H, C5H5), 5.58 (br d, J = 2 Hz, 1H, =CH), 4.56 (br d, J = 2 Hz, 1H, ZrCH), 3.14 (sept, J = 7 Hz, 1H, CH(CH3)2), 3.04 (sept, J = 7 Hz, 1H, CH(CH3)2), 2.10 (s, 0.5 H, C6H5CH3), 1.37 (d, J = 7 Hz, 3H, CH(CH3)2), 1.35 (d, J = 7 Hz, 3H, CH(CH3)2), 1.90 (d, J = 7 Hz, 3H, CH(CH3)2), 1.12 (d, J = 7 Hz, CH(CH3)2) ppm. 13C NMR (C6D6, 125 MHz): δ 149.7 (NC=C), 148.2 (Caryl), 144.3 (Caryl), 144.1 (Caryl), 140.6 (Caryl), 139.0 (Caryl), 129.7 (Caryl), 129.2 (Caryl), 128.9 (Caryl), 128.0 (Caryl), 127.7 (Caryl), 127.1 (Caryl), 125.9 (Caryl), 124.8 (Caryl), 123.9 (Caryl), 123.6 (Caryl), 121.4 (Caryl), 115.1 (C5H5), 113.6 (C5H5), 107.9 (=C(H)Ph), 53.1 (ZrC), 21.1 (CH(CH3)2), 28.7 (CH(CH3)2), 26.1 (CH(CH3)2), 26.0 (CH(CH3)2), 26.0 (CH(CH3)2, 25.9 (CH(CH3)2) ppm. FT-IR (Nujol): 3046 (w), 1592 (s), 1536 (m), 1379 (m), 1320 (s), 1254 (s), 1204 (m), 1115 (w), 1065 (w), 1015 (w), 910 (m), 801 (s), 768 (m), 688 (m), 629 (m) cm−1. Anal. Calcd for ZrNC37H39(C7H8)1/6: C, 75.86; H, 6.73; N, 2.32. Found: C, 76.20; H, 6.92; N, 2.21.
(R)-2,6-Dideutero-2,6-dimethyl-4-heptyn-3-ol (13-d2).
A flask containing Zn(OTf)2 (364 mg, 1 mmol) was heated with a heat gun under vacuum until gas evolution ceased and was then allowed to cool to room temperature. (+)-N-Methylephedrine (198 mg, 1.1 mmol) was added, and the flask was evacuated for 5 min. The flask was refilled with N2, and toluene (5 mL) and NEt3 (250 mg, 2.5 mmol) were added. After the mixture was stirred for 2 h, 3-methyl-1-butyne (0.767 mL, 5 mmol) was added, followed by isobutyraldehyde (0.460 mL, 5 mmol). The flask was stoppered, and the mixture heated to 60 ° C for 12 h. After being cooled to room temperature, saturated NH4Cl was added, and the mixture was extracted with Et2O (3×). The combined organic phases were washed with brine, dried (MgSO4), concentrated, and chromatographed (15% Et2O/pentane) to give the product as a colorless liquid (491 mg, 71%). 1H NMR matches literature values for 13-d0,53 minus the two methine protons, which were observed in the 2H NMR spectrum. 1H NMR (CDCl3, 400 MHz): δ 4.15 (s, 1H, CHOH), 1.69 (s, 1H, CHOH), 1.16 (s, 6H, CH(CH3)2), 0.98 (s, 3H, CH(CH3)2), 0.96 (s, 3H, CH(CH3)2) ppm. 2H NMR (CDCl3, 61.6 MHz): δ 2.65 (br s), 1.91 (br s) ppm.
(S)-2,6-Dideutero-2,6-dimethyl-3,4-heptadiene (5c-d2).
To a solution of PPh3 (1.55 g, 6 mmol) in THF (20 mL) at 0 ° C was added diethylazodicarboxylate (0.932 mL, 6 mmol), and the resulting mixture was stirred for 10 min. A solution of (R)-13-d2 (691 mg, 5 mmol) in THF (15 mL) was added, and the resulting mixture was stirred for 10 min. A solution of o-nitrobenzenesulfonylhydrazide54 (1.285 g, 6 mmol) in THF (15 mL) was added, and the resulting mixture was stirred for 1 h. The mixture was warmed to room temperature and stirred for 16 h. The solution was concentrated and chromatographed (pentane) to give the product as a colorless liquid (350 mg, 56%). 1H NMR (CDCl3, 300 MHz): δ 5.14 (s, 2H, =CH), 0.98 (s, 12H, CD(CH3)2). GC-MS (CI) m/z 126 (M+), 111 (MCH3+), >90% d2; GC: >95% ee (B-DM (30 m); 65 ° C; 0.8 mL/min) tr (R) = 5.6 min, tr (S) = 5.9 min. [α]D = + 91° (EtOH, c = 0.45).
Cp2ZrN(2,6-Me2C6H3)C(C4H9)=C(H)C(H)C2H5 (14).
To a solution of 3 (0.103 g, 0.25 mmol) in benzene (5 mL) was added a solution of 5b (0.031 g, 0.25 mmol) in benzene (2 mL). Evolution of a deep purple color occurred over several minutes at ambient temperature, followed by a slow fading to orange-brown over 2 d. Concentration of the reaction mixture gave a dark oil, which was extracted into pentane (10 mL). Filtration followed by solvent removal under reduced pressure afforded 14 as a yellow-brown oily solid (0.100 g, 83%). Several attempts to crystallize 14 to analytical purity were unsuccessful. 1H NMR (C6D6, 500 MHz): δ 7.09 (d, J = 7 Hz, 1H, m-ArH), 6.96 (d, J = 7 Hz, 1H, m-ArH), 6.90 (t, J = 7 Hz, 1H, p-ArH), 5.50 (s, 5H, C5H5), 5.34 (s, 5H, C5H5), 4.55 (d, J = 7 Hz, 1H, =CH), 2.45 (m, 1H, ZrCH), 2.37 (s, 3H, ArCH3), 2.12 (m, 1H, CH2), 1.86 (m, 2H, CH2), 1.74 (s, 3H, ArCH3), 1.38 (m, 2H, CH2), 1.27 (t, 3H, 7 Hz, CH3), 1.09 (m, 2H, CH2), 0.73 (t, J = 7 Hz, CH3), 0.49 (m, 1H, CH2) ppm. 13C NMR (C6D6, 125 MHz): δ 149.4 (NC=C), 133.2 (C6H3), 132.7 (C6H3), 132.3 (C6H3), 129.3 (C6H3), 129.1 (C6H3), 124.0 (C6H3), 107.0 (C5H5), 105.4 (=CH), 104.1 (C5H5), 69.0 (ZrC), 34.2 (CH2), 31.1 (CH2), 30.8 (CH2), 23.4 (CH3), 21.6 (CH2), 19.3 (CH3), 18.1 (CH3), 15.9 (CH3) ppm. FT-IR (Nujol): 1592 (w), 1522 (w), 1375 (s), 1281 (m), 1233 (m), 1189 (w), 1111 (w), 1095 (w), 1015 (m), 793 (s), 763 (m) cm−1. MS (EI) m/z = 463 (M+). HRMS (EI) calcd, m/z = 463.1821; found, m/z = 463.1817.
Cp2ZrN(2,6-Me2C6H3)C(CH2CH(CH3)2)=C(H)CH(CH3)2 (15).
To a solution of 3 (0.092 g, 0.22 mmol) in benzene (8 mL) was added a solution of 5c (0.033 g, 0.27 mmol) in benzene (2 mL). The mixture was heated at 75 ° C for 11 h. Solvent was removed under reduced pressure, and the resulting solid was precipitated from a concentrated ether solution at −30 ° C to afford spectroscopically pure 15 as a bright yellow solid (0.033 g, 33%). Several attempts to crystallize 15 to analytical purity were unsuccessful. 1H NMR (C6D6, 500 MHz): δ 7.04 (d, J = 7.5 Hz, 1H, m-ArH), 6.94 (d, J = 7.5 Hz, 1H, m-ArH), 6.89 (t, J = 7.5 Hz, 1H, p-ArH), 5.54 (s, 5H, C5H5), 5.42 (s, 5H, C5H5), 4.29 (s, 1H, =CH), 2.20 (s, 3H, ArCH3), 2.08 (s, 3H, ArCH3), 1.89 (s, 3H, ZrC(CH3)2), 1.68 (m, 3H, CH2CH), 1.34 (s, 3H, ZrC(CH3)2), 0.88 (d, J = 6.5 Hz, 3H, CH(CH3)2), 0.80 (d, J = 6.5 Hz, 3H, CH(CH3)2) ppm. 13 C NMR (C6D6, 125 MHz): δ 148.9 (NC=C), 138.6 (C6H5), 133.5 (C6H5), 132.8 (C6H5), 129.7 (C6H5), 129.4 (C6H5), 124.3 (C6H5), 108.3 (C5H5), 106.3 (=CH), 106.1 (C5H5), 60.0 (ZrC), 44.0 (CH2), 34.4 (ZrC-(CH3)2), 29.1 (ZrC(CH3)2), 27.2 (CH(CH3)2), 23.8 (CH(CH3)2), 23.4 (CH(CH3)2), 22.6 (ArCH3), 20.6 (ArCH3) ppm. FT-IR (Nujol): 3089 (w), 1591 (w), 1264 (m), 1236 (m), 1097 (w), 1080 (w), 1022 (w), 796 (s), 769 (m) cm−1. MS (EI) m/z = 463 (M+). HRMS (EI): calcd, m/z = 463.1814; found, m/z = 463.1817.
Cp2ZrN(2,6-Me2C6H3)C(CH2C6H11)=C(H)C(CH2)5 (16).
To a solution of 3 (0.112 g, 0.27 mmol) in benzene (8 mL) was added a solution of 5d (0.059 g, 0.29 mmol) in benzene (2 mL). The mixture was stirred at room temperature for 12 h and at 75 ° C for 27 h. The solvent was removed under vacuum to afford a yellow-brown sticky solid, which was precipitated from a concentrated ether solution to afford 16 as an orange solid (0.097 g, 65%). Several attempts to crystallize 16 to analytical purity were unsuccessful. 1H NMR (C6D6, 500 MHz): δ 7.06 (d, J = 7 Hz, 1H, m-ArH), 6.97 (d, J = 7 Hz, 1H, m-ArH), 6.90 (t, J = 7 Hz, 1H, p-ArH), 5.54 (s, 5H, C5H5), 5.38 (s, 5H, C5H5), 4.41 (s, 1H, =CH), 2.23 (s, 3H, ArCH3), 1.99 (s, 3H, ArCH3), 0.74–2.15 (m, 23H, C6H11, CH2C6H11) ppm. 13C NMR (C6D6, 125 MHz): δ 149.9 (NC=C), 137.6 (C6H3), 134.1 (C6H3), 133.7 (C6H3), 130.7 (C6H3), 130.5 (C6H3), 125.3 (C6H3), 113.2 (NC=C), 109.2 (C5H5), 106.7 (C5H5), 73.0 (ZrC), 44.1 (=CCH2), 43.6 (CH2), 39.1 (CH2), 38.2 (CH2), 35.8 (CH2), 35.3 (CH2), 34.5 (CH2), 30.2 (CH2), 29.4 (CH2), 28.2 (CH2), 28.1 (CH2), 27.9 (CH2), 23.5 (ArCH3), 21.0 (ArCH3) ppm. FT-IR (Nujol): 3071 (w), 1591 (m), 1513 (w), 1376 (m), 1265 (m), 1236 (m), 1102 (w), 1013 (m), 981 (w), 795 (s), 765 (m), 718 (w), 692 (w) cm−1. MS (EI): m/z = 543 (M+). HRMS (EI) calcd, m/z = 543.2442; found, m/z = 543.2443.
Cyclononadiene Metallacycle 17.
To a solution of 3 (0.103 g, 0.25 mmol) in C6D6 (1 mL) was added a solution of 1,2-cyclononadiene55 (0.033 g, 0.27 mmol) in C6D6 (1 mL). The resulting purple solution was stirred for 1 h at room temperature. The solvent was removed under vacuum, and the resulting purple solid was washed with pentane (2 × 2 mL) to give pure 17 by 1H NMR spectroscopy. An analytically pure sample was obtained by recrystallizing from saturated pentane. 1H NMR (C6D6, 500 MHz): δ 7.07 (d, J = 8 Hz, 1H, m-ArH), 7.06 (d, J = 8 Hz, 1H, m-ArH), 6.88 (t, J = 8 Hz, 1H, p-ArH), 5.82 (s, 5H, C5H5), 5.70 (s, 5H, C5H5), 4.87 (t, J = 8 Hz, 1H, =CH), 2.6–2.5 (m, 2H), 2.29 (t, J = 10 Hz, 1H, ZrCH), 2.23 (s, 3H, ArCH3), 2.2–2.1 (m, 2H), 2.00 (s, 3H, ArCH3), 2.0–1.9 (m, 2H), 1.9–1.8 (m, 1H), 1.8–1.7 (m, 1H), 1.5–1.3 (m, 4H) ppm. 13C NMR (C6D6, 125 MHz): δ 151.7 (NC= C), 136.3 (C6H3), 131.0 (C6H3), 129.8 (C6H3), 128.3 (C6H3), 121.6 (C6H3), 111.2 (C5H5), 110.9 (C5H5), 110.5 (=CH), 57.5 (ZrC), 36.0 (CH2), 33.0 (CH2), 32.1 (CH2), 27.7 (CH2), 25.2 (CH2), 25.0 (CH2), 21.4 (ArCH3), 20.1 (ArCH3), 15.5 (CH3), 14.8 (CH3) ppm. Anal. Calcd for ZrNC27H33: C, 70.1; H, 7.2; N, 3.0. Found: C, 70.36; H, 7.10; N, 2.99.
Cyclononadiene-Derived MAD Complex 18.
A solution of 17 (115 mg, 0.25 mmol) in benzene (4 mL) was heated at 75 ° C for 11 h. Solvent was removed under vacuum, and the resulting solid was crystallized by slow diffusion of Et2O into a THF solution to give pure bright yellow solid 18 as a 3:1 mixture of two isomers (65 mg, 57%). Major isomer, 1H NMR (C6D6, 500 MHz): δ 7.0–6.8 (m, 3H, ArH), 5.58 (s, 5H, C5H5), 5.28 (s, 5H, C5H5), 3.93 (s, 1H, J = 11 Hz, =CH), 2.98 (ddd, 1H, J = 5, 11, 13 Hz, ZrCH), 2.24 (s, 3H, ArCH3), 1.90 (s, 3H, ArCH3), 2.3–1.6 (m, 9H), 1.3–1.2 (m, 1H), 1.1–1.0 (m, 1H), 0.87 (q, 1H, J = 13 Hz, ZrCHCH2) ppm. 13C NMR (C6D6, 125 MHz): δ 150.4 (NC=C), 149.0 (C6H5), 134.0 (C6H5), 132.6 (C6H5), 129.3 (C6H5), 128.2 (C6H5), 124.5 (C6H5), 106.2 (C5H5), 104.6 (C5H5), 84.1 (=CH), 60.8 (ZrC), 34.4 (CH2), 31.2 (CH2), 31.1 (CH2), 30.0 (CH2), 30.1 (CH2), 21.4 (ArCH3), 18.5 (ArCH3) ppm. Minor isomer, 1H NMR (C6D6, 500 MHz): δ 7.08 (d, 1H, J = 8 Hz, m-ArH), 7.0–6.8 (m, 2H, ArH), 5.46 (s, 5H, C5H5), 5.28 (s, 5H, C5H5), 3.97 (s, 1H, J = 9 Hz, =CH), 2.65 (br d, 1H, J = 12 Hz, ZrCHCH2), 2.4–2.3 (m, 1H), 2.23 (s, 3H, ArCH3), 1.68 (s, 3H, ArCH3), 2.2–1.4 (m, 9H), −0.41 (br t, 1H, J = 10 Hz, ZrCH) ppm. 13C NMR (C6D6, 125 MHz): δ 148.7 (NC=C), 145.2 (C6H5), 132.6 (C6H5), 131.2 (C6H5), 128.7 (C6H5), 128.5 (C6H5), 123.8 (C6H5), 105.1 (C5H5), 102.0 (C5H5), 87.5 (=CH), 60.8 (ZrC), 36.6 (CH2), 35.4 (CH2), 32.8 (CH2), 32.6 (CH2), 30.1 (CH2), 26.1 (CH2), 21.5 (ArCH3), 21.3 (ArCH3) ppm. Anal. Calcd for ZrNC27H33: C, 70.1; H, 7.2; N, 3.0. Found: C, 70.39; H, 6.95; N, 2.98.
(R,R)-(ebthi)ZrN(2,6-Me2C6H3)C=(CHC6H5)(CHC6H5) (19).
To a suspension of (R,R)-(ebthi)ZrN(2,6-Me2C6H3)(THF) (1) (102 mg, 0.19 mmol) in benzene (8 mL) was added (R)-1,3-diphenylpropadiene (40 mg, 0.21 mmol), and the mixture was stirred for 1 h. The solvent was removed under vacuum, and the purple solid was recrystallized from a CH2Cl2/pentane mixture to yield the product as purple needles (100 mg, 81%). 1H NMR (C6D6, 400 MHz): δ 7.4–7.3 (m, 5H, ArH), 7.2–6.9 (m, 7H, ArH), 6.75 (t, J = 7.2 Hz, 1H, p-Ar), 5.80 (s, 1H, = CHPh), 5.60 (d, J = 2.8 Hz, 1H, ebthi), 5.48 (d, J = 2.8 Hz, 1H, ebthi), 5.28 (d, J = 2.8 Hz, 1H, ebthi), 4.90 (d, J = 2.8 Hz, 1H, ebthi), 4.56 (s, 1H, ZrCHPh), 2.67 (s, 3H, ArCH3), 2.6–2.2 (m, 14 H, ebthi), 2.03 (s, 3H, ArCH3), 1.9–1.8 (m, 1H), 1.6–1.5 (m, 1H), 1.3–1.0 (m, 3 H), 0.9–0.8 (m, 1H) ppm. 13C NMR (C6D6, 100 MHz): δ 152.9, 148.2, 140.8, 137.6, 136.4, 133.6, 132.5, 130.9, 130.4, 129.9, 129.6, 128.9, 128.3, 128.1, 126.6, 125.3, 123.4, 122.7, 122.5, 121.8, 121.2, 116.7, 112.7, 110.2, 106.5, 105.6, 60.5, 28.2, 26.9, 24.8, 24.2, 23.7, 23.5, 23.5, 23.1, 22.9, 22.6, 22.5, 21.0 ppm. IR (Nujol): 3066, 2879, 1587, 1507, 1270, 1030, 944, 778, 699 cm−1. Anal. Calcd for C43H45- ZrN·CH2Cl2: C, 70.28; H, 6.30; N, 1.86. Found: C, 70.17; H, 6.69; N, 1.86.
After a solution of this compound was heated in C6D6 at 105 ° C for 24 h, an equilibrium mixture of 19 and a new compound, assigned as 21 on the basis of signals at 6.54 (d), 6.49 (d, =CHPh), 5.61 (d), 5.54 (d), 4.84 (d), and 3.81 (d, ZrCHPh), were formed in a 3:1 ratio.
(R,R)-(ebthi)ZrN(2,6-Me2C6H3)C=(CHC6H5)(CHC6H5) (20).
To a solution of (R,R)-(ebthi)ZrN(2,6-Me2C6H3)(THF) (27 mg, 0.05 mmol) in toluene (2 mL) was added (S)-1,3-diphenylpropadiene (12 mg, 0.0625 mmol), and the mixture was stirred for 1 h. The solvent was removed under vacuum. The solid was redissolved in toluene (2 mL), and the solvent was removed under vacuum three more times to remove the THF. 1H NMR (C6D6, 500 MHz): δ 7.45 (d, J = 7 Hz, 1H, ArH), 7.2–6.8 (m, 12 H, ArH), 6.31 (d, J = 3 Hz, 1H, ebthi), 5.85 (d, J = 2.5 Hz, 1H, ebthi), 5.81 (s, 1H, =CHPh), 5.30 (d, J = 2.5 Hz, 1H, ebthi), 5.01 (d, J = 3.0 Hz, 1H, ebthi), 4.94 (s, 1H, ZrCHPh), 2.7–2.4 (m, 8H), 2.33 (s, 3H, ArCH3), 2.19 (s, 3H, ArCH3), 2.1–2.0 (m, 4H), 1.3–0.8 (m, 8H).
Kinetic Study of the Rearrangement of 20.
After heating at 45 ° C for 8 h, in addition to signals corresponding to 19, 20, and 21, signals at 6.53 (d, =CHPh), 6.36 (d), 6.14 (d), 5.44 (d), 5.07 (d), and 4.92 (d, ZrCHPh), assigned as 22, appeared. The concentrations of 19, 20, 21, and 22 were determined by 1H NMR spectroscopy relative to an internal standard (Cp2Fe) and were monitored as a function of time at 45, 55, and 65 ° C. The data were fitted to the kinetic scheme in Scheme 7 with the aid of the Gepasi kinetic modeling program.31–33
Cp2Zr(SnMe3)N(2,6-Me2C6H3)C=CHPh(CH2Ph) (23).
To a solution of metallacycle 6 (133 mg, 0.25 mmol) in benzene (2 mL) was added Me3SnH (49 mg, 0.3 mmol), and the resulting mixture was stirred for 18 h at room temperature. The volatile materials were removed under vacuum to give a yellow powder. Recrystallization from toluene/pentane gave pure 23 as yellow blocks (89 mg, 51%). 1H NMR (C6D6, 500 MHz): δ 7.48 (d, J = 7 Hz, 2H, ArH), 7.2–7.1 (m, 2H, ArH), 7.01 (t, J = 8 Hz, 1H, ArH), 6.97 (br s, 1H, ArH), 6.81 (t, J = 8 Hz, 1H, ArH), 6.8–6.7 (m, 3H, ArH), 6.52 (br s, 1H, ArH), 6.38 (br s, 2H, ArH), 5.85 (br s, 1H, =CH), 5.68 (br s, 5H, C5H5), 5.35 (br s, 5H, C5H5), 4.10 (br d, J = 11 Hz, 1H, NCCH2), 2.57 (br d, J = 11 Hz, NCCH2), 2.26 (br s, 3H, ArCH3), 1.65 (br s, 3H, ArCH3), 0.33 (s, 9H, JSn-H = 15 Hz) ppm. 13C NMR (C6D6, 125 MHz): δ 152.0, 151.3, 138.3, 136.1, 132.0, 130.2, 129.0, 128.9, 128.5, 128.4, 128.3, 127.6, 126.1, 125.6, 125.1, 107.6 (C5H5), 105.7 (C5H5), 101.6 (=CHPh), 35.6 (NCCH2), 21.4 (ArCH3), 18.8 (ArCH3), −5.8 (Sn(CH3)3). Anal. Calcd for C36H41ZrNSn: C, 62.0; H, 5.9; N, 2.0. Found: C, 62.19; H, 6.07; N, 2.04.
Cp2Zr(SC6H4CF3)N(2,6-Me2C6H3)C=CHPh(CH(SC6H4CF3)-Ph) (24).
To a solution of metallacycle 6 (67 mg, 0.25 mmol) in benzene (2 mL) was added bis(4-trifluoromethylphenyl) disulfide (44 mg, 0.3 mmol), and the resulting mixture was stirred for 18 h at room temperature. The volatile materials were removed under vacuum to give an orange powder. Recrystallization from toluene/pentane gave pure 24 as an orange powder (52 mg, 47%). 1H NMR (C6D6, 500 MHz): δ 7.42 (br s, 2H, ArH), 7.28 (s, 1H, ArSCHPh), 7.2–7.0 (m, 5H, ArH), 6.96 (br s, 4H, ArH), 6.9–6.8 (m, 2H, ArH), 6.80 (d, J = 8 Hz, 2H, ArH), 6.74 (br s, 5H, ArH), 6.63 (d, J = 8 Hz, 2H, ArH), 6.20 (br s, 5H, C5H5), 5.47 (br s, 5H, C5H5), 4.97 (s, 1H, =CH), 2.55 (s, 3H, ArCH3), 2.10 (s, 3H, ArCH3) ppm. 13C NMR (C6D6, 125 MHz): δ 155.9, 151.8, 151.3, 143.8, 138.0, 137.5, 137.4, 134.9, 134.1, 130.2, 129.9, 128.8, 126.5, 126.3, 126.2, 125.8, 125.4, 125.1, 113.6 (C5H5), 112.1 (C5H5), 47.9 (ArSCHPh), 20.1 (ArCH3), 19.9 (ArCH3) ppm. Anal. Calcd for C47H39ZrNS2F6: C, 63.6; H, 4.4; N, 1.6. Found: C, 63.61; H, 4.56; N, 1.77.
Cp2Zr(OCH2Ph)N(2,6-Me2C6H3)C9H13 (25).
To a solution of metallacycle 14 (115 mg, 0.25 mmol) in toluene (2 mL) was added benzaldehyde (27 mg, 0.25 mmol), and the resulting solution was stirred for 1 h at room temperature. The solvent was removed under vacuum to give an orange oil, which was dissolved in pentane (1 mL) and kept at −30 ° C for 5 d. Orange crystals of 25 were isolated after decanting the solvent and washing with cold pentane (80 mg, 56%). 1H NMR (C6D6, 400 MHz): δ 7.4–7.2 (m, 4H, ArH), 7.2–7.1 (m, 3H, ArH), 6.99 (t, J = 7 Hz, 1H, p-ArH), 5.97 (br s, 5H, C5H5), 5.80 (br s, 5H, C5H5), 5.54 (br d, J = 16 Hz, 1H, =CH), 5.42 (br s, 1H, =CH), 5.28 (br s, 1H, =CH), 4.92 (s, 2H, OCH2Ph), 2.4–2.3 (m, 1H), 2.39 (s, 3H, ArCH3), 2.31 (s, 3H, ArCH3), 2.1–1.9 (m, 2H), 1.8–1.6 (m, 2H), 1.6–1.4 (m, 2H), 1.3–1.1 (m, 2H) ppm. 13C NMR (C6D6, 125 MHz): δ 156.8, 153.5, 143.6, 135.6, 135.2, 128.7, 128.5, 128.4, 127.1, 126.8, 124.1, 114.0 (=CH), 113.0 (=CH), 112.2 (C5H5), 111.9 (C5H5), 111.8 (NC=CH), 75.9 (OCH2Ph), 33.1, 31.3, 28.7, 28.5, 20.3 (ArCH3), 19.9 (ArCH3) ppm. Anal. Calcd for C34H37ZrON: C, 72.0; H, 6.6; N, 2.5. Found: C, 71.81; H, 6.74; N, 2.49.
Cp2Zr(OCHMePh)N(2,6-Me2C6H3)C9H13 (26).
To a solution of metallacycle 14 (115 mg, 0.25 mmol) in toluene (2 mL) was added acetophenone (33 mg, 0.27 mmol), and the resulting solution was stirred for 1 h at room temperature. The solvent was removed under vacuum to give an orange oil, which was dissolved in pentane (1 mL) and kept at −30 ° C for 5 d. Orange crystals of 26 were isolated after decanting the solvent and washing with cold pentane (87 mg, 60%). 1H NMR (C6D6, 400 MHz): δ 7.31 (d, J = 7 Hz, 2H, ArH), 7.26 (t, J = 7 Hz, 2H, ArH), 7.2–7.0 (m, 3H, ArH), 6.99 (t, J = 7 Hz, 1H, ArH), 5.88 (br s, 5H, C5H5), 5.75 (br s, 5H, C5H5), 5.55 (br s, 1H, =CH), 5.4–5.2 (m, 2H, =CH), 4.89 (q, J = 6 Hz, 1H, OCHCH3), 2.5–2.4 (m, 1H), 2.36 (s, 3H, ArCH3), 2.28 (s, 3H, ArCH3), 2.1–2.0 (m, 2H), 1.7–1.6 (m, 2H), 1.6–1.4 (m, 3H), 1.37 (br s, 3H, OCHCH3), 1.3–1.1 (m, 2H) ppm. 13C NMR (343 K, C6D6, 100 MHz): δ 153.6 (NC=CH), 147.9, 135.5, 135.2, 134.9, 134.0, 129.3, 128.5, 127.3, 126.3, 124.1, 111.8 (C5H5), 81.7 (OCHCH3Ph), 33.0, 31.3, 28.8, 28.4, 28.3, 26.9 (OCHCH3-Ph), 20.1 (ArCH3) ppm. Anal. Calcd for C35H41ZrON: C, 72.1; H, 7.1; N, 2.4. Found: C, 71.91; H, 7.09; N, 2.70.
Cp2Zr(OCH(C6H4Cl)2)N(2,6-Me2C6H3)C9H13 (27).
To a solution of metallacycle 14 (58 mg, 0.13 mmol) in toluene (2 mL) was added 4,4′ -dichlorobenzophenone (63 mg, 0.25 mmol), and the resulting mixture was stirred for 1 h at room temperature. The solvent was removed under vacuum to give an orange oil, which was dissolved in pentane (1 mL) and kept at −30 ° C for 5 d. Orange crystals of 27 were isolated after decanting the solvent and washing with cold pentane (40 mg, 45%). 1H NMR (C6D6, 500 MHz): δ 7.2–7.1 (m, 3H, ArH), 7.1–7.0 (m, 5H, ArH), 6.99 (t, J = 8 Hz, 1H, p-ArH), 5.82 (br s, 5H, C5H5), 5.72 (br s, 5H, C5H5), 5.64 (s, 1H, OCHAr2), 5.4–5.2 (m, 3H, =CH), 2.36 (s, 3H, ArCH3), 2.30 (s, 3H, ArCH3), 2.0–1.8 (m, 2H), 1.6–1.5 (m, 2H), 1.5–1.3 (m, 3H), 1.2–1.1 (m, 2H) ppm. 13C NMR (343 K, C6D6, 125 MHz): δ 156.8 (NC=CH), 153.6, 145.5, 135.2, 134.6, 133.3, 131.8, 128.8, 128.6, 128.6, 128.1, 124.4, 119.2, 112.3 (C5H5), 86.2 (OCHAr2), 33.0, 31.1, 28.8, 28.5, 28.4, 20.0 (ArCH3) ppm. Anal. Calcd for C40H40ZrONCl2: C, 67.4; H, 5.7; N, 2.0. Found: C, 67.65; H, 5.70; N, 2.26.
Dicyclopropylallene Metallacycle 29.
To a solution of imido complex 6 (103 mg, 0.25 mmol) in toluene (2 mL) was added 1,3-dicyclopropylallene (33 mg, 0.27 mmol), and the resulting mixture was stirred for 1 h at room temperature. The solvent was removed under vacuum to give an orange-red powder (136 mg, quant.). An analytically pure sample of 29 was isolated by crystallization from toluene/pentane. 1H NMR (C6D6, 500 MHz): δ 7.12 (d, J = 7 Hz, 1H, m-ArH), 7.08 (d, J = 7 Hz, 1H, m-ArH), 6.97 (t, J = 7 Hz, 1H, p-ArH), 6.65 (d, J = 15 Hz, 1H, NCCH=CH), 5.92 (ddd, J = 3, 12, 15 Hz, 1H, NCCH= CH), 5.75 (s, 5H, C5H5), 5.37 (s, 5H, C5H5), 3.04 (dd, J = 1, 8 Hz, 1H, NC=CH), 2.67 (ddd, J = 3, 9, 12 Hz, 1H, ZrCH2CH2), 2.28 (s, 3H, ArCH3), 2.13 (s, 3H, ArCH3), 2.10 (dq, J = 7, 12 Hz, 1H, ZrCH2CH2), 1.26 (quin, J = 7 Hz, 1H, ZrCH2CH2), 0.64 (q, J = 11 Hz, 1H, ZrCH2CH2), 0.7–0.6 (m, 1H, C3H5), 0.6–0.5 (m, 1H, C3H5), 0.3–0.2 (m, 1H, C3H5), 0.2–0.1 (m, 1H, C3H5), 0.1–0.0 (m, 1H, C3H5) ppm. 13C NMR (C6D6, 125 MHz): δ 154.5 (NC=CH), 147.4, 134.3, 134.2, 129.7, 128.9, 128.8, 123.8 (=CH), 120.7 (=CH), 110.7 (C5H5), 108.2 (C5H5), 96.0 (NC=CH), 30.9 (ZrCH2CH2), 20.3 (ArCH3), 20.1 (ArCH3), 17.1 (ZrCH2CH2), 10.2 (CH(CH2)2), 7.9 (CH(CH2)2), 7.7 (CH-(CH2)2) ppm. Anal. Calcd for C27H31ZrN: C, 70.4; H, 6.8; N, 3.0. Found: C, 70.09; H, 7.09; N, 3.13.
Dicyclopropylallene Metallacycle 30.
To a solution of Cp2Zr= N(2,6-i-Pr2C6H3)(THF)52 (117 mg, 0.25 mmol) in toluene (2 mL) was added 1,3-dicyclopropylallene (33 mg, 0.27 mmol), and the resulting mixture was stirred for 1 h at room temperature. The solvent was removed under vacuum to give an orange-red powder. Orange/red crystals of pure 30 were isolated by crystallization from pentane at −30 ° C (89 mg, 69%). 1H NMR (C6D6, 500 MHz): δ 7.22 (dd, J = 3, 7 Hz, 1H, m-ArH), 7.2–7.1 (m, 2H, ArH), 6.62 (d, J = 15 Hz, 1H, NCCH=CH), 6.01 (t, J = 15 Hz, 1H, NCCH=CH), 5.83 (s, 5H, C5H5), 5.53 (s, 5H, C5H5), 3.48 (quin, J = 7 Hz, 1H, ArCH(CH3)2), 3.17 (quin, J = 7 Hz, 1H, ArCH(CH3)2), 2.82 (dd, J = 1, 8 Hz, 1H, ZrCH2CH2), 2.69 (t, J = 9 Hz, 1H, ZrCH2CH2), 2.13 (dq, J = 7, 12 Hz, 1H, ZrCH2-CH2), 1.41 (d, J = 7 Hz, 3H, ArCH(CH3)2), 1.32 (d, J = 7 Hz, 3H, ArCH(CH3)2), 1.31 (d, J = 7 Hz, 3H, ArCH(CH3)2), 1.28 (d, J = 7 Hz, 3H, ArCH(CH3)2), 0.72 (q, J = 11 Hz, 1H, ZrCH2CH2), 0.7–0.6 (m, 1H, C3H5), 0.6–0.5 (m, 1H, C3H5), 0.3–0.2 (m, 1H, C3H5), 0.2–0.1 (m, 1H, C3H5), 0.1–0.0 (m, 1H, C3H5) ppm. 13C NMR (C6D6, 125 MHz): δ 151.6, 149.6, 144.8, 144.5, 127.6, 124.8, 124.7, 123.7, 120.5, 110.6 (C5H5), 108.0 (C5H5), 98.3 (NC=CH), 31.1 (ZrCH2CH2), 27.7, 27.0, 26.6, 25.6, 24.6, 23.9, 17.9 (ZrCH2CH2), 10.1 (CH(CH2)2), 7.6 (CH(CH2)2), 7.4 (CH(CH2)2) ppm. Anal. Calcd for C31H39ZrN: C, 72.0; H, 7.6; N, 2.7. Found: C, 71.83; H, 7.82; N, 2.73.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health through Grant no. GM-25459 and a National Research Service Award Fellowship to Dr. Forrest Michael. We thank Dr. Anna Davis and Professor Kenneth Raymond for assistance with CD spectroscopy, Professor Armin deMeijere for the generous gift of 1,3-dicyclopropylallene, and Dr. Fred Hollander and Dr. Allen Oliver of the UC Berkeley CHEXRAY facility for the X-ray structure determinations.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for synthesis of allenes, CD spectra of 6–8, and crystallographic data for 10, 18a, 23, and 30 (PDF, CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Muller TE, Beller M. Transition Met Org Synth. 1998;2:316. [Google Scholar]
- 2.Pohlki F, Doye S. Chem Soc Rev. 2003;32:104. doi: 10.1039/b200386b. [DOI] [PubMed] [Google Scholar]
- 3.Nobis M, Driessen-Holscher B. Angew Chem, Int Ed. 2001;40:3983. doi: 10.1002/1521-3773(20011105)40:21<3983::aid-anie3983>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Bytschkov I, Doye S. Eur J Org Chem. 2003:935. [Google Scholar]
- 5.Roesky PW, Mueller TE. Angew Chem, Int Ed. 2003;42:2708. doi: 10.1002/anie.200301637. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Shi Y, Odom AL. J Am Chem Soc. 2004;126:1794. doi: 10.1021/ja038320g. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Schafer LL. Org Lett. 2003;5:4733. doi: 10.1021/ol0359214. [DOI] [PubMed] [Google Scholar]
- 8.Khedkar V, Tillack A, Beller M. Org Lett. 2003;5:4767. doi: 10.1021/ol035653+. [DOI] [PubMed] [Google Scholar]
- 9.Knight PD, Munslow I, O’Shaughnessy PN, Scott P. Chem Commun. 2004:894. doi: 10.1039/b401493f. [DOI] [PubMed] [Google Scholar]
- 10.Ackermann L. Organometallics. 2003;22:4367. [Google Scholar]
- 11.Ackermann L, Bergman RG, Loy RN. J Am Chem Soc. 2003;125:11956. doi: 10.1021/ja0361547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackermann L, Bergman RG. Org Lett. 2002;4:1475. doi: 10.1021/ol0256724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson JS, Bergman RG. J Am Chem Soc. 2001;123:2923. doi: 10.1021/ja005685h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708. [Google Scholar]
- 15.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michael FE, Duncan AP, Sweeney ZK, Bergman RG. J Am Chem Soc. 2003;125:7184. doi: 10.1021/ja0348389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elsevier CJ, Vermeer P. J Org Chem. 1989;54:3726. [Google Scholar]
- 18.Frantz DE, Faessler R, Carreira EM. J Am Chem Soc. 2000;122:1806. [Google Scholar]
- 19.Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 20.Boyall D, Frantz DE, Carreira EM. Org Lett. 2002;4:2605. doi: 10.1021/ol026282k. [DOI] [PubMed] [Google Scholar]
- 21.Myers AG, Zheng B. J Am Chem Soc. 1996;118:4492. [Google Scholar]
- 22.Davis JM, Whitby RJ, Jaxa-Chamiec A. J Chem Soc, Chem Commun. 1991:1743. [Google Scholar]
- 23.Scholz J, Nolte M, Krueger C. Chem Ber. 1993;126:803. [Google Scholar]
- 24.Scholz J, Kahlert S, Goerls H. Organometallics. 1998;17:2876. [Google Scholar]
- 25.Scholz J, Kahlert S, Goerls H. Organometallics. 2004;23:1594. [Google Scholar]
- 26.Scholz J, Goerls H. Organometallics. 2004;23:320. [Google Scholar]
- 27.Enders D, Kroll M, Raabe G, Runsink J. Angew Chem, Int Ed. 1998;37:1673. doi: 10.1002/(SICI)1521-3773(19980703)37:12<1673::AID-ANIE1673>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 28.Lorenz V, Gorls H, Scholz J. Angew Chem, Int Ed. 2003;42:2253. doi: 10.1002/anie.200250116. [DOI] [PubMed] [Google Scholar]
- 29.Erker G, Kruger C, Muller G. Adv Organomet Chem. 1985;24:1. [Google Scholar]
- 30.Yasuda H, Tatsumi K, Nakamura A. Acc Chem Res. 1985;18:120. [Google Scholar]
- 31.Mendes P. Comput Appl Biosci. 1993;9:563. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 32.Mendes P. Trends Biochem Sci. 1997;22:361. doi: 10.1016/s0968-0004(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 33.Mendes P, Kell DB. Bioinformatics. 1998;14:869. doi: 10.1093/bioinformatics/14.10.869. [DOI] [PubMed] [Google Scholar]
- 34.Hoyt H, Michael F, Bergman R. J Am Chem Soc. 2004;126:1018. doi: 10.1021/ja0385944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bazan GC, Rodriguez G, Cleary BP. J Am Chem Soc. 1994;116:2177. [Google Scholar]
- 36.Keaton RJ, Koterwas LA, Fettinger JC, Sita LR. J Am Chem Soc. 2002;124:5932. doi: 10.1021/ja017294o. [DOI] [PubMed] [Google Scholar]
- 37.Kissounko DA, Sita LR. J Am Chem Soc. 2004;126:5946. doi: 10.1021/ja0493897. [DOI] [PubMed] [Google Scholar]
- 38.This method has been used to synthesize MAD complexes from allylamido-(methyl)zirconium complexes, see ref 23.
- 39.Alibrandi G, Scolaro LM, Minniti D, Romeo R. Inorg Chem. 1990;29:3467. [Google Scholar]
- 40.Bryndza HE. J Chem Soc, Chem Commun. 1985:1696. [Google Scholar]
- 41.Evans J, Schwartz J, Urquhart PW. J Organomet Chem. 1974;81:C37. [Google Scholar]
- 42.Jordan RF, LaPointe RE, Bajgur CS, Echols SF, Willett R. J Am Chem Soc. 1987;109:4111. [Google Scholar]
- 43.Harlan CJ, Hascall T, Fujita E, Norton JR. J Am Chem Soc. 1999;121:7274. [Google Scholar]
- 44.Brown KL, Zhou L. Inorg Chem. 1996;35:5032. doi: 10.1021/ic9603666. [DOI] [PubMed] [Google Scholar]
- 45.Brown KL, Li J. J Am Chem Soc. 1998;120:9466. [Google Scholar]
- 46.Amaudrut J, Wiest O. J Am Chem Soc. 2000;122:3367. [Google Scholar]
- 47.Hunter WE, Hrncir DC, Bynum RV, Penttila RA, Atwood JL. Organometallics. 1983;2:750. [Google Scholar]
- 48.Schock LE, Marks TJ. J Am Chem Soc. 1988;110:7701. [Google Scholar]
- 49.Diogo HP, De Alencar Simoni J, Minas da Piedade ME, Dias AR, Martinho Simoes JA. J Am Chem Soc. 1993;115:2764. [Google Scholar]
-
50.DFT calculations at the BP86/LACVP level of theory were performed on the compounds in the following equation using Jaguar 5.5, Schrödinger, Inc., Portland, OR, 1991–2000.
- 51.Walsh PJ, Hollander FJ, Bergman RG. Organometallics. 1993;12:3705. [Google Scholar]
- 52.Zuckerman RL, Bergman RG. Organometallics. 2000;19:4795. doi: 10.1021/om010091o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baudin JB, Julia SA, Wang Y. Bull Soc Chim Fr. 1995;132:739. [Google Scholar]
- 54.Myers AG, Zheng B, Movassaghi M. J Org Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]
- 55.Skatteboel L, Solomon S. Org Synth. 1969;49:35. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.