

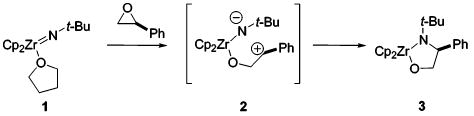
Imidometal complexes of zirconium and titanium often exhibit high levels of reactivity, activating carbon–hydrogen bonds1–3 and adding to carbon–carbon4 and carbon–heteroatom5 double and triple bonds. Several important questions remain about the mechanistic details of the reactions of these complexes. In particular, little is known about the roles of the nitrogen nucleophilicity and metal Lewis acidity. Epoxides proved to be ideal for our examination of these characteristics, reacting with zirconium imido complexes to provide cyclic ring-opened products, as well as providing a potential route to new synthetic methods. The results of our reactivity studies provide strong evidence for a stepwise mechanism and suggest that the Lewis acidity of the zirconium center and the nucleophilic behavior of the imido nitrogen play an important role in epoxide-opening reactions.
We have investigated the reactions of epoxides with two imido complexes: the achiral biscyclopentadienyl(tert-butylimido) zirconium complex 1, and its chiral analogue (ebthi)Zr=NAr(THF)4 (ebthi = ethylenebis(tetrahydroindenyl), Ar = 2,6-dimethylphenyl) 4. The reaction of styrene oxide with 1 proceeded with ring-opening and high regioselectivity, yielding metallacycle 3 (eq 1). The reaction of (rac)-4 with (S)-styrene oxide proceeded with similar regiochemistry. Notably, product (rac)-5 was formed as a single diastereomer, which implicated a pathway that allowed for scrambling of the original (S)-styrene oxide stereocenter. The structure of product 5 was confirmed by single-crystal X-ray diffraction (eq 2).
(1).
(2).
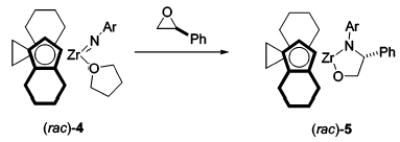
The high degree of regioselectivity contrasted with the much less selective, and probably synchronous, reactions of 1 with unsymmetrically substituted alkynes.6 On the basis of this contrast, and the fact that (rac)-5 was formed as a single diastereomer, it seemed likely that the epoxide reactions were proceeding by a stepwise mechanism involving coordination of the epoxide oxygen to zirconium, followed by Zr-assisted ring-opening to give zwitterion 2, and then ring-closure to 3.
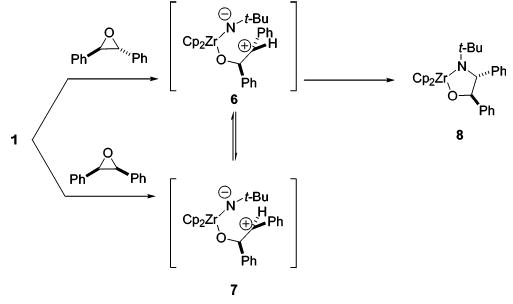
We sought additional evidence for this pathway by further examining the stereochemistry of the addition reaction. In separate experiments, compound 1 was added to a C6D6 solution of trans-stilbene oxide and to a C6D6 solution of cis-stilbene oxide (Scheme 1). 1H NMR spectra of both solutions obtained after 4 h at ambient temperature showed formation of the same metallacycle. We initially assigned this metallacycle as the trans isomer, since we theorized that the lower steric interaction between the adjacent phenyl groups would make the trans transition state more favorable. This stereochemical assignment was later confirmed for the ebthi system (vide infra). Monitoring the reaction by 1H NMR spectroscopy showed that the unreacted epoxides did not isomerize under the reaction conditions. These data provided strong evidence for rate-limiting ring-opening to form intermediates 6 and 7, which undergo rapid interconversion by rotation about the C–C σ-bonds, followed by ring-closure to form 8 at a rate much faster than reversion to epoxides and 1.
Scheme 1.
The reaction of chiral ebthi complex (rac)-4 with cis- or trans-stilbene oxide was also studied (eq 3). This reaction could
(3).
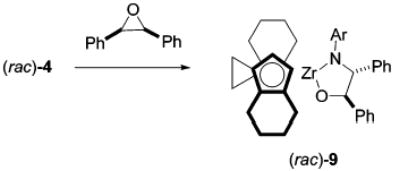
theoretically have resulted in the production of four different diastereomers, arising both from the planar chirality of the ebthi ligand and from the benzylic stereocenters. A 1H NMR spectrum of the crude reaction mixture showed that product (rac)-9 was formed as the same single diastereomer, regardless of the starting stereochemistry of the epoxide. An X-ray diffraction study revealed that (rac)-9 was the R,R,R,R/S,S,S,S racemate, confirming the trans stereochemistry (Figure 1). This offered support for our initial hypothesis that 8 was the trans isomer (vide supra) and additional evidence that this reaction involved the intermediacy of zwitterions. The high diastereoselectivity of this reaction suggested the possibility of a kinetic resolution of the racemic imido compound (rac)-4 by the readily available enantiopure trans-stilbene oxide. Addition of 0.55 equiv of (R,R)-trans-stilbene oxide (>98% ee)7 to a solution of 1.0 equiv of (rac)-4 in C6H6 at ambient temperature resulted in a modest product diastereoselectivity of 3:1, corresponding to 50% ee of unreacted 4.
Figure 1.
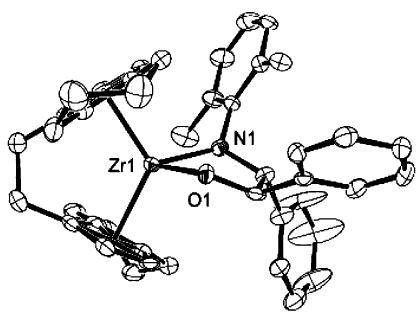
ORTEP structure of 9, showing R,R,R,R/S,S,S,S racemate.
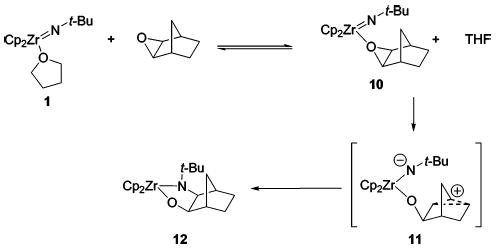
Although epoxide complexes with unopened styrene oxide or stilbene oxide bound to zirconium were not observed by 1H NMR spectroscopy in the reactions with 1 or 4, we presumed that these complexes were intermediates. Therefore, we sought a system with a higher barrier for ring-opening that would allow for observation of an epoxide complex. A C6D6 solution of N-tert-butylimidozirconocene complex 13 and exo-norbornene oxide exhibited a broad 1H NMR spectrum, consistent with exchange of the THF and exo-norbornene oxide ligands at ambient temperature (Scheme 2). Removal of the THF in vacuo from this solution produced exo-norbornene oxide complex 10 quantitatively, as observed by 1H NMR spectroscopy. Crystals of 10 were obtained at low temperature and characterized by X-ray diffraction (Figure 2a). This is one of the few examples of intermediate epoxide complexes that have been isolated and structurally characterized.8,9
Figure 2.
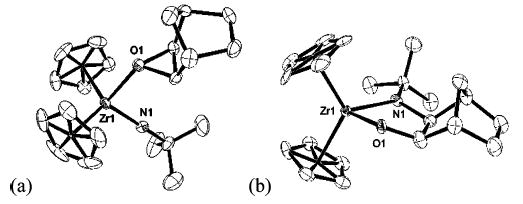
ORTEP structures of (a) epoxide complex 10 and (b) cis-metallacycle 12.
Heating either a solution of 1 and exo-norbornene oxide (45 °C for 24 h) or a solution of 10 (45 °C for 5 h) yielded cis-metallacycle 12. The cis stereochemistry of 12 is consistent with the conversion of 10 to the (presumably carbon-bridged or “nonclassical”) zwitterion 11, followed by trapping of the cation by the anionic amide ligand. Interestingly, no trapping product from the rearranged norbornyl framework is observed, perhaps because closure to form 12 proceeds through a kinetically favored five-membered-ring transition state, whereas trapping at the adjacent carbon atom requires a six-membered-ring transition state. The stereo- and regio-chemistry of 12 were confirmed by X-ray diffraction (Figure 2b).
Consistent with the proposed mechanism, conversion of 10 to 12 exhibited first-order kinetics (k = 0.017 (59.8 °C), 0.051 (69.8 °C), 0.084 (79.8 °C), and 0.22 s−1 (89.9 °C)),10 independent of the concentration of excess epoxide. Rates measured over this temperature range gave activation parameters ΔH‡ = 19 ± 2 kcal mol−1 and ΔS‡ = 10 ± 5 cal mol−1 K−1, in agreement with rate-determining ring-opening by acid-assisted ionization.11
Finally, preliminary results indicate that it is possible to cleave the amido alkoxide portion of these complexes under mild conditions, effecting a net group transfer. Addition of 2 equiv of benzoic acid to 9 at ambient temperature resulted in a rapid and quantitative (>95%) cleavage to the amino alcohol and the zirconium dibenzoate complex, as observed by 1H NMR spectroscopy.
In conclusion, imido complexes 1 and 4 reacted cleanly with epoxides. Regio- and stereochemical probes have provided an understanding of the reaction mechanism, and the results indicate that a stepwise pathway operates in which epoxide ring-opening is the rate-determining step, leading to zwitterions with substantial carbocation character. The results demonstrate the important bifunctionality of the Lewis acidity of the zirconium center and the nucleophilicity of the imido moiety in promoting the ring-opening reaction.
Scheme 2.
Acknowledgments
This work was supported by the NIH through Grant No. GM-25459 to R.G.B., and by the NSF though a graduate fellowship to S.A.B. We thank Dr. Fred Hollander and Dr. Allen Oliver of the UC Berkeley CHEXRAY facility for the X-ray structure determinations. We also thank Prof. Donna G. Blackmond for help in analyzing kinetic data. The Center for New Directions in Organic Synthesis is supported by Bristol-Meyers Squibb as a Sponsoring Member and Novartis as a Supporting Member.
Footnotes
Supporting Information Available: Experimental procedures, kinetic data analysis, and characterization of all new compounds (PDF). X-ray crystallographic data for 5, 9, 10, and 12 (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schaller CP, Bonanno JB, Wolczanski PT. J Am Chem Soc. 1994;116:4133. [Google Scholar]
- 2.Cummins CC, Baxter SM, Wolczanski PT. J Am Chem Soc. 1988;110:8731. [Google Scholar]
- 3.Walsh PJ, Hollander FJ, Bergman RG. J Am Chem Soc. 1988;110:8729. [Google Scholar]
- 4.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer KE, Walsh PJ, Bergman RG. J Am Chem Soc. 1995;117:974. [Google Scholar]
- 6.Lee SY, Bergman RG. Tetrahedron. 1995;51:4255. [Google Scholar]
- 7.Tu Y, Wang ZX, Shi Y. J Am Chem Soc. 1996;118:9806. [Google Scholar]
- 8.Darensbourg DJ, Wildeson JR, Lewis SJ, Yarbrough JC. J Am Chem Soc. 2002;124:7075. doi: 10.1021/ja020184c. [DOI] [PubMed] [Google Scholar]
- 9.Groves JT, Han Y, Engen DV. J Chem Soc, Chem Commun. 1990:436. [Google Scholar]
- 10.The standard deviation in the fit for each k was extremely small. We conservatively estimate the actual error in reproducibility to be ± 0.006 s−1 at 69.5 °C, based on a triplicate run. A detailed error analysis can be found in the Supporting Information.
- 11.Long FA, Pritchard JG, Stafford FE. J Am Chem Soc. 1957;79:2362. [Google Scholar]