

Abstract
Genetic analysis in zebrafish has been instrumental in identifying genes necessary for visual system development and function. Recently, a large-scale retroviral insertional mutagenesis screen, in which 315 different genes were mutated, that resulted in obvious phenotypic defects by 5 days postfertilization was completed. That the disrupted gene has been identified in each of these mutants provides unique resource through which the formation, function, or physiology of individual organ systems can be studied. To that end, a screen for visual system mutants was performed on 250 of the mutants in this collection, examining each of them histologically for morphological defects in the eye and behaviorally for overall visual system function. Forty loci whose disruption resulted in defects in eye development and/or visual function were identified. The mutants have been divided into the following phenotypic classes that show defects in: (1) morphogenesis, (2) growth and central retinal development, (3) the peripheral marginal zone, (4) retinal lamination, (5) the photoreceptor cell layer, (6) the retinal pigment epithelium, (7) the lens, (8) retinal containment, and (9) behavior. The affected genes in these mutants highlight a diverse set of proteins necessary for the development, maintenance, and function of the vertebrate visual system.
THE zebrafish has been an important model through which genes necessary for visual system development and function have been identified (reviewed in Easter and Malicki 2002 and Neuhauss 2003). Zebrafish eyes are large, easily accessible, and structurally similar to the human eye. Eye formation in zebrafish is analogous to that observed in other vertebrate embryos, thus providing an excellent model system with which the understanding of vertebrate eye development can be advanced. Additionally, many disrupted genes and pathways identified as integral to the formation of the zebrafish eye produce phenotypes that resemble disorders of the human visual system. Thus, characterization of the molecular mechanisms of eye development in zebrafish should facilitate a better understanding of these human pathologies (Goldsmith and Harris 2003).
Eye development in zebrafish first becomes morphologically obvious at the 6 somite stage (SS), the time at which the optic lobes evaginate from the diencephalon (Schmitt and Dowling 1994). Thereafter, eye development proceeds rapidly with lens induction occurring around the 14–15 SS and morphological distinction between the retina and retinal pigment epithelium (RPE) apparent by the 18–19 SS. The first postmitotic neurons of the retina are generated at 28 hr postfertilization (hpf) and by 72 hpf the retina is functional (Easter and Nicola 1996; Hu and Easter 1999; Schmitt and Dowling 1999). Retinas of many fish and amphibians also possess a specialized region at their margins, termed peripheral or ciliary marginal zones, that perpetually adds cells to the retina during the lifetime of the animal (Johns 1977).
Several generations of chemically based forward genetic screens have been undertaken in zebrafish (Driever et al. 1996; Haffter et al. 1996; Matsuda and Mishina 2004), some of which have focused on eye development and function (Malicki et al. 1996; Fadool et al. 1997; Neuhauss et al. 1999). While these chemically based screens have been instrumental in generating interesting mutant phenotypes, positional cloning of these mutations is still quite laborious, despite the genomic advances of the last few years. Retrovirus-mediated insertional mutagenesis provides an attractive alternative to chemical mutagenesis techniques since the affected gene can be rapidly identified using the proviral insert as a molecular tag to localize the site of insertion in the genome and thereby to identify the mutated gene (Gaiano et al. 1996; Amsterdam et al. 1999; Amsterdam 2003). Indeed, a large-scale insertional mutagenesis screen performed over the last 6 years has generated >500 insertional mutants of which 315 different affected loci have been identified (Golling et al. 2002; Amsterdam et al. 2004). This collection of mutants presents a wealth of possible analyses since studies are not limited to gross phenotypic characterizations; one can target for study specific physiological processes and biochemical pathways in which the mutated gene's protein product would normally function.
It is estimated that this collection of 315 insertional mutants represents ∼22% of the vertebrate gene complement that can be mutated to result in a visible embryonic phenotype (Amsterdam et al. 2004). With that utility in mind, a shelf screen was performed on 250 different insertional loci for which the affected genes have been cloned to identify those possessing defects in the development and function of the visual system. Forty mutations were identified that affected the visual system and this article reports the identification and initial characterization of each of these mutants.
MATERIALS AND METHODS
Animals:
The methods for the generation and identification of insertional mutants have been reported previously in Gaiano et al. (1996) and Amsterdam et al. (1999). Embryos were obtained from the natural spawning of heterozygous carriers setup in pairwise crosses. Embryos were collected and raised at 28.5° after Westerfield (1995) and were staged according to Kimmel et al. (1995).
Histology:
Mutants and wild-type siblings were collected and fixed overnight at 4° in a solution of 1% (w/v) paraformaldehyde, 2.5% glutaraldehyde, and 3% sucrose in PBS. They were washed 3 × 5 min in PBS and refixed for 90 min at 4° in a 2% OsO4 solution, washed 3 × 5 min in PBS at RT, and dehydrated through a graded ethanol series (50, 70, 80, 90, 2 × 100%). Embryos were further dehydrated 2 × 10 min in propylene oxide and infiltrated 1–2 hr in a 50% propylene oxide/50% Epon/Araldite mixture (Polysciences). Embryos were then incubated overnight at RT in 100% Epon/Araldite resin with caps open to allow for propylene oxide evaporation and resin infiltration, embedded and baked at 60° for 2–3 days. Sections 1–1.25 μm were cut, mounted on glass slides, and stained in a 1% methylene blue/1% borax solution. Sections were mounted in DPX (Electron Microscopy Sciences, Fort Washington, PA) and photographed on a Leica DMRB microscope mounted with a QImaging Retiga EXi digital camera. Images were subsequently processed using Adobe Photoshop 5.0.
Acridine orange staining:
Acridine orange (Molecular Probes, Eugene, OR) was diluted in fish water to a final concentration of 1 μg/ml. Embryos were placed in this solution for 10 min, washed briefly five times in fish water, and immediately observed under GFP optics on a Leica dissecting scope.
Immunohistochemistry:
Mutants and wild-type siblings were collected and fixed overnight at 4° in a solution of 4% paraformaldehyde in PBS. Embryos were washed at RT 3 × 5 min in PBS and then infiltrated by 35% sucrose for 1–2 hr at RT. Embryos were then arranged and embedded in plastic molds containing TBS cryopreservation media (Triangle Biomedical Sciences, Durham, NC). Cryosections 12 μm in thickness were cut on a Leica CM1900 cryostat and adhered to gelatin-coated slides. After drying for 1–2 hr at RT, slides were lined with a hydrophobic marker (PAP pen), rehydrated briefly in PBS, and blocked for 1–2 hr in 5% NGS. Primary antibodies, diluted in 5% NGS, were added and slides were incubated overnight at 4°. Slides were then washed in PBS at RT 2 × 5 min and 1 × 30 min and incubated in secondary antibody, diluted in 5% NGS, for 90 min at RT. Slides were washed 3 × 15 min in PBS at RT and mounted in Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). The following antibodies and dilutions were used: 1d1 (1:30), 5e11 (1:100), zn5 (1:100), and goat anti-mouse Cy3 secondary (1:500). Imaging was performed in a Zeiss 510 laser scanning confocal microscope. A total of 3–5 optical sections (1 μm in thickness) were collected and projected using Zeiss confocal software.
Optokinetic response assay:
Optokinetic response (OKR) assays were performed after Brockerhoff et al. (1995). For testing, two to four embryos at a time were immobilized in a small Petri dish containing 5% methylcellulose. The Petri dish was positioned in the center of a drum lined with vertical black and white stripes, each 1 cm wide. The drum was illuminated with a tungsten light source attenuated by up to 3.5 log units with neutral density filters. The drum rotated at ∼8 rpm and, during 30-sec trials, the direction of rotation was changed four to six times. Beginning at full light intensity, embryos were tested to see if they could respond to the moving stripes. A response was defined as the demonstration of either a smooth pursuit-saccade cycle or eye tracking movements in both the counterclockwise and the clockwise directions depending on the rotation of the drum.
Electroretinography:
Isolated whole-eye electroretinograms (ERGs) were obtained using methods described in Kainz et al. (2003). Briefly, 5 days postfertilization (dpf), light-adapted larvae were placed onto filter paper moistened with Mangel's Ringer solution with 20 mm dextrose (pH 7.8). A pair of forceps held the animal and a small loop made of tungsten wire gently removed the eye, which was placed onto the filter paper, cornea side up. A pulled glass micropipette (tip 10 μm in diameter) containing Ringer solution and a chloride-coated silver wire was inserted into the eye at the border of the marginal zone and the lens. A ground wire was placed under the moistened filter paper within the recording chamber. The eye was bathed in Ringers throughout the course of a recording session, which lasted 30–75 min. ERGs were recorded at 24°–25°. Responses were amplified by a Dagan Cornerstone amplifier bandpass filtered (0.1–100 Hz), total gain ∼10,000, and collected using PClamp software (Axon Instruments, Burlingame, CA). A 1409 W/cm2 background light attenuated by a −1.6 log unit ND filter was used. The stimulus was produced by a tungsten halogen light, 9503 μW/cm2 unattenuated intensity and was adjusted with neutral density filters. The duration of the stimulus was 1000 msec, while the interstimulus time was 15 sec.
RESULTS
Insertional mutagenesis screen:
The generation, screening, and cataloging of zebrafish insertional mutants have been described elsewhere (Gaiano et al. 1996; Amsterdam et al. 1999, 2004; Golling et al. 2002). In this study, 250 different insertional mutants were screened for defects in eye morphology and for deficits in visual behavior. Figure 1 provides a general summary of the screen and its results. The morphology screen sought to identify mutants with abnormal development or maintenance of eye structures at 5 dpf. Of the initial 250 lines, 192 survived until 5 dpf and these were further studied. To assay eye development, several mutant embryos from each of these 192 lines were fixed, processed, and sectioned for histological examination.
Figure 1.—

Summary of the screen. The affected loci in 315 insertional mutants have been identified (Amsterdam et al. 2004). This screen studied 250 of these, 192 of which survived until 5 dpf and whose histology was examined. Of these 192 mutants, 81 showed a nonspecific morphological phenotype in the eye likely as a secondary result of more general systemic defects; 38 showed a morphological phenotype in the eye that appeared direct, i.e., not a secondary result of general systemic defects; 40 mutants showed small eyes with normal morphology; and 33 total mutants had wild-type eye size and morphology, 2 of which, however, failed behavioral testing for visual function.
Obvious phenotypic defects in eye development were observed in >60% of the mutant lines sectioned. These phenotypes mostly manifest as severe retinal degeneration evident by the presence of many pyknotic nuclei scattered through all retinal cell layers as well as large regions of acellular holes, indicative of prior cell death. In addition, many mutants exhibited severe lens degeneration in conjunction with the above retinal degeneration. Thus, it became necessary to separate those phenotypes likely resulting from a direct role of the affected gene product in the eye from those resulting from multisystem defects and, therefore, presumably secondary to a more general set of physiological problems. A set of criteria was established to score the overall health of the mutant embryos up to 5 dpf and to correlate these with the nature of the eye phenotype observed after histological analysis. These criteria included daily observations of overall embryo health as well as a variety of physical features such as the amount of unconsumed yolk, overall head and body size, presence of cardiac edema, blood circulation, general or localized necrosis, and locomotion. Analysis of the histological data in conjunction with these physical observations identified a subset of mutant lines that might represent an eye phenotype directly related to the mutated gene's normal cellular role rather than those phenotypes resulting from general physiological causes. In addition, roughly one-fifth of the mutants that presented severe eye degeneration phenotypes at 5 dpf showed a more subtle phenotype at earlier stages. These mutants were rescreened at 3 dpf since it was possible that relevant eye defects were obscured by overall eye degeneration by 5 dpf. All rescreens entailed a second round of histology for phenotypic verification, as well as acridine orange staining at 3 dpf to provide an indication of the amount of cell death occurring in the mutants. Acridine orange staining enabled a distinction between generalized CNS degeneration and cell death localized to the eye, possibly in conjunction with other limited CNS structures.
Of the 192 mutant loci histologically examined, 38 were ultimately identified that fit our criteria for possessing a relevant morphological eye phenotype (Table 1). Eighty-one mutant loci displayed eye phenotypes that were deemed likely to be secondary to more general system-wide defects (see supplementary Table 1 at http://www.genetics.org/supplemental/). In addition, 33 other mutants displayed wild-type eye size and morphology (see supplementary Table 2 at http://www.genetics.org/supplemental/; Figure 10), whereas 40 mutants displayed smaller eyes with no other apparent morphological defects in eye development (see supplementary Table 3 at http://www.genetics.org/supplemental/). In the latter mutants, all cell layers had formed and were properly patterned but the size of the eye was smaller overall. These mutants were not pursued further from the morphological standpoint of the screen.
TABLE 1.
Gene identities of zebrafish insertional mutations
| Mutanta | Allelesb | Zebrafish gene disrupted | Locus link no. |
GenBank accession no. |
ENU mutant |
OKRc |
|---|---|---|---|---|---|---|
| Early eye morphogenesis | ||||||
| ndr2 | 2718A | ndr2 (cyclops) | 4838 | AF002219 | cycd | ND |
| Growth retardation and central retina defects | ||||||
| smarca5 | 550 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 5 |
8467 | AF506217, AF506218 | − | |
| ccna2 | 2696, 3594A | Cyclin A2 | 890 | AF234784 | − | |
| fen1 | 4026 | FEN1b (flap structure-specific endonuclease 1) | 2237 | AY648844 | − | |
| Peripheral marginal zone defects | ||||||
| mcm2 | 1244, 3205 | MCM2 | 4171 | AY099531 | + | |
| cpsf4 | 1, 1364B | Cleavage and polyadenylation specificity factor 4 (no arches) |
10898 | U70479 | − | |
| fgr | 1532B | Novel (FLJ12716-like; foie gras) | 60684 | AY648743 | + | |
| pat | 1858A | Novel (FLJ20291-like; pate) | 54883 | AY648753 | − | |
| rfc5 | 1887 | Replication factor C, subunit RFC5 | 5985 | AY648755 | + | |
| rfc4 | 2877A | Replication factor C, subunit RFC4 | 5984 | AY648805 | + | |
| mcm3 | 319, 3068 | MCM3 | 4172 | AF506208 | + | |
| znf162 | 3282 | Zinc finger protein 162 | 7536 | AY391459 | +/− | |
| polr3f | 1942, 3290B | RNA polymerase III 39-kDa subunit | 10621 | AY648756 | + | |
| dmap1 | 3721 | DNA methyltransferase 1-associated protein 1 | 55929 | BC048054 | + | |
| Retinal lamination defects | ||||||
| nfyc | 962, 2019 | Nuclear transcription factor Y, gamma | 4802 | BC045364 | + | |
| mpp5 | 2226 | MAGUK p55 subfamily member 5 (nagie oko) | 64398 | AY648770 | noke | + |
| prkci | 3208 | Protein kinase C, iota (heart and soul) | 5584 | AF390109 | hasf | ND |
| tyms | 3510 | Thymidylate synthase | 7298 | AY005804 | + | |
| Photoreceptor defects | ||||||
| nrf1 | 399 | NRF-1 | 4899 | NM_131680 | + | |
| ixl | 428 | Intersex | 55588 | AF506210 | + | |
| pwi | 1482 | Novel (WRB-like; pinball wizard) | 7485 | AY648739 | − | |
| odc | 1841B | Ornithine decarboxylase | 4953 | AF290981 | +/− | |
| ift172 | 2211 | IFT172/LIM-interacting protein | 26160 | AY618923 | +/− | |
| ift57 | 3417 | IFT57/HIPPI | 55081 | AY618924 | + | |
| Retinal pigment epithelium defects | ||||||
| atp6ap1 | 112 | v-ATPase Ac45 | 537 | NM_173265 | − | |
| atp6v1e1 | 577A | v-ATPase subunit e | 529 | AF506222 | − | |
| atp6v1h | 923 | v-ATPase SDF/54-kDa subunit | 51606 | AF506234 | − | |
| atp6v0c | 1207 | v-ATPase 16-kDa proteolipid subunit | 527 | AY099523 | − | |
| atp6v1f | 1988 | v-ATPase subunit F | 9296 | AY648759 | − | |
| atp6v0d1 | 2188B | v-ATPase AC39 subunit | 9114 | AY648766 | − | |
| paics | 2688 | Phosphoribosylaminoimidazole carboxylase | 10606 | BC048051 | + | |
| Lens defects | ||||||
| uhrf1 | 272, 3020 | Ubiquitin-like, containing PHD and RING finger domains, 1 |
29128 | AY648713 | − | |
| copz1 | 528 | Cotamer zeta1 | 51644 | NM_131508 | Variableg | |
| dkfzp434168 | 1548, 3649 | Novel (DKFZP434B168-like) | 25896 | AY099529 | Variableg | |
| mgc10433l | 2735A | Novel (MGC10433-like) | 79171 | AY648798 | − | |
| Blowout mutants | ||||||
| lamb1 |
1113B, 1297, 3181 |
Laminin B1 (grumpy) | 3912 | AF468049 | guph | + |
| ncad |
1389, 3096, 3644 |
N-cadherin | 1000 | NM_131081 | ncadi | + |
| lamc1 | 1193, 2557, 3890, 4131 |
Laminin C1 (sleepy) | 3915 | NM_173277 | slyh | ND |
| Behavioral mutants | ||||||
| lman2l | 472 | Lectin, mannose-binding 2-like | 81562 | AF506214 | − | |
| slc25a5 | 526A | Solute carrier family 25 alpha, member 5 | 292 | AF506216 | − | |
Images of mutants can be found at http://web.mit.edu/ccr/pnas_zebrafish_mutant_images/ and information on other phenotypes in these mutants can be found in supplemental data from Amsterdam et al. (2004).
Hopkins allele designation (hi number). Italic type indicates allele from which histology is pictured.
OKR results were as follows: −, no response; +, normal response; +/−, weak response; ND, not tested.
Horne-Badovinac et al. (2001); Peterson et al. (2001).
Variable OKR results were dependent on the severity of lens defects. Those embryos with severe lens defects did not respond while those with less severe defects responded weakly to normally.
Figure 10.—
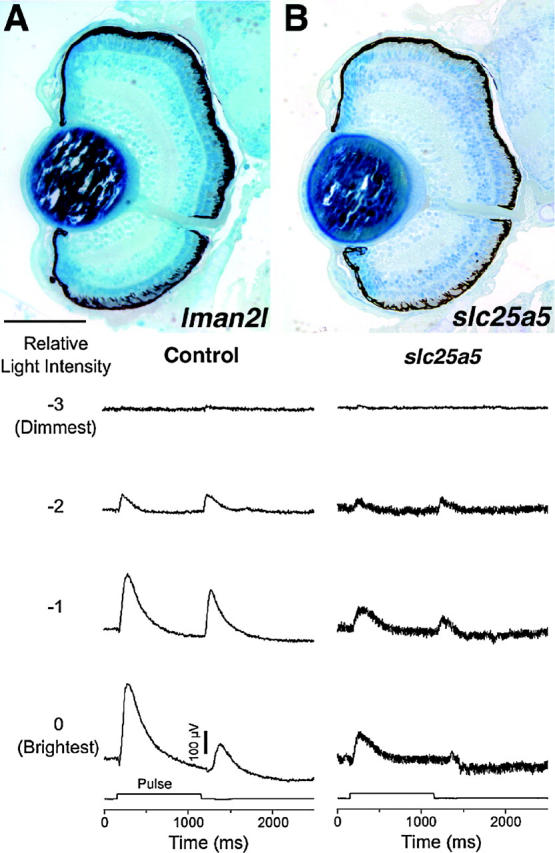
Behavioral mutants with normal eye histology. (A) lman2l mutants do not move their eyes in response to OKR stimuli but behave normally in ERG testing. (B) slc25a5 mutants respond weakly to OKR stimuli and exhibit defective ERG responses. ERG recordings from a wild-type 5-dpf sibling (control) and recordings from an slc25a5 mutant are presented. Even at the brightest light levels (0, −1 log units), slc25a5 embryos respond much less robustly to visual stimuli than do their control siblings. Dorsal is up. Bar, 120 μm.
For each of the 38 morphological mutants described herein, the phenotypes were observed in multiple embryos from a minimum of two independent crosses. The histology presented is representative of that observed in all insertional mutants at this locus. Because of the large number of mutants screened, this report describes only the preliminary phenotypic characterization of their eye defects. A more detailed molecular and ultrastructural analysis of each will be required to fully characterize their defects. Additionally, detailed information about defects in other organ systems in each of these mutants can be found in Amsterdam et al. (2004). The mutants have been grouped into eight phenotypic categories on the basis of defects observed in: (1) morphogenesis, (2) growth and central retinal development, (3) the peripheral marginal zone, (4) retinal lamination, (5) the photoreceptor cell layer, (6) the retinal pigment epithelium, (7) the lens, and (8) retinal containment.
A second screen performed on the insertional mutants assayed the function of their visual systems using the OKR, a robust assay of visual behavior (reviewed in Neuhauss 2003). This assay has proven useful in identifying many visually deficient zebrafish mutants (Brockerhoff et al. 1995, 1998; Neuhauss et al. 1999). As detailed above, many of the insertional mutants presented system-wide defects that would obviously prevent them from behaving normally in this screen. These have not been included in this report as their behavioral deficit is likely secondary to more general defects. Additionally, many of the morphological mutants also showed deficits in visual function (Table 1). For the behavioral mutant category, however, behaviorally deficient fish with morphologically normal eyes were specifically being sought. Thus, screening of the 33 mutant lines with wild-type eye size/morphology and the 40 small eyed/normal morphology lines resulted in the identification of two behavioral mutants (Table 1).
Histology of wild-type 3-dpf and 5-dpf zebrafish eyes is presented in Figure 2 for comparison with that of the mutants described below. At 3 dpf, wild-type embryos have formed the five principal laminae in the retina: the three cellular laminae [the ganglion cell (GCL), the inner nuclear (INL), and outer nuclear (ONL) layers] and the two plexiform layers [the inner (IPL) and the outer (OPL)] (Figure 2A). By 5 dpf these laminae are more fully formed such that only small, proliferative regions at the retinal margin remain nonlaminated (Figure 2, B and C). The GCL is composed mainly of the retinal ganglion cells, the output neurons of the retina, along with so-called “displaced” amacrine cells. The INL is composed of amacrine, bipolar, and horizontal cells, whereas the ONL is composed of the photoreceptor cells. Zebrafish possess four types of photoreceptors that mature in a specific order: UV cones first, blue cones next, red/green double cones next, and finally rods. A higher-magnification view of the ONL, dorsal to the optic nerve, shows clear morphological differentiation of photoreceptors with well-defined inner and outer segments at 5 dpf (Figure 2D). The most mature and prominent photoreceptors in this image are the short UV-sensitive cones lying in the innermost region of the ONL (asterisks in Figure 2D), whereas the more distal photoreceptors are blue cones, red/green double cones, and a few rods (arrows in Figure 2D).
Figure 2.—
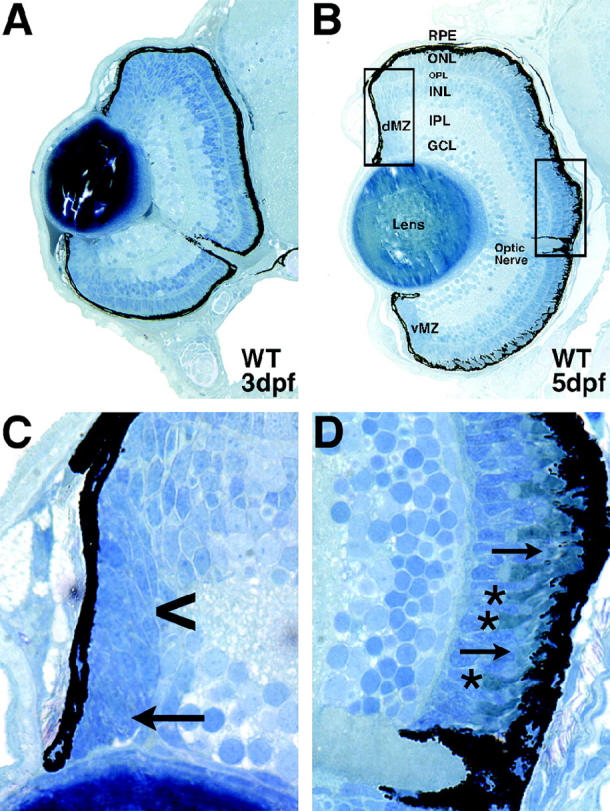
Wild-type development of the zebrafish retina. (A) A 3-dpf retina with well-formed laminae in the central retina and large regions of undifferentiated cells at the retinal periphery. (B) A 5-dpf retina exhibiting nuclear laminae and cell types characteristic of the mature retina. (C) High-magnification view of the dorsal peripheral retina. Note the change in cell shape as epithelial progenitor cells divide at the peripheral-most dMZ (arrow) and are subsequently displaced into the central retina to differentiate into more rounded appearing retinal neurons (arrowhead). (D) High-magnification view of the central retina and optic nerve. Note the prominent short single cones in the innermost region of the ONL (asterisks) and the elongated rod outer segments in the outer ONL, protruding into the RPE (arrows). Dorsal is up in all panels. Bar, 100 μm. RPE, retinal pigment epithelium; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; dMZ, dorsal marginal zone; vMZ, ventral marginal zone.
Mutations affecting early eye morphogenesis:
One mutant was identified, ndr2, that displayed abnormal morphogenesis of the eye field (Table 1). This mutant is an allele of cyclops (cyc), which encodes a nodal related signaling factor (Rebagliati et al. 1998; Sampath et al. 1998). ndr2 mutants, like cycb16, develop two partial eyes, fused at the ventral midline (data not shown; Hatta et al. 1991; Fulwiler et al. 1997).
Mutations resulting in growth retardation and central retinal defects:
Three mutants were identified that displayed a small eye phenotype at 5 dpf, the retinas of which appeared to lack most retinal cell types (Table 1). Histological sections of eyes from these mutants are presented in Figure 3, B–D. Each of these mutants was easily identifiable at 3 dpf by their much smaller eyes relative to sibling control embryos. By 5 dpf, their eyes were roughly half the size of their wild-type siblings. By histology, retinas from these lines show evidence of some lamination and have likely generated retinal ganglion cells (RGCs) as a rudimentary cell layer has formed in the inner retina where the GCL normally resides. They have also formed what appears to be an optic nerve, although it is possible this is a remnant of the optic stalk. The INL and ONL of these mutants are poorly formed. It is difficult to morphologically identify many of the cells in these layers as specific neuronal subtypes purely by histological means.
Figure 3.—
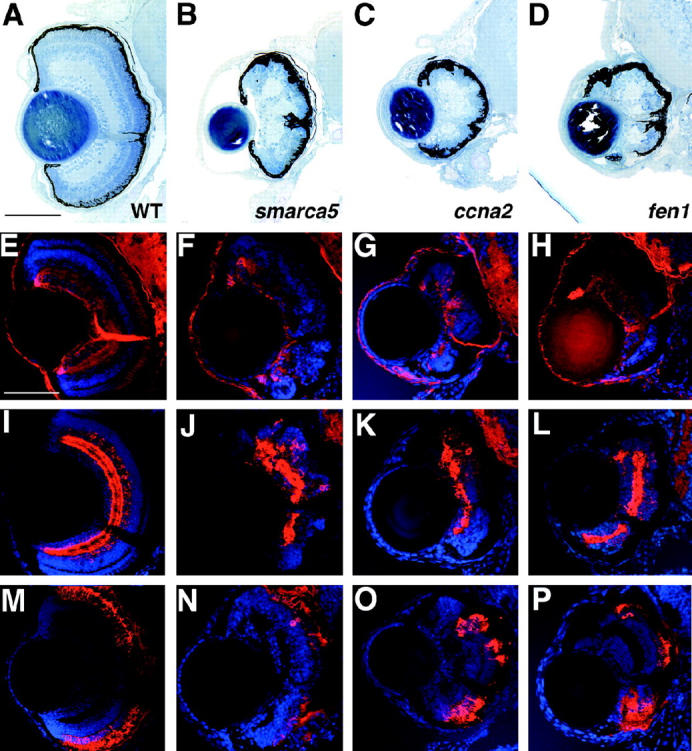
Mutants with growth retardation and retinal degeneration. (A) Wild type, (B) smarca5, (C) ccna2, and (D) fen1 have drastically smaller eyes with defects in the number and organization of retinal neurons at 5 dpf. Retinal patterning is severely affected with the most obvious defects in the outer retina. (E–P) Immunohistochemical analysis of wild-type (E, I, and M), smarca5 (F, J, and N), ccna2 (G, K, and O), and fen1 (H, L, and P) mutants at 5 dpf. (E–H) zn5 staining of RGCs. Each of the mutants differentiated a population of RGCs, the axons of which form an optic nerve of significantly less diameter than that of wild-type siblings. (I–L) 5e11 staining of amacrine cells and their processes. Each of the mutants has differentiated amacrine cells, although their number is much reduced and their distribution is chaotic relative to wild-type siblings. (M–P) 1d1 staining of rod photoreceptors. Each of the mutants has formed rods but in numbers reduced relative to wild-type siblings. Dorsal is up. Bars, 100 μm in A–E, I, and M and 70 μm in F–H, J–L, and N–P.
To begin to understand what cell types are present in retinas of these mutants, immunohistochemistry was performed on 5-dpf retinal sections using markers for RGCs (zn5; Figure 3, E–H), amacrine cells (5e11; Figure 3, I–L), and rods (1d1; Figure 3, M–P). It is apparent from this marker staining that neurons composing each of the three main cellular laminae are present, although they are observed in much reduced numbers relative to sibling controls and are highly disorganized. For example, relative to the wild-type retina at 5 dpf and normalized to their smaller size, these mutants, on average, have 48% less 5e11-stained amacrine cells in their retinas (data not shown). Localized regions of cell death in the brains of these mutants were obvious at 2 dpf and acridine orange staining at 3 dpf identified apoptotic nuclei scattered throughout their retinas. At later days, however, continuing cell death in the CNS was no longer obvious and the mutants appeared to develop normally (data not shown). By 5 dpf, they were vigorous swimmers and displayed robust touch responses and, other than having much smaller heads and eyes, they appeared generally healthy. That the eyes in these mutants are small and appear to lack large numbers of retinal neurons suggests that a defect in progenitor cell maintenance might have occurred. While more detailed experiments will be necessary to ascertain the nature of their phenotype, that these loci encode proteins involved in transcriptional and cell cycle regulation supports this hypothesis (Table 1).
Mutations affecting the peripheral marginal zone:
As noted above, the teleost retina possesses a peripheral marginal zone (MZ) that in many cold-blooded species perpetually adds cells to the retina during the animal's life (Figure 2C). Cells of the marginal zone retain stem cell-like characteristics and can continually generate all types of retinal cells (Johns 1977; Wetts and Fraser 1988). Some higher vertebrates, such as birds and mice, may also possess a similar region between the neural retina and the ciliary epithelium at their retinal periphery (Fischer and Reh 2000; Tropepe et al. 2000). Several mutations that disrupt the formation or maintenance of the peripheral marginal zone have been reported in zebrafish, but the affected loci have not yet been identified (Fadool et al. 1997; Link and Darland 2001). Ten insertional mutations were identified in this screen that affect loci involved in marginal zone formation or maintenance (Table 1). Retinal histology from each of these mutants is presented in Figure 4.
Figure 4.—
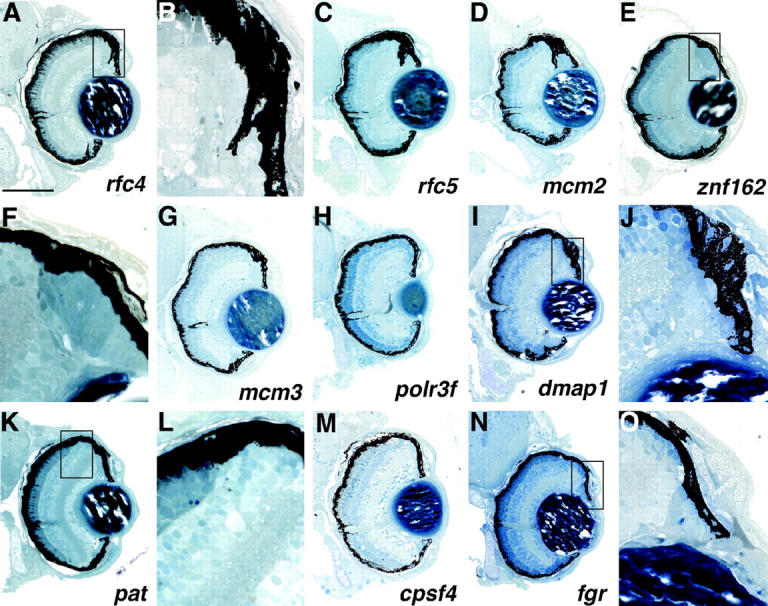
Mutants with defects in the formation and/or maintenance of the peripheral marginal zones. Each of these mutants shows reduced or abnormal marginal zones normally present at the retinal periphery. (A, C, and D) Several of the mutants exhibit RPE protrusions invading the region normally solely occupied by the marginal zone cells. (B) A high-magnification view of the boxed region in A. (E and F) The marginal zones of znf162 are filled with dense, columnar tissue possibly reflecting a large block of cells that have failed to differentiate. (G–N) Mutants with decreased marginal zones. (K and L) pat mutants exhibit an abrupt termination of the outer retina prior to reaching the retinal periphery. (L) A high-magnification view of the boxed region in K illustrating this region. Note that the ONL terminates prematurely and the INL and IPL abut the RPE. (M and N) Mutants with nearly absent marginal zones. (O) High-magnification view of N showing that only the peripheral-most marginal zone cells remain in the fgr mutant. Dorsal is up. Bar, 100 μm.
Mutants in this category appear quite similar in that central retinal development and patterning is normal whereas peripheral marginal zones are greatly reduced both dorsally and ventrally (Figure 4). Differing levels of phenotypic severity are observed between the lines in this group. For example, the rfc4, rfc5, and mcm2 mutants show abnormal RPE invasion into regions of the retinal periphery, resulting in the retinal tissue there being nearly encased in RPE (Figure 4, A–D). On the basis of their morphology, the cells in these regions appear to be largely undifferentiated (Figure 4B) although the significance of the RPE protrusions is currently unknown. The marginal zones of one mutant, znf162, are filled with dense, columnar cells possibly reflecting a large block of cells that have failed to differentiate (Figure 4, E and F). The mcm3, polr3f, and dmap1 mutants all exhibit reduced marginal zones (Figure 4, G–I). The pat mutant shows an abrupt termination between the cells of the outer retina and the region normally occupied by the MZ (Figure 4K). A high-magnification view of this region highlights the abrupt outer retina termination at the retinal periphery (Figure 4L). Here, the INL, OPL, and ONL extend to the retinal margin with no intervening cells (compare Figure 4L with Figure 2C). The two remaining mutants, cpsf4 and fgr, have even more reduced marginal zone regions (Figure 4, M–O). In addition, fgr mutants also frequently have malformed lenses (data not shown). Several of the marginal zone mutants show localized regions of cell death in their MZs when viewed at earlier days (data not shown). In general, this class of mutants frequently showed phenotypic defects in other proliferative regions of the body, including the brain, liver, and gut (Amsterdam et al. 2004; data not shown). Thus, it is possible that these mutants represent a more general growth inhibition at later stages of development rather than specific marginal zone functions (see discussion). Two of the loci affected, fgr and pat, encode proteins of unknown biochemical function, while the other eight loci encode proteins involved in various aspects of DNA replication and mRNA modification (Table 1).
Mutations affecting retinal lamination:
Lamination in the zebrafish retina becomes evident by 3 dpf when the three principal cellular laminae can be readily observed (Figure 2A). As the retina grows and new neurons are generated by the MZs, the new cells maintain appropriate laminar positions for their cell types such that only the MZs remain nonlaminated at later days of development (Figure 2, B and C). Four insertional mutants with defects in retinal lamination were identified (Table 1). Retinal histology from each of these mutants is presented in Figure 5.
Figure 5.—
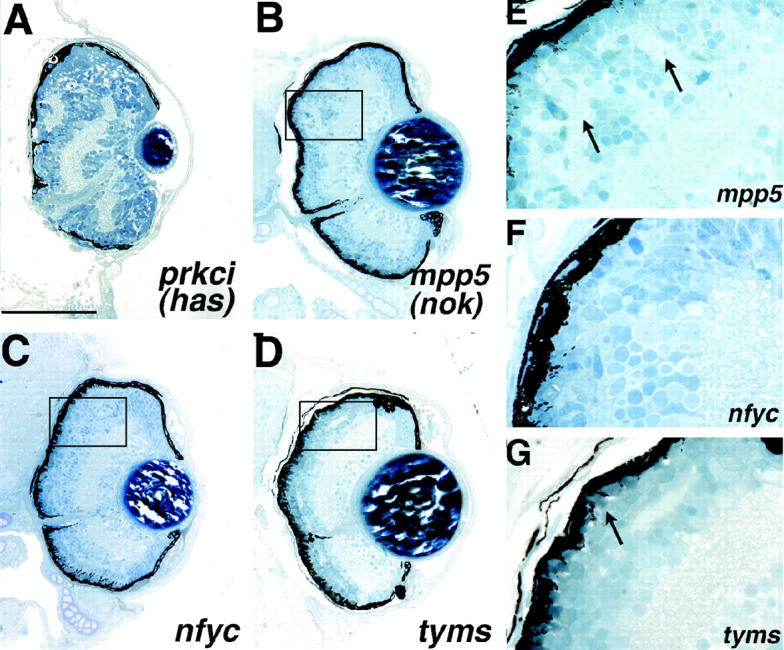
Mutants with retinal lamination defects. (A) prkci (has) and (B) mpp5 (nok) mutants. (E) A high-magnification view of the boxed region in B illustrating the ectopic IPL regions in the outer retina of this mutant (arrows). (C) nfyc and (D) tyms mutants show more subtle defects in the lamination of the outer retina. (F) A high-magnification view of the boxed region in C illustrating the outer retina lamination defect in nfyc. Lamination is normal in the central-most retina and breaks down toward the periphery. It does not appear that photoreceptors have formed in the peripheral retina. (G) A high-magnification view of the boxed region in D illustrating the intermingling of cells between the INL and ONL in this mutant with some regions showing no OPL. Differentiated photoreceptors are present in this mutant (arrow). Dorsal is up. Bar, 100 μm.
prkci and mpp5 mutants both present defects in retinal lamination within their outer retinas (Figure 5, A and B). prkci mutants also showed aberrant RPE formation. A high-magnification view of the mpp5 retina highlights the breakdown in lamination observed in this mutant where ectopic plexiform layers can be readily observed (arrows in Figure 5E). mpp5 is allelic to the ENU-induced nagie oko (nok) locus that encodes a membrane-associated guanylate kinase (MAGUK) scaffolding factor (Wei and Malicki 2002). prkci is allelic to heart and soul (has), an ENU-induced locus encoding an atypical protein kinase C (Horne-Badovinac et al. 2001; Peterson et al. 2001). The defects described here for the insertional alleles are consistent with those reported for nok and has, respectively, although mpp5 (nok) insertional mutants are not as severely affected as those reported for nokm227 and nokm520 (Wei and Malicki 2002).
Two other mutants, nfyc and tyms showed subtle retinal lamination defects (Figure 5, C and D). The nfyc phenotype is manifest as a lack of photoreceptor formation peripherally with either a breakdown in lamination between the ONL and INL or a lack of OPL formation in these peripheral regions (Figure 5C). The central retina appears unaffected in nfyc mutants. A higher-magnification view of the dorsal retina highlights their defect, as regions of the INL can be seen intermingling with the ONL in conjunction with a lack of OPL formation (Figure 5F). Like nfyc, tyms mutants also show either regions where a breakdown in lamination between cells of the INL and ONL has occurred or a lack of OPL formation (Figure 5D). In tyms mutants, INL regions throughout the retina come into contact with the ONL with no OPL regions evident between them (Figure 5G). The gene disrupted in nfyc is NFY-γ, part of a trimeric transcription factor complex (Table 1; Maity and de Crombrugghe 1998). The mutation in tyms is in the thymidylate synthase gene that encodes an enzyme involved in metabolism, DNA synthesis, and DNA repair (Table 1).
Mutations affecting photoreceptor development and survival:
Six mutants were identified with phenotypic defects in the formation and/or maintenance of the photoreceptor cell layer. Two mutants appear to have defects in ONL formation, ixl and odc. ixl mutants present an interesting ONL phenotype in which the photoreceptors have formed but are morphologically abnormal, resulting in a distortion of overall retinal shape to a more oval appearance in mutant eyes (Figure 6, A and A′). In this mutant, it appears as if the photoreceptors (PRs) might have precociously developed such that their outer segments are much more robust in appearance (Figure 6A′) when compared with a wild-type control (Figure 2D). Morphologically, the ONL in ixl mutants appears to be composed of all photoreceptor subtypes but conclusive identification of these cells will require a molecular or electron microscopic characterization. ixl encodes a zebrafish ortholog of Intersex, a transcriptional cofactor (Table 1). odc mutants display a well-defined ONL but the photoreceptors in these mutants extend little to no outer segment material (Figure 6, B and B′). ONL development otherwise appears grossly normal (Figure 6B′). This locus encodes ornithine decarboxylase, an enzyme involved in polyamine biosynthesis (Table 1).
Figure 6.—
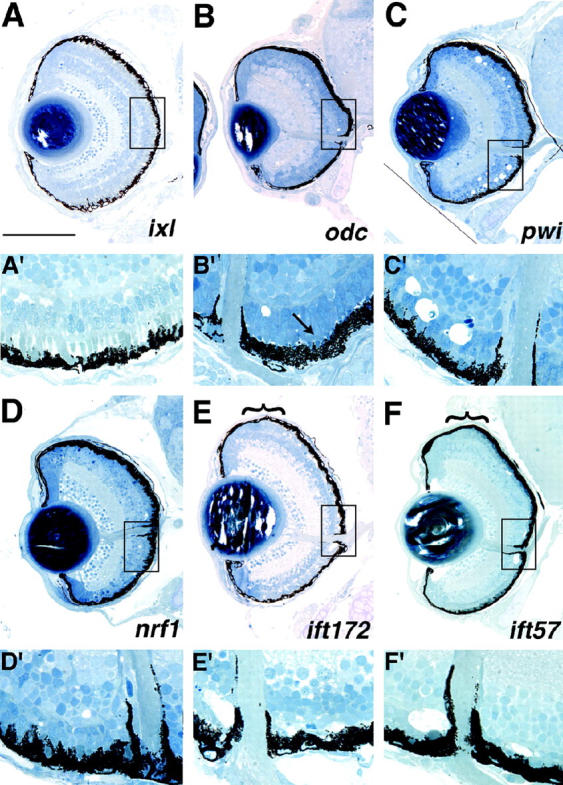
Mutants with defects in the formation and/or maintenance of the photoreceptor layer. (A and A′) ixl mutants exhibit abnormal photoreceptor development and morphology. When compared with wild-type morphology in Figure 2D, the outer segments in this mutant appear larger and more robust (A′). (B and B′) odc photoreceptors, while clearly present, form little outer segment material (arrow in B′ pointing to small outer segment). (C and C′) pwi mutants show acellular holes in both the central and the peripheral retina. (D and D′) nrf1 mutants have fewer photoreceptors likely as a result of photoreceptor-specific degeneration (D′). (E and E′) ift172 and (F and F′) ift57 mutants exhibit acellular holes in the central ONL, likely representing photoreceptor degeneration as photoreceptors are clearly observed in the retinal periphery (PR regions in the periphery are highlighted by brackets in E and F). Note that below the low-magnification image of each section (A–F), a high-magnification image is presented to highlight the boxed region of the ONL (A′–F′) compared to that of a wild-type sibling in Figure 2D. In A–F, dorsal is up. For A′–F′, the images are rotated 90° clockwise relative to their respective retinal sections. Bar, 100 μm.
Four mutants displayed what is likely to be photoreceptor-specific retinal degeneration: pwi, nrf1, ift172, and ift57 (Figure 6, C–F). pwi mutants show ONL defects both centrally and peripherally (Figure 6C). A high-magnification view of the retina from this mutant highlights the acellular regions observed in the central retina (Figure 6C′). In these mutants there are also a few acellular holes in the INL (Figure 6C) but otherwise their inner retinas are relatively normal. Since both the central and the peripheral retinas are affected in pwi mutants, it is unknown whether PRs initially form and subsequently degenerate or whether defects are present at the onset of PR formation. The pwi locus encodes a protein of unknown biochemical function (Table 1). nrf1 mutants exhibit extensive photoreceptor degeneration by 5 dpf (Figure 6 D and D′). This locus encodes an ortholog of human nuclear respiratory factor 1 (nrf1), which has been previously characterized (Becker et al. 1998). ift172 and ift57 also exhibit photoreceptor degeneration throughout the ONL (Figure 6 E, E′, F, and F′). That the PRs of the peripheral retina are unaffected in these mutants implies that the oldest PRs in the central retina are formed normally but degenerate at later days of development. ift172 and ift57 mutants are particularly interesting as the affected loci encode proteins that are involved in intraflagellar transport (IFT) a process required for the assembly and maintenance of cilia and flagella in vertebrates (reviewed in Pazour and Rosenbaum 2002). Indeed, Tsujikawa and Malicki (2004) have recently reported a role for IFT proteins in photoreceptor maintenance in zebrafish. In their study, IFT88 was shown to be encoded by the oval locus and IFT57 was implicated in PR maintenance on the basis of morpholino antisense oligonucleotide targeting of its transcript.
Mutations affecting the retinal pigment epithelium:
A variety of functions in the eye are served by the RPE (reviewed in Marmorstein 2001). Much of the blood supply for the retina is provided by the choroid and cells of the RPE function as a blood-retina barrier to regulate the bidirectional flow of materials into and out of the photoreceptor cells. RPE cells also carry out the isomerization of all-trans retinal to 11-cis retinol during the visual cycle. Additionally, photoreceptors shed roughly 10% of their outer segment material daily and it is the cells of the RPE that phagocytose this material and degrade it. A number of human pathologies that result in visual impairment affect the RPE, underscoring the importance of this cell layer in photoreceptor function and maintenance.
Eight loci that affect the formation and/or maintenance of the RPE were identified (Table 1) and histology from seven of these lines is presented in Figure 7. The eighth mutant line, copz1, is discussed below in a category devoted to lens mutants since embryos from this line display prominent defects in lens formation in conjunction with RPE defects. Six lines with RPE defects, atp6ap1, atp6v1e1, atp6v1h, atp6v0c, atp6v1f, and atp6v0d1 exhibited pigmentation defects and abnormal RPE structure (Figure 7, A–I). These mutants also displayed differing degrees of retinal degeneration. The loci affected in each of these mutant lines encode subunits or accessory proteins of the lysosomal vacuolar ATPase complex (Table 1). Each of these mutants also has decreased melanophore pigmentation along the length of its body axis (Golling et al. 2002; Amsterdam et al. 2004). RPE morphology is abnormal in these mutants, and thus the defect is not solely manifest as a lack of RPE pigmentation (Figure 7F). atp6v1h and atp6v1f mutants show a severe retinal degeneration at 5 dpf with pyknotic nuclei observed throughout the ONL and scattered throughout the INL and GCL (Figure 7, H and I). The brains of these mutants also frequently show signs of cell death (data not shown). Retinas of atp6ap1, atp6v0c, and atp6v0d1 contain pyknotic nuclei but, in these mutant retinas, the foci are mainly limited to the transition region between newly generated neurons of the peripheral marginal zone and the retina proper (Figure 7, A, D, and G; high-magnification view of atp6ap1 in Figure 7B). Additionally, atp6ap1 and atp6v0c mutants also have distorted retinal shapes with dorsal regions extending farther back into the adjacent forebrain (Figure 7, A and D). Interestingly, in these mutants, where there is pigmented RPE, photoreceptor outer segments are readily observed (Figure 7C). These outer segments, however, are morphologically abnormal in length and appearance (high-magnification view of atp6ap1 in Figure 7C). In atp6v1e1 mutants, the ONL has degenerated significantly centrally (Figure 7, E and F), while the dorsal peripheral retina shows little overt photoreceptor differentiation and the ventral peripheral retina appears grossly normal. Finally, the seventh mutant in this category, paics, exhibits patchy pigmentation of the RPE and abnormal RPE morphology but unlike the v-ATPase mutants, this mutant shows no obvious degeneration in the adjacent retina (Figure 7J). The paics locus encodes an enzyme involved in purine biosynthesis, phosphoribosylaminoimidazole carboxylase (Table 1).
Figure 7.—
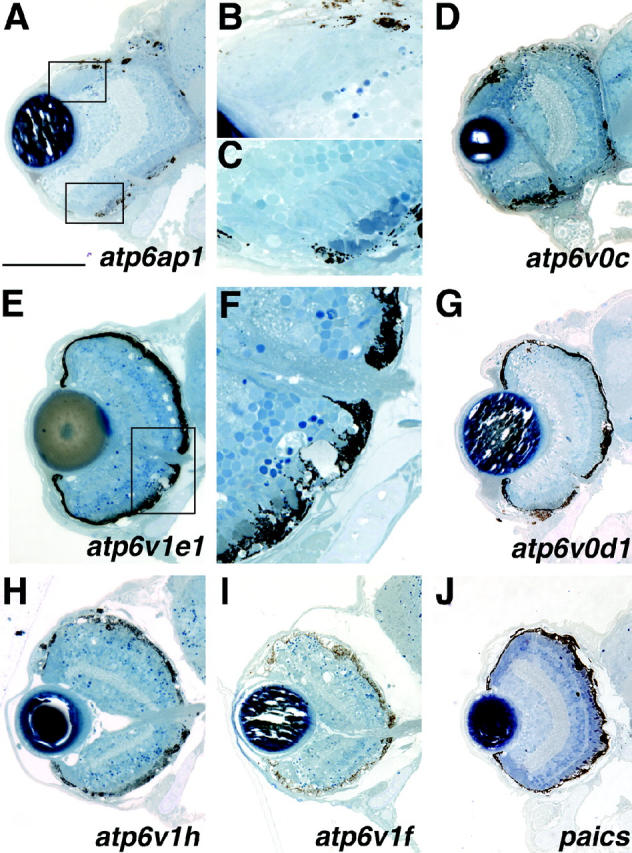
Mutants with defects in the formation and/or maintenance of the retinal pigment epithelium. All mutants in this class exhibit patchy RPE pigmentation and abnormal RPE morphology and most exhibit RPE degeneration. Additional defects are as follows: (A) atp6ap1 and (D) atp6v0c mutants show abnormal retinal morphology with the dorsal-central retina extending into the forebrain. The retina has not expelled through the RPE or Bruch's membrane, however. In several mutants (A, D, and G) pyknotic nuclei are observed at the transition between the MZ and the central retina. (B) A high-magnification view of the top boxed region in A. (C) Photoreceptor outer segments are present where pigmented epithelium is present but their morphology appears abnormal. (E) The ONL of atp6v1e1 has significantly degenerated as pyknotic nuclei and acellular holes are easily observed. (F) A high-magnification view of the boxed region in E. Note the abnormal RPE morphology. (H and I) RPE mutants with extensive retinal degeneration. (J) paics defects are mainly limited to RPE pigmentation and morphology. Dorsal is up. Bar, 100 μm.
Mutations affecting the lens:
The development and maintenance of the lens in zebrafish has not yet been well characterized although several mutants with lens defects have been reported (Heisenberg et al. 1996; Vihtelic et al. 2001). Lens formation in zebrafish initiates with a thickening of the surface ectoderm overlying the optic vesicle at the 14–15 SS. By the 18 SS, the lens placode has thickened and its cells adopt a radial orientation relative to the adjacent neural retina (Schmitt and Dowling 1994). Subsequently, cell differentiation commences and generates two populations of lens cells: primary lens fibers and lens epithelial cells. The lens fiber cells compose the lens proper while the lens epithelial cells form a monolayer and surround the lens at its distal and lateral perimeters.
Four mutations that affected the development of the lens were identified in our screen (Table 1). Two mutations, uhrf1 and dkfzp434168, result in severe disorganization of lens structure (Figure 8, A and B). The uhrf1 mutant lenses are composed of a disordered mass of cells and possibly reflect lens degeneration rather than a defect in lens morphogenesis (Figure 8A). The lens in dkfzp434168 mutants is of normal size but displays loosely packed, disorganized cells (Figure 8B). In addition, the distal-most region of the lens, underlying the corneal epithelium, appears to have degenerated as only a mass of tissue remains. Defects in marginal zone formation are also frequently observed at these loci. In uhrf1, the MZ territory is occupied by columnar cells characteristic of epithelial tissue (Figure 8A, asterisk). The nature of these cells is not known at this time. Retinas from dkfzp434168 mutants do not form MZs (Figure 8B). The locus disrupted in dkfzp434168 encodes a protein of unknown biochemical function while that of uhrf1 encodes a zebrafish ortholog of UHRF1, a nuclear phosphoprotein involved in cell cycle progression (Table 1; Muto et al. 1995). The other two mutants in this category, copz1 and mgc104331, both show normal formation of the central lens nucleus with apparent defects in the surrounding regions of the lens cortex (Figure 8, C and D). The cells of the lens nucleus are the oldest cells in the lens, with growth proceeding circumferentially around the lens perimeter in the cortex. Thus, these mutations possibly reflect defects in the formation or differentiation of newly generated lens cells. Like uhrf1, the MZs of mgc104331 are also composed mainly of columnar cells that appear to be an extension of the abnormal cells originating in the lens cortex (Figure 8D, asterisk). mgc104331 encodes a protein of unknown biochemical function (Table 1).
Figure 8.—
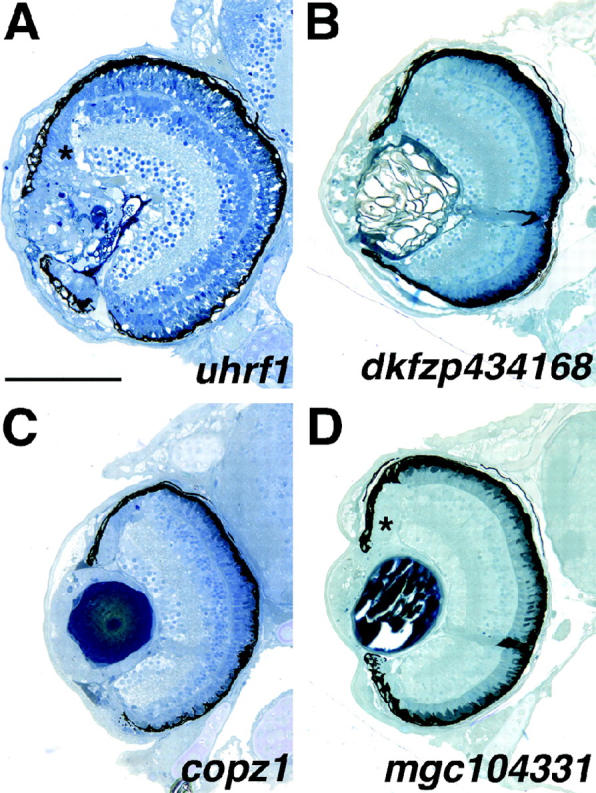
Mutants with lens defects. (A) uhrf1 mutants have severe lens disorganization as well as expansion of epithelial-like tissue into the marginal zones (asterisk). (B) dkfzp434168 mutants exhibit gross lens disorganization and lack both dorsal and ventral marginal zones. (C) copz1 mutants show abnormal formation of the peripheral lens cortex. These mutants also exhibit patchy RPE pigmentation. (D) mgc104331 mutants exhibit defects in the lens cortex and lens tissue expands and fills the anterior chamber space between the lens and cornea as well as forming a continuum with the peripheral marginal zones (asterisk). Dorsal is up. Bar, 100 μm.
“Blowout” mutants:
Mutations in three loci resulted in a retinal blowout: the expulsion of retinal cells through the RPE into the adjacent forebrain (Figure 9). These loci, lamb1, ncad, and lambc1, each exhibit retinal blowout by 3 dpf. All three of these mutants are alleles of known ENU-induced loci: lamb1:grumpy (gup; Parsons et al. 2002), ncad:ncad (Lele et al. 2002; Malicki et al. 2003) and lambc1:sleepy (sly; Parsons et al. 2002). Two mutant alleles of the ncad locus [parachute (pac) and glass onion (glo)] have been characterized in detail and shown to encode the N-cadherin protein (Pujic and Malicki 2001; Lele et al. 2002; Erdmann et al. 2003; Malicki et al. 2003). Histologically, insertional ncad mutant retinas look similar to these reported alleles in that, in addition to the retinal blowout, there is some disruption of regular lamination (Figure 9B). The lamb1 mutant, an allele of gup, encodes laminin β1 while lambc1, an allele of sly, encodes laminin γ1 (Table 1). gup and sly mutants have not been reported to have a blowout phenotype; however, they were identified in a behavioral screen for zebrafish mutants with defective visual function and shown to form disorganized optic nerves (Neuhauss et al. 1999). Beyond the blowout in lamb1, gross retinal morphology looks normal at 5 dpf (Figure 9A) while that of lambc1 is disrupted. At 5 dpf, lambc1 mutants show minor lamination defects and significant lens and corneal malformations (Figure 9C). Several other insertional alleles of the laminin γ1 locus have also been isolated (Table 1; Amsterdam et al. 2004). While the phenotypes are similar between these alleles, one of them, hi2557, presents a more severe retinal phenotype than that of hi3890 (data not shown).
Figure 9.—

Blowout mutants. (A) lamb1 (gup) retinas expel through the RPE and into the forebrain. Retinal lamination and pattern are grossly normal, however. (B) ncad mutants also exhibit retinal blowout into the forebrain and these mutants show some defects in retinal lamination. (C) lambc1 (sly) retinas expel through the RPE and these mutants have clear lens and cornea malformations. Dorsal is up. Bar, 90 μm.
Behavioral mutants:
As discussed above, many mutant loci were identified that were behaviorally blind or exhibited severely attenuated responses when tested by OKR assays (data not shown). In total, 33 of the 250 insertional mutants showed morphologically normal eyes of wild-type size and 40 mutants displayed smaller eyes with no other apparent morphological defects. When tested by OKR, 2 of these 73 loci showed distinct behavioral defects, both of the wild-type eye size group, and these mutants were further examined (Figure 10). Of 21 lman2l mutants tested, only 6 responded to OKR stimuli at 0 log unit light intensity (full light), while at −1 log unit intensity, none of the 21 mutants responded. The mutants showed no eye movements to track the moving OKR stripe while wild-type siblings displayed normal tracking behaviors down to −3 log unit intensity in these assays (data not shown). slc25a5 mutants also did not track properly in OKR assays at all light levels tested; i.e., they showed weak to no eye movements to track the moving stripes (n = 19). That both of these mutants did not track properly in the OKR assay indicates that they are behaviorally deficient but does not assess whether their retinal circuitry functions properly. The underlying defects in these mutants could reflect a motor or tectal defect rather than one in retinal physiology. To begin to differentiate between these possibilities and to assay outer retina function, ERGs were performed. ERGs measure the summed field potential of the outer retina in response to a pulse of light and are extensively used in zebrafish to assess retinal function (e.g., Van Epps et al. 2001; Kainz et al. 2003). ERGs from lman2l mutants were normal at all light levels (data not shown). lman2l mutants are touch insensitive (Golling et al. 2002) and thus, this mutant possibly reflects a more general sensory/motor defect rather than one based in retinal physiology. It is also possible, however, that defects in lman2l mutants lie in the inner retina or the optic tectum, possibilities not addressed by ERG recordings. Further experiments will be necessary to ascertain the cellular basis of their functional deficits. The locus affected in lman2l encodes a vip36-like mannose-binding lectin that is thought to be involved in trafficking of glycoproteins between the Golgi and the cell surface (Table 1; Fiedler et al. 1994; Fiedler and Simons 1996). Conversely, ERGs from slc25a5 show a clear deficit in outer retinal function (Figure 10). At all light levels tested, slc25a5 ERG recordings were significantly attenuated relative to sibling controls. The locus affected in slc25a5 encodes a mitochondrial ADP/ATP carrier protein (Table 1) and the ERG deficits therefore may possibly reflect an underlying problem in energy production.
DISCUSSION
Insertional mutagenesis has emerged as a powerful technique to rapidly identify genes important in vertebrate development (Golling et al. 2002; Zambrowicz et al. 2003; Amsterdam et al. 2004). To date, insertional mutagenesis in zebrafish has identified 315 loci that when disrupted, produce visible embryonic phenotypes by 5 dpf and for which the disrupted genes have been identified (Golling et al. 2002; Amsterdam et al. 2004). Of 250 insertional loci screened, 40 were identified that directly affected the development and function of the visual system. Of these loci, only 8 have been previously ascribed a role in vertebrate eye development, nearly all from mutational and functional studies in zebrafish.
In addition to these 40 mutants, 40 additional mutants were identified that had smaller eyes but were morphologically normal (see supplementary Table 3 at http://www.genetics.org/supplemental/). These mutants likely reflect defects in eye growth, possibly in the context of overall CNS growth defects. MZs in these mutants were proportional for the reduced eye size. Since we were seeking mutants with clear morphological defects, and to limit the scope of the screen, these mutants were not further studied. Morphological defects were observed in 81 other insertional mutants in the collection (see supplementary Table 1 at http://www.genetics.org/supplemental/). These mutants were not included in this report, however, as their eye phenotypes were in the context of massive CNS degeneration and/or accompanied by multiple defects in other organ systems. Eye phenotypes observed in these lines could not be separated from these more general system-wide mutation effects. The morphological portion of this screen sought to identify eye mutations that were generally healthy at 5 dpf and thus likely reflecting the loss of a direct cellular function for the affected locus in the eye. Changing this parameter and thereby the threshold for inclusion in the screen would certainly increase the number of mutants identified on the basis of these new criteria.
Indeed, several zebrafish mutants with reported eye defects were excluded from our screen. For example, one such insertional mutation, that at the dead eye locus (dye), results in severe necrosis in the eyes and tectum visible at 2 dpf (Allende et al. 1996). dye mutants die at 5 dpf and show severe necrosis in all regions of the brain in addition to lacking most of their pharyngeal skeleton. The pleiotropic nature of the dye phenotype made it too severe to be included in this screen for relatively visual system-specific mutants. Another zebrafish mutant with eye defects, no tectal neuron (ntn), generated in a trimethylpsoralen mutagenesis screen (Matsuda and Mishina 2004), affects the cct3 locus that encodes the γ-subunit of chaperonin containing TCP-1 (CCT). The ntn mutants show extensive cell death in the eye and tectum by 2 dpf and this death continues such that by 4 dpf the embryo has very small eyes with protruding lenses, a further deteriorated tectum, reduced jaw structures, and small pectoral fins. Two insertional mutant alleles at the cct3 locus, hi383A and hi1867, look phenotypically very similar to ntn (J. M. Gross, unpublished observations). These mutants and the pleiotropic nature of their phenotype did not fit the criteria of our screen as possessing eye-specific defects and, thus, they were not included in this report. The CCT complex is composed of eight subunits (reviewed in Dunn et al. 2001) and in addition to the γ-subunit, insertional mutants at four other CCT subunit-encoding loci have been identified (Golling et al. 2002; Amsterdam et al. 2004). Much like cct3, each of these mutants has small eyes and overall CNS degeneration (J. M. Gross, unpublished observations). By our screening criteria, these did not represent eye-specific phenotypes and so these mutants were not included in this report. These examples are meant to highlight the importance of specifically defining the screening criteria utilized herein. Other screening parameters might identify as eye mutants some of the insertional mutants excluded here and proceed in characterizing them as such.
From this screen, a diverse set of genes has been identified as playing important roles in eye development and visual function. Many of these have not yet been implicated in or well studied during eye development and therefore their molecular characterization should greatly increase our understanding of these processes as well as human pathologies that affect the eye. For example, two mutants, ift172 and ift57, show significant degrees of photoreceptor degeneration in their central retinas (Figure 6, E, E′, F, and F′). These loci encode members of the IFT protein family that are required in cilia for anterograde transport of proteins from the cytoplasm, along the ciliary axoneme to the distal cilia (reviewed in Pazour and Rosenbaum 2002). The disruption of several IFT proteins has been associated with retinitis pigmentosa-like pathologies, both in mouse and in zebrafish (Pazour et al. 2002; Tsujikawa and Malicki 2004). Additionally, insertional mutations at these two loci also result in polycystic kidneys, implicating a common pathway in the zebrafish embryo for the generation and/or maintenance of ciliated cell types (Sun et al. 2004). Thus, these mutants should provide an excellent in vivo model for understanding intraflagellar transport and its potential role in PR degeneration.
We identified six insertional loci with abnormal RPE morphology and pigmentation and varying degrees of retinal degeneration that encode components of the vacuolar ATPase (v-ATPase) protein complex or v-ATPase-associated proteins (Table 1). The v-ATPase is composed of two domains: the peripheral V1 complex consisting of eight subunits and the integral V0 complex consisting of five subunits (reviewed in Nishi and Forgac 2002). v-ATPases are best known for their roles in proton transport through which they play important roles in acidification of endosomes, protein degradation, endocytosis, intracellular transport, and membrane fusion. Several human pathologies have been attributed to defects in v-ATPases, such as osteoporosis, renal tubular acidosis, deafness, and possibly age-related macular degeneration (reviewed in Alper 2002; Bergmann et al. 2004). Age-related macular degeneration (AMD) affects approximately one-fourth of the population over age 65, resulting in varying degrees of visual impairment. The molecular mechanisms underlying AMD, however, are largely unknown. A hallmark of AMD progression is the accumulation of drusen, storage bodies composed mainly of undegraded photoreceptor outer segment lipids (Feeney-Burns et al. 1980; Crabb et al. 2002). A major component of drusen, A2E, accumulates in lysosomes and blocks their ability to degrade outer segment material (Finnemann et al. 2002). The v-ATPase pump is inhibited by high concentrations of A2E, suggesting a possible link between v-ATPase function and AMD progression (Bergmann et al. 2004). The six v-ATPase mutants identified in this screen show differing degrees of retinal degeneration, ranging from minor degeneration in atp6v0d1 (V0 subunit d; Figure 7G) to severe degeneration in atp6v1h (V1 subunit H; Figure 7H) and atp6v1f (V1 subunit F; Figure 7I). It will be interesting to look for adult phenotypes in heterozygous animals at these loci to determine if these are applicable animal models for the study of AMD and AMD-related pathologies in humans, several of which are autosomal dominant disorders.
We identified several mutations that affected the development of the central retina or the development of the peripheral MZs and that resulted from the disruption of genes encoding proteins that function in aspects of transcription, translation, and cell cycle regulation (Table 1). It is interesting to speculate on the nature of these mutations with respect to early retinal development as well as stem cell maintenance in the retinal periphery. It is possible that the central retinal defects manifest in the smarca5, ccna2, and fen1 mutants result from specific roles of these proteins in early events of retinal development, i.e., in the formation of specific retinal cell types. It is also possible, however, that these phenotypes result simply from a block in retinoblast proliferation such that most retinal cell types are missing, since their progenitors die at earlier phases of development. Thus, in these mutants, the role for the disrupted protein may not be specifically in retinal patterning, but rather may be in maintaining progenitor populations such that an appropriate number of progenitors are present to generate all retinal cell neurons. This logic can be applied to the mutants that displayed reduced MZs as well. Most of these resulted from disruption of a gene encoding a factor involved in DNA replication or mRNA modification (Table 1). Are these factors specifically required only for maintenance of the MZ stem cell population? It is doubtful that such proteins are not utilized in early retinal development. This suggests that a significant maternal complement of mRNA or protein must be present, and/or maternally supplied proteins must have unusually high stability such that they persist through several days of development. Early aspects of retinal development are thereby normal, but upon depletion of these maternal stores, a phenotype is manifest in the continually proliferative MZs. Indeed, a significant role for maternal factors in morphogenesis and establishment of the embryonic body plan after the zygotic transition has recently been reported (Wagner et al. 2004). These phenotypes therefore might represent a continuum, not of specific gene product function but actually of maternal load. Many mutants died or showed pleiotropic defects casting doubt on the specificity of their retinal phenotype and were not included in this report. Many of these mutants might represent one end of this continuum: low maternal load. The three mutants that displayed growth retardation and central retinal defects likely due to progenitor cell death might reside somewhere in the middle of the continuum. These mutants may have possessed enough maternal mRNA or protein to develop generally well but they manifest defects in slightly later aspects of retinal development when these maternal loads were depleted. Finally, the MZ mutants would reside at the other end of the continuum where a high maternal complement enabled them to develop quite normally and phenotypic defects were obvious only in the latest aspects of retinal development, i.e., in MZ maintenance. Thus, are these proteins specifically involved in retinal development or the maintenance of MZ cells in the retina or are they actually required in all proliferating cells, a function masked to differing degrees by maternal stores? A survey of gene expression in mammalian neural progenitors relative to differentiated neurons has pointed to a significant enrichment of transcripts in progenitors that encode proteins involved in DNA replication, protein synthesis, protein turnover, and chromatin remodeling (Livesey et al. 2004). This is not surprising given the high transcriptional activity and unique developmental functions of neural progenitor cells, suggesting that the latter of the above scenarios might be the case. Further characterization of these mutants will clearly be necessary however, to differentiate between these possibilities.
In summary, in this screen we have identified a diverse set of genes that are involved in visual system development and function. Many of these have not yet been implicated in, or studied during, eye development and therefore their molecular characterization should greatly increase the understanding of these processes as well as human pathologies that affect the eye. Additionally, multiple screens of this collection of insertional mutants, similar in scope to the one reported here, are currently underway to assay a variety of developmental, physiological, and behavioral parameters. As more of these screens are completed, an integrated picture should emerge as to the roles that each of these loci play during embryonic development. The synthesis of data from several such screens will likely reveal previously unidentified similarities between the formation and maintenance of multiple organ systems and physiological processes.
Acknowledgments
We are grateful to Steve Earle for assistance with histology and also to Dave Smith of the Harvard Imaging Facility. This work was supported by National Eye Institute grants EY015064 to J.M.G., EY013502 to B.D.P., EY014790 to J.I.M., and EY00811 to J.E.D. and a grant from the National Center for Research Resources to N.H.
References
- Allende, M. L., A. Amsterdam, T. Becker, K. Kawakami, N. Gaiano et al., 1996. Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes Dev. 10: 3141–3155. [DOI] [PubMed] [Google Scholar]
- Alper, S. L., 2002. Genetic diseases of acid-base transporters. Annu. Rev. Physiol. 64: 899–923. [DOI] [PubMed] [Google Scholar]
- Amsterdam, A., 2003. Insertional mutagenesis in zebrafish. Dev. Dyn. 228: 523–534. [DOI] [PubMed] [Google Scholar]
- Amsterdam, A., S. Burgess, G. Golling, W. Chen, Z. Sun et al., 1999. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 13: 2713–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam, A., R. M. Nissen, Z. Sun, E. C. Swindell, S. Farrington et al., 2004. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 101: 12792–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, T. S., S. M. Burgess, A. H. Amsterdam, M. L. Allende and N. Hopkins, 1998. not really finished is crucial for development of the zebrafish outer retina and encodes a transcription factor highly homologous to human nuclear respiratory factor-1 and avian initiation binding repressor. Development 125: 4369–4378. [DOI] [PubMed] [Google Scholar]
- Bergmann, M., F. Schutt, F. G. Holz and J. Kopitz, 2004. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 18(3): 562–564. [DOI] [PubMed] [Google Scholar]
- Brockerhoff, S. E., J. B. Hurley, U. Janssen-Bienhold, S. C. Neuhauss, W. Driever et al., 1995. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc. Natl. Acad. Sci. USA 92: 10545–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockerhoff, S. E., J. E. Dowling and J. B. Hurley, 1998. Zebrafish retinal mutants. Vision Res. 38: 1335–1339. [DOI] [PubMed] [Google Scholar]
- Crabb, J. W., M. Miyagi, X. Gu, K. Shadrach, K. A. West et al., 2002. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 99: 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever, W., L. Solnica-Krezel, A. F. Schier, S. C. Neuhauss, J. Malicki et al., 1996. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123: 37–46. [DOI] [PubMed] [Google Scholar]
- Dunn, A. Y., M. W. Melville and J. Frydman, 2001. Review: cellular substrates of the eukaryotic chaperonin TRiC/CCT. J. Struct. Biol. 135: 176–184. [DOI] [PubMed] [Google Scholar]
- Easter, S. S., Jr., and J. J. Malicki, 2002. The zebrafish eye: developmental and genetic analysis. Results Probl. Cell Differ. 40: 346–370. [DOI] [PubMed] [Google Scholar]
- Easter, S. S., Jr., and G. N. Nicola, 1996. The development of vision in the zebrafish (Danio rerio). Dev. Biol. 180: 646–663. [DOI] [PubMed] [Google Scholar]
- Erdmann, B., F. P. Kirsch, F. G. Rathjen and M. I. More, 2003. N-cadherin is essential for retinal lamination in the zebrafish. Dev. Dyn. 226: 570–577. [DOI] [PubMed] [Google Scholar]
- Fadool, J. M., S. E. Brockerhoff, G. A. Hyatt and J. E. Dowling, 1997. Mutations affecting eye morphology in the developing zebrafish (Danio rerio). Dev. Genet. 20: 288–295. [DOI] [PubMed] [Google Scholar]
- Feeney-Burns, L., E. R. Berman and H. Rothman, 1980. Lipofuscin of human retinal pigment epithelium. Am. J. Ophthalmol. 90: 783–791. [DOI] [PubMed] [Google Scholar]
- Fiedler, K., and K. Simons, 1996. Characterization of VIP36, an animal lectin homologous to leguminous lectins. J. Cell Sci. 109(Pt. 1): 271–276. [DOI] [PubMed] [Google Scholar]
- Fiedler, K., R. G. Parton, R. Kellner, T. Etzold and K. Simons, 1994. VIP36, a novel component of glycolipid rafts and exocytic carrier vesicles in epithelial cells. EMBO J. 13: 1729–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnemann, S. C., L. W. Leung and E. Rodriguez-Boulan, 2002. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 99: 3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, A. J., and T. A. Reh, 2000. Identification of a proliferating marginal zone of retinal progenitors in postnatal chickens. Dev. Biol. 220: 197–210. [DOI] [PubMed] [Google Scholar]
- Fulwiler, C., E. A. Schmitt, J. M. Kim and J. E. Dowling, 1997. Retinal patterning in the zebrafish mutant cyclops. J. Comp. Neurol. 381: 449–460. [PubMed] [Google Scholar]
- Gaiano, N., A. Amsterdam, K. Kawakami, M. Allende, T. Becker et al., 1996. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature 383: 829–832. [DOI] [PubMed] [Google Scholar]
- Goldsmith, P., and W. A. Harris, 2003. The zebrafish as a tool for understanding the biology of visual disorders. Semin. Cell Dev. Biol. 14: 11–18. [DOI] [PubMed] [Google Scholar]
- Golling, G., A. Amsterdam, Z. Sun, M. Antonelli, E. Maldonado et al., 2002. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat. Genet. 31: 135–140. [DOI] [PubMed] [Google Scholar]
- Haffter, P., M. Granato, M. Brand, M. C. Mullins, M. Hammerschmidt et al., 1996. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123: 1–36. [DOI] [PubMed] [Google Scholar]
- Hatta, K., C. B. Kimmel, R. K. Ho and C. Walker, 1991. The cyclops mutation blocks specification of the floor plate of the zebrafish central nervous system. Nature 350: 339–341. [DOI] [PubMed] [Google Scholar]
- Heisenberg, C. P., M. Brand, Y. J. Jiang, R. M. Warga, D. Beuchle et al., 1996. Genes involved in forebrain development in the zebrafish, Danio rerio. Development 123: 191–203. [DOI] [PubMed] [Google Scholar]
- Horne-Badovinac, S., D. Lin, S. Waldron, M. Schwarz, G. Mbamalu et al., 2001. Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr. Biol. 11: 1492–1502. [DOI] [PubMed] [Google Scholar]
- Hu, M., and S. S. Easter, 1999. Retinal neurogenesis: the formation of the initial central patch of postmitotic cells. Dev. Biol. 207: 309–321. [DOI] [PubMed] [Google Scholar]
- Johns, P. R., 1977. Growth of the adult goldfish eye. III. Source of the new retinal cells. J. Comp. Neurol. 176: 343–357. [DOI] [PubMed] [Google Scholar]
- Kainz, P. M., A. R. Adolph, K. Y. Wong and J. E. Dowling, 2003. Lazy eyes zebrafish mutation affects Muller glial cells, compromising photoreceptor function and causing partial blindness. J. Comp. Neurol. 463: 265–280. [DOI] [PubMed] [Google Scholar]
- Kimmel, C. B., W. W. Ballard, S. R. Kimmel, B. Ullmann and T. F. Schilling, 1995. Stages of embryonic development of the zebrafish. Dev. Dyn. 203: 253–310. [DOI] [PubMed] [Google Scholar]
- Lele, Z., A. Folchert, M. Concha, G. J. Rauch, R. Geisler et al., 2002. parachute/n-cadherin is required for morphogenesis and maintained integrity of the zebrafish neural tube. Development 129: 3281–3294. [DOI] [PubMed] [Google Scholar]
- Link, B. A., and T. Darland, 2001. Genetic analysis of initial and ongoing retinogenesis in the zebrafish: comparing the central neuroepithelium and marginal zone. Prog. Brain Res. 131: 565–577. [DOI] [PubMed] [Google Scholar]
- Livesey, F. J., T. L. Young and C. L. Cepko, 2004. An analysis of the gene expression program of mammalian neural progenitor cells. Proc. Natl. Acad. Sci. USA 101: 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity, S. N., and B. de Crombrugghe, 1998. Role of the CCAAT-binding protein CBF/NF-Y in transcription. Trends Biochem. Sci. 23: 174–178. [DOI] [PubMed] [Google Scholar]
- Malicki, J., S. C. Neuhauss, A. F. Schier, L. Solnica-Krezel, D. L. Stemple et al., 1996. Mutations affecting development of the zebrafish retina. Development 123: 263–273. [DOI] [PubMed] [Google Scholar]
- Malicki, J., H. Jo and Z. Pujic, 2003. Zebrafish N-cadherin, encoded by the glass onion locus, plays an essential role in retinal patterning. Dev. Biol. 259: 95–108. [DOI] [PubMed] [Google Scholar]
- Marmorstein, A. D., 2001. The polarity of the retinal pigment epithelium. Traffic 2: 867–872. [DOI] [PubMed] [Google Scholar]
- Matsuda, N., and M. Mishina, 2004. Identification of chaperonin CCT gamma subunit as a determinant of retinotectal development by whole-genome subtraction cloning from zebrafish no tectal neuron mutant. Development 131: 1913–1925. [DOI] [PubMed] [Google Scholar]
- Muto, M., M. Utsuyama, T. Horiguchi, E. Kubo, T. Sado et al., 1995. The characterization of the monoclonal antibody Th-10a, specific for a nuclear protein appearing in the S phase of the cell cycle in normal thymocytes and its unregulated expression in lymphoma cell lines. Cell Proliferation 28: 645–657. [DOI] [PubMed] [Google Scholar]
- Neuhauss, S. C., 2003. Behavioral genetic approaches to visual system development and function in zebrafish. J. Neurobiol. 54: 148–160. [DOI] [PubMed] [Google Scholar]
- Neuhauss, S. C., O. Biehlmaier, M. W. Seeliger, T. Das, K. Kohler et al., 1999. Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J. Neurosci. 19: 8603–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi, T., and M. Forgac, 2002. The vacuolar (H+)-ATPases—nature's most versatile proton pumps. Nat. Rev. Mol. Cell. Biol. 3: 94–103. [DOI] [PubMed] [Google Scholar]
- Parsons, M. J., S. M. Pollard, L. Saude, B. Feldman, P. Coutinho et al., 2002. Zebrafish mutants identify an essential role for laminins in notochord formation. Development 129: 3137–3146. [DOI] [PubMed] [Google Scholar]
- Pazour, G. J., and J. L. Rosenbaum, 2002. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 12: 551–555. [DOI] [PubMed] [Google Scholar]
- Pazour, G. J., S. A. Baker, J. A. Deane, D. G. Cole, B. L. Dickert et al., 2002. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 157: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, R. T., J. D. Mably, J. N. Chen and M. C. Fishman, 2001. Convergence of distinct pathways to heart patterning revealed by the small molecule concentramide and the mutation heart-and-soul. Curr. Biol. 11: 1481–1491. [DOI] [PubMed] [Google Scholar]
- Pujic, Z., and J. Malicki, 2001. Mutation of the zebrafish glass onion locus causes early cell-nonautonomous loss of neuroepithelial integrity followed by severe neuronal patterning defects in the retina. Dev. Biol. 234: 454–469. [DOI] [PubMed] [Google Scholar]
- Rebagliati, M. R., R. Toyama, P. Haffter and I. B. Dawid, 1998. cyclops encodes a nodal-related factor involved in midline signaling. Proc. Natl. Acad. Sci. USA 95: 9932–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath, K., A. L. Rubinstein, A. M. Cheng, J. O. Liang, K. Fekany et al., 1998. Induction of the zebrafish ventral brain and floorplate requires cyclops/nodal signalling. Nature 395: 185–189. [DOI] [PubMed] [Google Scholar]
- Schmitt, E. A., and J. E. Dowling, 1994. Early eye morphogenesis in the zebrafish, Brachydanio rerio. J. Comp. Neurol. 344: 532–542. [DOI] [PubMed] [Google Scholar]
- Schmitt, E. A., and J. E. Dowling, 1999. Early retinal development in the zebrafish, Danio rerio: light and electron microscopic analyses. J. Comp. Neurol. 404: 515–536. [PubMed] [Google Scholar]
- Sun, Z., A. Amsterdam, G. J. Pazour, D. G. Cole, M. S. Miller et al., 2004. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development 131: 4085–4093. [DOI] [PubMed] [Google Scholar]
- Tropepe, V., B. L. Coles, B. J. Chiasson, D. J. Horsford, A. J. Elia et al., 2000. Retinal stem cells in the adult mammalian eye. Science 287: 2032–2036. [DOI] [PubMed] [Google Scholar]
- Tsujikawa, M., and J. Malicki, 2004. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 42: 703–716. [DOI] [PubMed] [Google Scholar]
- Van Epps, H. A., C. M. Yim, J. B. Hurley and S. E. Brockerhoff, 2001. Investigations of photoreceptor synaptic transmission and light adaptation in the zebrafish visual mutant nrc. Invest. Ophthalmol. Visual Sci. 42: 868–874. [PubMed] [Google Scholar]
- Vihtelic, T. S., Y. Yamamoto, M. T. Sweeney, W. R. Jeffery and D. R. Hyde, 2001. Arrested differentiation and epithelial cell degeneration in zebrafish lens mutants. Dev. Dyn. 222: 625–636. [DOI] [PubMed] [Google Scholar]
- Wagner, D. S., R. Dosch, K. A. Mintzer, A. P. Wiemelt and M. C. Mullins, 2004. Maternal control of development at the midblastula transition and beyond: mutants from the zebrafish II. Dev. Cell 6: 781–790. [DOI] [PubMed] [Google Scholar]
- Wei, X., and J. Malicki, 2002. nagie oko, encoding a MAGUK-family protein, is essential for cellular patterning of the retina. Nat. Genet. 31: 150–157. [DOI] [PubMed] [Google Scholar]
- Westerfield, M., 1995 The Zebrafish Book. University of Oregon Press, Eugene, OR.
- Wetts, R., and S. E. Fraser, 1988. Multipotent precursors can give rise to all major cell types of the frog retina. Science 239: 1142–1145. [DOI] [PubMed] [Google Scholar]
- Zambrowicz, B. P., A. Abuin, R. Ramirez-Solis, L. J. Richter, J. Piggott et al., 2003. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl. Acad. Sci. USA 100: 14109–14114. [DOI] [PMC free article] [PubMed] [Google Scholar]