

Abstract
Recently, we reported that two siblings presenting with the clinical syndrome congenital disorders of glycosylation (CDG) have mutations in the gene encoding Cog7p, a member of the conserved oligomeric Golgi (COG) complex. In this study, we analyzed the localization and trafficking of multiple Golgi proteins in patient fibroblasts under a variety of conditions. Although the immunofluorescent staining pattern of several Golgi proteins was indistinguishable from normal, the staining of endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC)-53 and the vesicular-soluble N-ethylmaleimide-sensitive factor attachment protein receptors GS15 and GS28 was abnormal, and the steady-state level of GS15 was greatly decreased. Retrograde transport of multiple Golgi proteins to the ER in patient fibroblasts via brefeldin A-induced tubules was significantly slower than occurs in normal fibroblasts, whereas anterograde protein trafficking was much less affected. After prolonged treatment with brefeldin A, several Golgi proteins were detected in clusters that colocalize with the microtubule-organizing center in patient cells. All of these abnormalities were normalized in COG7-corrected patient fibroblasts. These results serve to better define the role of the COG complex in facilitating protein trafficking between the Golgi and ER and provide a diagnostic framework for the identification of CDG defects involving trafficking proteins.
INTRODUCTION
The congenital disorders of glycosylation (CDG) are a group of autosomal recessive multisystem disorders typically defined by defects in proteins directly involved in the N-linked glycosylation pathway (Freeze, 2001; Marquardt and Denecke, 2003). The molecular defects resulting in CDG were recently expanded to include proteins involved in overall Golgi function. Specifically, two siblings were described with a fatal form of CDG (classified as CDG-IIe) caused by mutation in the gene encoding Cog7p, a member of the conserved oligomeric Golgi (COG) complex (Wu et al., 2004). Fibroblasts from these patients are deficient in COG subunits 5-8 that compromise lobe B of the complex and exhibit subtle decreases in the sialylation of both N- and O-linked oligosaccharides (Wu et al., 2004; Oka et al., 2005). The glycosylation defects were COG dependent because they were effectively rescued by transduction of CDG-IIe fibroblasts with wild-type COG7 cDNA.
The vesicle tethering COG complex is thought to mediate numerous steps in anterograde and retrograde transport within the secretory apparatus (reviewed in Oka and Krieger, 2005). A role for the COG complex in maintaining the fidelity of retrograde transport within the Golgi has been supported by recent evidence indicating that acute depletion of COG3 prevents tethering of retrograde vesicles within the Golgi resulting in the accumulation of Golgi proteins, including the soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) GS15 and GS28 in COG complex-dependent (CCD) vesicles (Zolov and Lupashin, 2005). Because resident Golgi proteins such as glycosylation enzymes are known to recycle within the Golgi stack and between the Golgi and ER, these findings support the general hypothesis that altered trafficking of these proteins results in their mislocalization, altered activity, possible degradation, or a combination. Indeed, a role for the COG complex in controlling the abundance of a subset of Golgi proteins (including glycosylation enzymes) in Chinese hamster ovary (CHO) cells has been recently demonstrated by Krieger and colleagues (Oka et al., 2004).
In contrast, a role for the COG complex in trafficking events between the endoplasmic reticulum (ER) and Golgi is less established (Sztul and Lupashin, 2006). Studies in COG-deficient yeast mutants suggest that COG may play a role in ER export and ER-to-Golgi transport (VanRheenen et al., 1998, 1999; Morsomme et al., 2003). In support of a functional role for the COG complex in trafficking between the ER and Golgi, we observed slower recovery of Golgi localization of a green fluorescent protein (GFP)-sialyltransferase chimera after photobleaching in patient fibroblasts (Wu et al., 2004). Despite the fact that slower recovery of Golgi localization can most directly be interpreted by a block in anterograde transport, the directionality of transport affected was not conclusive from these experiments because preexisting protein in intermediate compartments (i.e., ER-Golgi intermediate compartment [ERGIC]) may move slowly back to the ER before entering the Golgi.
In the present study, we have analyzed the localization and trafficking of multiple Golgi- and ERGIC-localized proteins in control and COG7-deficient patient fibroblasts under several different conditions with the goal of better defining the intracellular membrane trafficking pathways perturbed by the Cog7p defect. Our findings reveal a number of alterations in protein trafficking and stability in the COG7-deficient cells under steady-state conditions. Furthermore, brefeldin A (BFA) treatment of COG7-deficient fibroblasts unveils several additional defects in the localization and trafficking of Golgi proteins. The implications of these findings on the function of the COG complex, the clinical features in CDG-IIe patients, and the diagnosis of other COG-deficient patients are discussed.
MATERIALS AND METHODS
Reagents and Antibodies
Reagents and sources were as follows: ECL Plus detection kit (Amersham Biosciences, Piscataway, NJ), methionine- and cysteine-free Ham's F-12 medium (Invitrogen, Carlsbad, CA), Trans 35S protein labeling mix (MP Biomedicals, Irvine, CA), MG132 (Calbiochem, San Diego, CA), protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), and BFA (Calbiochem) prepared in stock solutions (5 mg/ml) with dimethyl sulfoxide. All other reagents including leupeptin and pepstatin A were obtained from Sigma-Aldrich (St. Louis, MO).
Antibodies used for immunoblot (IB) and immunofluorescence (IF) studies were obtained from standard commercial sources and as gifts from generous individual investigators (see below). Antibodies (and their appropriate dilutions) were as follows: rabbit polyclonal antibodies: anti-GPP130 antibody (IB, 1:500; IF, 1:250; Covance, Berkeley, CA), anti-giantin antibody (IB, 1:2000; IF, 1:1000; Covance), and anti-β COP antibody (IF, 1:2000; Abcam, Cambridge, MA); and murine monoclonal antibodies: anti-ERGIC-53 antibody (IF, 1:500; a generous gift from Dr. Hans-Peter Hauri, University of Basel, Basel, Switzerland), anti-β1,4-galactosyltransferase (βGalT) antibody (clone 36/118; IB, 1:20; IF, 1:50; a generous gift from Dr. Eric Berger, University of Zurich, Zurich, Switzerland), anti-GM130 antibody (IF, 1:100; IB, 1:250; BD Biosciences, San Jose, CA), anti-GS28 antibody (IB, 1:3000; IF, 1:1000; BD Biosciences), anti-GS15 antibody (IB, 1:250, IF, 1:100; BD Biosciences), anti-GS27 antibody (IB, 1:1000; IF, 1:250; BD Biosciences), anti-γ-adaptin antibody (IF, 1:500; BD Biosciences), anti-γ-tubulin (IF, 1:1000; Sigma-Aldrich), and anti-β-tubulin (IB, 1:5000; Sigma-Aldrich). Alexa Fluor-conjugated secondary antibodies for immunofluorescence were obtained from Invitrogen, and species-specific horseradish peroxidase-conjugated secondary antibodies used for Western blotting were purchased from Amersham Biosciences.
Cell Lines
Fibroblasts were maintained in DMEM supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 U/ml streptomycin. COG7-corrected cells were prepared by retroviral transduction of patient fibroblasts as described previously (Wu et al., 2004). In experiments where cells were grown at 15°C, plates were sealed with parafilm and incubated in a temperature-controlled environment inside a cold room. The proteasome inhibitor MG132 was added to cultures to a final concentration of 25 μM for 10 h before Western blot analysis. Inhibition of lysosomal proteases was achieved by addition of 150 μM leupeptin and 100 μM pepstatin A to the culture medium for 16 h before analysis.
BFA-induced Retrograde and Anterograde Transport Experiments
Cells were treated with various concentrations of BFA (1 or 5 μg/ml) for the retrograde and coat release experiments. Cell populations for any given experiment were routinely quantitated in a blinded manner. For the recovery experiments, BFA-containing media were removed by aspiration followed by three washes with phosphate-buffered saline (PBS)+ (PBS containing 1 mM Ca2+ and Mg2+) to prevent cell loss. Coverslips were then removed from the wells and washed carefully on both sides with PBS+ before being placed in new wells containing fresh media warmed to 37°C. A lower concentration of BFA (0.25 μg/ml) was used for these experiments to achieve washout of the drug in fibroblasts. Likewise, longer treatment times were necessary at this concentration to fully redistribute Golgi proteins to the ER. Reversal of the effects of BFA could not be accomplished in a shorter time scale in these cells despite exhaustive measures (described above) to washout the drug and may be attributed to differences in the sensitivity to the drug and/or cell permeability.
Immunoblotting and Indirect Immunofluorescence Microscopy
For the determination of protein levels, cells were grown at 37°C in culture medium to near confluence, harvested, and solubilized by addition of SDS lysis buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 10% glycerol) with scraping, and heated at 95°C for 5-10 min. The lysates (60 μg of protein) were then fractionated by SDS-polyacrylamide (8 and 12%) gel electrophoresis, and the proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA) and immunoblotted with the indicated antibodies using the ECL Plus detection kit (Amersham Biosciences) according to the manufacturer's instructions. Protein concentration in lysates was determined using the MicroBCA protein assay reagent kit (Pierce Chemical, Rockford, IL). Cells grown on coverslips were fixed with 3% paraformaldehyde, stained, and analyzed by immunofluorescence microscopy as described previously (Ghosh et al., 2003). The standard dilution buffer for primary and secondary antibodies contained PBS, pH 7.4, 0.1% Triton X-100, and 1 mg/ml bovine serum albumin. The slides were viewed with an Eclipse fluorescence microscope (model E-800; Nikon, Tokyo, Japan), and images were acquired using a Magnafire camera system from Optronics (Goleta, CA). Images were prepared using Adobe Photoshop (Adobe Systems, Mountain View, CA), and only linear adjustments were made.
Confocal Microscopy
Confocal images were acquired using an Olympus FV500 laser scanning microscope equipped with a 60× oil immersion (numerical aperture 1.4) objective. Stacks of 0.25 μM (based on calculated optimums) optical sections were collected in the z-dimension. Each stack was subsequently collapsed into a single image (maximum intensity or z-projection), and further analyses were performed offline using the MetaMorph software (Molecular Devices, Sunnyvale, CA).
CD59 Immunoprecipitation Experiments
Cells grown at 37°C until 80-90% confluent were pulse labeled with [35S]cysteine in cysteine-free media for 20 min and chased for various times. Lysates were prepared by scraping cells on ice in cell lysis buffer (CLB; 10 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.05% SDS, and protease inhibitor cocktail). Newly synthesized CD59 was immunoprecipitated overnight using a monoclonal antibody to human CD59 (Serotec, Oxford, United Kingdom). After addition of protein A-agarose beads, samples were washed four times with CLB before elution of labeled CD59 molecules and analysis by SDS-PAGE and autoradiography.
RESULTS
Retrograde Trafficking of Golgi Proteins Is Altered in COG7-deficient Fibroblasts
We first sought to clarify the direction of transport between the ER and Golgi that was affected in COG7-deficient cells. For these studies, we used the fungal metabolite BFA, a selective inhibitor of the GTP-exchange factor for ADP-ribosylation factor (ARF) that induces rapid collapse of the Golgi apparatus into the ER via tubule-like structures (Lippincott-Schwartz et al., 1989, 1990). In the initial experiments, control and patient (P1) fibroblasts were treated with BFA and the localization of the Golgi matrix protein giantin was observed at various times in fixed cells (Figure 1). No detectable differences were noted in the localization of giantin in untreated cells (Figure 1A). After 12-min treatment with BFA, nearly all the control fibroblasts exhibited a staining pattern consistent with ER localization. In contrast, giantin remained predominantly localized to the Golgi ribbon at this time point in P1 fibroblasts indicating a delay in the effects of BFA. To gain a more quantitative assessment of the delayed response to BFA observed in the mutant fibroblasts, cells within a given population were classified into one of four staining patterns that reflect the various stages of Golgi collapse into the ER (Figure 1B). Whereas the majority of control cells exhibited ER staining for giantin (pattern 4) after 12-min treatment with BFA, only a small proportion of P1 cells displayed complete redistribution of giantin to the ER at this time point (70 versus 6%). The majority (79%) of P1 cells showed incomplete Golgi collapse (patterns 2 and 3), whereas a small fraction (15%) exhibited no observable alteration in Golgi ribbon structure (pattern 1; Figure 1B). Fibroblasts from the sibling of P1 (termed P2) displayed an identical delay in the response to BFA (our unpublished data).
Figure 1.

BFA-induced retrograde transport of the Golgi matrix protein giantin is delayed in COG-deficient patient fibroblasts. (A) Control, patient, and COG7-corrected patient fibroblasts were treated with 5 μg/ml BFA for 12 min, and the localization of giantin was analyzed by immunofluorescence. Bar, 20 μm. (B) Stained cells within a population (200-300) were classified into one of four typical staining patterns (1-4) reflecting the BFA-induced collapse of the Golgi into the ER. Pattern 1, intact Golgi ribbon; pattern 2, early tubule formation; pattern 3, overt tubulation and loss of ribbon structure; and pattern 4, ER localization. The relative percentage of cells in a representative experiment within these classifications is shown. Highly similar percentages were observed in independent experiments. The arrows indicate BFA-induced tubules. (C) The percentage of cells with an exclusive ER staining pattern was plotted as a function of time. The data represent the average of three independent experiments; errors bars indicate standard deviations. The symbols are as follows: control, closed circles; P1, closed squares; and COG7-corrected P1, closed triangles.
The kinetics of the Golgi collapse is shown in Figure 1C, where the percentage of cells with an exclusive ER staining pattern is plotted as a function of time. Extrapolation of t1/2 values (time at which 50% of the cells exhibit an ER staining pattern) reveals that Golgi collapse is roughly 2.5 times slower in P1 fibroblasts (t1/2 of 24 min) relative to control cells (t1/2 of 10 min). The delayed response to BFA was COG7 dependent because BFA-treated COG7-corrected P1 fibroblasts are nearly indistinguishable from control cells (Figure 1, A and B) and displayed similar kinetics of Golgi collapse (t1/2 of 13 min). The lack of complete correction of the delay in COG7-corrected P1 cells is likely due to not all cells within the population being transduced with retrovirus containing wild-type COG7 cDNA. Similar results were observed with the Golgi enzyme βGalT (Supplemental Figure S1), demonstrating that the block in Golgi-to-ER retrograde transport in COG7-deficient cells is not selective for matrix proteins.
Release of coat proteins from Golgi membranes is one of the earliest detectable changes in cells treated with BFA and is necessary for tubule formation (Donaldson et al., 1990; Lippincott-Schwartz, 1993). The delayed onset of BFA-induced Golgi collapse could therefore be due to slower coat protein release after ARF inactivation. The initial release of two coat proteins, the Golgi COPI component βCOP and the trans-Golgi network adaptor AP-1, was examined after BFA treatment, and the results are shown in Figure 2. Whereas βCOP is localized primarily to the Golgi in control and patient cells, a shift in its distribution to the cytoplasm occurred rapidly in both cell types and was complete after only 3-min exposure to BFA. The release of AP-1 from the trans-Golgi network was somewhat slower compared with βCOP in control and patient fibroblasts. As seen with βCOP, no difference in the initial kinetics of AP-1 release could be detected. Therefore, the delayed Golgi collapse after BFA treatment in patient fibroblasts occurs after ARF-guanine-nucleotide exchange factor inhibition and coat protein release.
Figure 2.

Initial kinetics of coat protein release are not altered in patient fibroblasts after BFA treatment. Control and patient fibroblasts were treated with or without 1 μg/ml BFA for various times and the localization of βCOP and AP-1 was followed by immunofluorescence. Bar, 20 μm.
We next examined anterograde movement of giantin from the ER back to the Golgi after treatment and washout of BFA. After 60 min. treatment, >95% of both control and P1 fibroblasts exhibited a predominantly ER staining pattern for giantin. Importantly, single giantin-positive clusters were observed in nearly all patient cells (Table 1, see arrows at 0 h recovery time). These clusters, detected in a small percentage of control cells (<5%) as well, became most apparent after prolonged BFA treatment. At each time point of recovery, qualitatively similar staining patterns were observed (Figure 3A). To obtain a quantitative assessment, cells were classified into one of four typical staining patterns that reflect the emergence of giantin from the ER back to the Golgi (Figure 3B). Complete recovery of the localization of giantin into a ribbon-like Golgi structure (pattern 4r) was slightly delayed in P1 fibroblasts (73 ± 2 versus 55 ± 5% at 4-h washout). This delay, however, was much less striking compared with the differences observed for BFA-induced retrograde transport (Figure 1). Similar results were obtained with βGalT (Supplemental Figure S2). βGalT-positive membrane clusters were also noted in patient fibroblasts, although these unique structures were often less defined, seen in fewer cells compared with giantin, and not detectable in control cells (Table 1).
Table 1.
Quantitation of clusters in control and P1 fibroblasts after BFA treatment
| % of cells with clusters
|
|||
|---|---|---|---|
| Golgi protein | CTRL | P1 | P1[COG7] |
| GM130 | 0 | 2 ± 1 | N.D. |
| Giantin | 3 ± 2 | 87 ± 1 | 7 ± 2 |
| GPP130 | 0 | 46 ± 4 | 3 ± 1 |
| GS27 | 0 | 0 | N.D. |
| GS28 | 1 ± 1 | 38 ± 4 | N.D. |
| βGalT | 0 | 30 ± 7 | 0 |
Cells were treated with 0.25 μg/ml BFA for 60 min, and cells with single juxtanuclear clusters within populations of 200–300 were quantitated. Values represent the average of two to three independent experiments. Errors represent SDs. N.D., not determined.
Figure 3.

Recovery of Golgi localization of giantin is not substantially altered in COG-deficient patient fibroblasts after BFA washout. (A) Control, patient, and COG7-corrected patient fibroblasts were treated with 0.25 μg/ml BFA for 60 min followed by washout of the drug as described in Materials and Methods. The localization of giantin at various recovery times was followed by immunofluorescence. The arrows indicate the stable, residual Golgi-like staining of giantin after BFA treatment. Bar, 20 μm (B) Stained cells within a population (200-300) were classified into one of four typical staining patterns (1-4r) reflecting the recovery of Golgi localization. Pattern 1r, ER localization; pattern 2r, ERGIC localization; pattern 3r, Golgi mini-stacks; and pattern 4r, Golgi ribbon. The average percentage of cells within these classifications in two independent experiments is shown. Errors represent SEs of the mean. a, cells with single, concentrated remnant-like structures were counted as exhibiting full ER localization (#1r; see Materials and Methods).
Because a small delay in the return of resident proteins to the Golgi after BFA washout was observed, we investigated whether the transport of cargo proteins from the ER-to-Golgi might be altered in COG7-deficient fibroblasts. Indeed, experiments in COG-deficient yeast cells have suggested that subunits of the COG complex play a role in protein sorting upon ER export of glycosylphosphatidylinositol (GPI)-linked proteins (Morsomme and Riezman, 2002). Pulse-chase experiments were performed to follow the forward transport of the GPI-linked protein CD59 in control and P1 cells. The addition of N- and O-linked oligosaccharides on CD59 results in changes in the size of this molecule and indicates its transport from the ER into the Golgi (Rudd et al., 1997). No detectable difference in the maturation of CD59 was observed between control and P1 fibroblasts providing further support for the selective nature of protein transport defect in COG7-deficient cells (Supplemental Figure S3). This observation is consistent with our previous finding that the sorting of the lysosomal enzyme cathepsin D (a measure of both ER-to-Golgi and Golgi-to-lysosome transport) was not detectably altered in COG7-deficient fibroblasts (Wu et al., 2004).
Localization and Stability of Golgi Proteins in Control and Patient Fibroblasts
The localization of resident Golgi proteins has been examined previously in COG1- and COG2-deficient CHO cells as well as in COG3-depleted HeLa cells (Oka et al., 2004; Zolov and Lupashin, 2005). In these studies, a subset of Golgi proteins (termed GEARs) were identified whose intracellular distribution and stability was COG sensitive. These proteins included the Golgi matrix components GM130 and giantin; the Golgi SNAREs GS15, GS27, and GS28; and the cis-Golgi glycoprotein GPP130. Because no gross alteration in the localization of the cis/medial Golgi marker giantin and the trans-Golgi marker βGalT was observed in P1 fibroblasts, we examined the steady-state localization of the COG-sensitive Golgi proteins by immunofluorescence microscopy in an effort to determine whether their localization was affected. GM130 and GPP130 exhibited a typical Golgi ribbon-like staining pattern in control cells and this distribution was similar in patient fibroblasts (Figure 4). GS27 had a diffuse Golgi staining pattern in both cell types, consistent with both Golgi and ERGIC localization. Although GS15 exhibits a polarized Golgi localization in control cells, the staining of this protein was substantially lower and more dispersed in P1 cells. The abnormal localization of GS15 in these cells was COG7-dependent because its distribution and intensity in COG7-corrected P1 cells was comparable with control cells (Figure 5A). The localization of GS28 in COG7-deficient fibroblasts was also abnormal but to a lesser extent compared with GS15.
Figure 4.

Immunofluorescence localization of Golgi proteins in the presence and absence of BFA. Control and patient fibroblasts were treated with or without 0.25 μg/ml BFA for 60 min, and the localization of the marker proteins was followed by immunofluorescence. Arrows indicate the presence of remnant-like structures for some proteins. Note that some Golgi proteins redistribute to the ER while others (GM130 and GS27) redistribute predominantly to ER exit sites. Bar, 20 μm.
Figure 5.

Abnormal localization of ERGIC and Golgi proteins is normalized in COG7-corrected patient fibroblasts. The localization of GS15, ERGIC-53, and GPP130 were followed by immunofluorescence. Golgi localization of GS15 (A) and ERGIC localization of ERGIC-53 (B) is restored in COG7-corrected P1 fibroblasts. (C) P1 and COG7-corrected P1 fibroblasts were treated with 0.25 μg/ml BFA for 60 min. Note the lack of BFA-induced clusters in the COG7-corrected cells. Quantitation of the frequency of GPP130-positive remnants observed in COG7-corrected cells is shown in Table 1.
The presence of clusters containing giantin and βGalT predominantly in P1 but not control fibroblasts after prolonged BFA treatment indicated that differences in the localization of Golgi resident proteins may emerge at nonequilibrium conditions. Therefore, the localization of the GEAR proteins was examined after BFA treatment to determine whether formation of these clusters was a general phenomenon. As shown in Figure 4, GPP130 redistributed completely to the ER in control fibroblasts after BFA treatment. GS28 and GS15 exhibit a more granular ER-like staining pattern in these cells upon BFA treatment. Although the majority of these proteins also redistributed to the ER in P1 fibroblasts, clusters similar to that observed for giantin and βGalT were noted for GPP130 and GS28 after treatment with BFA. Because of the weak staining of GS15 in untreated P1 fibroblasts, it is unclear whether these structures are present. In contrast, GS27 and GM130 redistribute primarily to punctate structures uniformly dispersed toward the cell periphery in response to BFA treatment in both control and patient fibroblasts. These punctate structures are characteristic of protein accumulation in ER exit sites and have been previously observed for GM130 as well as other proteins such as ERGIC-53 (Lippincott-Schwartz et al., 1990; Ward et al., 2001; Puri and Linstedt, 2003). Importantly, no juxtanuclear, cluster staining of GM130 and GS27 was detected in either control or P1 fibroblasts. In addition, clusters were not readily detected in BFA-treated COG7-corrected patient fibroblasts (Figure 5C), demonstrating the COG-dependent nature of these structures. The percentage of cells that exhibit clusters upon BFA treatment is summarized in Table 1. Although the relative number of cells with these structures varies between proteins, only those proteins that redistribute completely within the ER upon BFA treatment in control cells (βGalT, GPP130, giantin, and GS28) develop these unique structures in P1 fibroblasts.
To determine whether differences in localization correlate with decreases in steady-state levels, the relative amounts of the various Golgi proteins was assessed by immunoblotting. We found a striking reduction in GS15 in both P1 and P2 fibroblasts (Figure 6A). This decrease was COG7 dependent because GS15 levels were normalized in COG7-corrected P1 fibroblasts (Figure 6B). Consistent with their normal staining pattern, the levels of GS27 and βCOP were not substantially reduced in P1 fibroblasts (Figure 6B). Treatment of P1 cells with either the proteasome inhibitor MG132 or the lysosomal protease inhibitors leupeptin and pepstatin A failed to increase the levels of GS15 (Figure 6C). Although we found decreased levels of multiple Golgi proteins, including GPP130, giantin, βGalT, and GS28 in some experiments, the reduction in these proteins was highly variable, and, in some cases did not seem to be dependent on COG7 because the levels were also decreased in COG7-corrected P1 fibroblasts. Furthermore, little or no decrease in the levels of these proteins was observed in P2 fibroblasts (our unpublished data).
Figure 6.
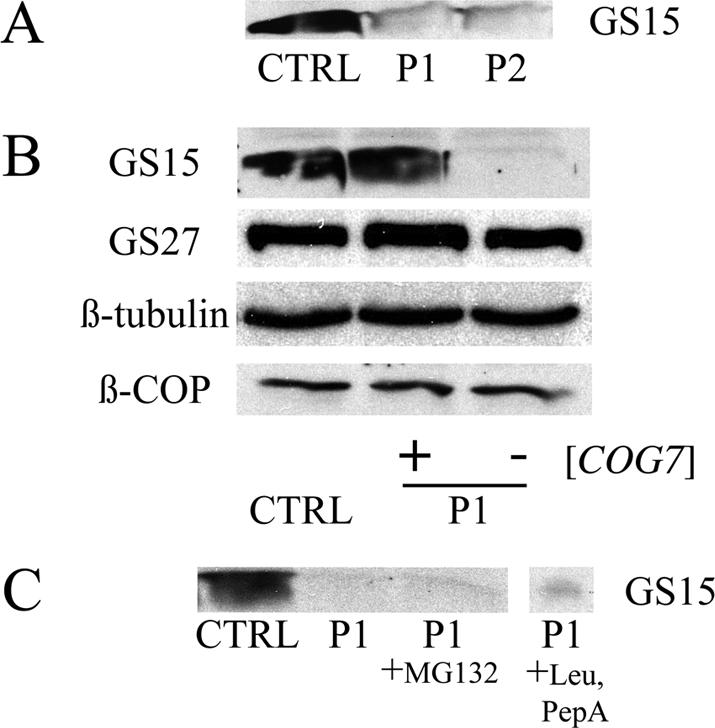
Steady-state level of GS15 is reduced in patient fibroblasts. Representative immunoblots are shown for the various proteins. In all cases, 100 μg of total protein was resolved on 15% SDS-PAGE gels. (A) Western blot analysis of GS15 levels in control, P1, and P2 fibroblasts. (B) Western blot analysis of control, P1, and COG7-corrected P1 fibroblast lysates. GS15 levels are normalized in COG7-corrected patient fibroblasts. (C) P1 cells were treated with either 25 μM MG132 for 10 h or 150 μM leupeptin and 100 μM pepstatin A for 16 h before sample preparation. Note that neither lysosomal or proteasomal inhibition restored GS15 levels.
The Nature of the Unique Membrane Clusters in P1 Fibroblasts
Because of the singular juxtanuclear location of the clusters, we investigated the possibility that the accumulation of giantin observed in BFA-treated P1 cells occurred at the microtubule-organizing center (MTOC). The MTOC, which can be specifically labeled by anti-γ-tubulin, serves to organize pre-Golgi membranes after their exit from the ER (Oakley and Akkari, 1999; Rios and Bornens, 2003). The γ-tubulin staining in patient and control cells was not altered in the presence or absence of BFA (Figure 7A). There is little or no colocalization between γ-tubulin and giantin in untreated control and P1 cells consistent with the distinct apposition of the Golgi stack and the MTOC. On BFA treatment, giantin, which redistributes exclusively to the ER in control cells, fails to localize with γ-tubulin. In contrast, the giantin-positive clusters observed in P1 cells show strong colocalization with γ-tubulin. These results indicate that the accumulation of Golgi markers in clusters is coincident with the MTOC. The clusters are sensitive to nocodazole treatment, providing further support for the presence of microtubules (Figure 7B). ERGIC-53 is largely excluded from these structures upon BFA treatment, indicating that they are not ER export sites (our unpublished data).
Figure 7.

Giantin colocalizes with the MTOC marker γ-tubulin in BFA-treated P1 but not control fibroblasts (A) Control and patient fibroblasts were treated with or without 0.25 μg/ml BFA for 60 min and stained for either giantin or γ-tubulin. The localization of these proteins was analyzed by confocal immunofluorescence. The asterisk denotes the single, juxtanuclear staining of the MTOC. Bar, 20 μm. (B) Cells were treated with 0.25 μg/ml BFA for 60 min followed by 30-min exposure to 10 μM nocodazole. Fixed cells were then stained for giantin using a specific polyclonal antibody. Note the sensitivity of the remnant-like structures to nocodazole indicating the presence of microtubules.
The Steady-State Localization of ERGIC-53 Is Altered in P1 Fibroblasts
We next examined the localization of the cargo receptor ERGIC-53, a protein that defines the ERGIC and actively recycles between the ER and Golgi (Itin et al., 1995). If the retrograde transport of ERGIC-53 was impaired in the COG7-deficient fibroblasts, the steady-state pattern of this protein might be altered. ERGIC-53 staining of control cells revealed a pattern that has characteristics of both the Golgi and the ER and is herein referred to as a classic ERGIC pattern. In contrast, ERGIC-53 staining in many P1 fibroblasts more strongly resembled a Golgi-like ribbon structure (abnormal ERGIC pattern). This abnormal ERGIC pattern was seen in 50 ± 7% of P1 cells versus only 5 ± 2% of control cells. The abnormal ERGIC pattern was COG-dependent because ERGIC-53 localization in COG7-corrected cells was indistinguishable from control cells (Figure 5B).
To determine whether the abnormal localization of ERGIC-53 in P1 cells colocalizes with the Golgi, double immunofluorescence studies with ERGIC-53 and giantin were performed. There was no overlap between the two proteins in control fibroblasts (Figure 8, second row). In contrast, there was a high degree of colocalization in P1 cells. The majority of ERGIC-53 clustered in regions surrounding giantin-positive Golgi membranes, indicating that the localization of the protein was shifted to the cis-Golgi face. Prolonged exposure of cells to lower temperature has been shown to result in an accumulation of ERGIC-53 in the ERGIC compartment and a shift of the compartment closer to the cis-Golgi membranes (Klumperman et al., 1998). Because the intracellular distribution of ERGIC-53 in P1 cells bears similarity to the effects of lower temperature on ERGIC-53 localization, control and P1 cells were cultured at 15°C for 3 h and ERGIC-53 localization analyzed by immunofluorescence. As shown in Figure 8, the localization of ERGIC-53 in control cells kept at 15°C is consistent with an accumulation of ERGIC-53 in the ERGIC compartment and a shift toward the cis-Golgi. In contrast, ERGIC-53 staining in P1 fibroblasts was not detectably altered at lower temperature, indicating that ERGIC-53 had already undergone a shift in its localization. The effects of lower temperature are distinct from the action of BFA as demonstrated in Figure 8. Therefore, proteins that actively recycle between the Golgi and ER are altered by COG7 deficiency.
Figure 8.

ERGIC-53 localization is altered in COG7-deficient fibroblasts. Control and patient fibroblasts were either treated with 1 μg/ml BFA for 30 min. or incubated at 15°C for 3 h as indicated. The localization of ERGIC-53 (first, third, and fourth rows) was followed by immunofluorescence. Arrows indicate representative cells with abnormal concentration of ERGIC-53 in structures resembling the cis-Golgi. For the colocalization studies (second row), giantin is stained in red and ERGIC-53 in green. Bar, 20 μm.
DISCUSSION
These studies have uncovered a number of alterations in Golgi protein localization, stability and trafficking in COG7-deficient fibroblasts. One of the most striking findings in this study was the delay in retrograde trafficking of giantin and βGalT upon treatment of the COG7-deficient fibroblasts with BFA (Figure 1 and Supplemental Figure S1). Although much attention has been focused on the role of the COG complex in intra-Golgi and ER-to-Golgi transport, our results indicate that COG7 is also required for efficient Golgi-to-ER retrograde transport. The block in mainly retrograde (but not anterograde) transport between the ER and Golgi is clearly evidenced by the redistribution of ERGIC-53 to the cis-Golgi compartment in patient fibroblasts (Figure 8).
One can envision multiple mechanisms whereby Cog7p deficiency results in a delayed response to BFA. It is known that after treatment with BFA, the coat proteins COPI and AP-1 are rapidly released from the Golgi membrane into the cytosol, allowing tubules to form that subsequently fuse with the ER (Lippincott-Schwartz et al., 1990; Lippincott-Schwartz and Zaal, 2000; Altan-Bonnet et al., 2004). Therefore, the slower tubule formation in BFA-treated patient fibroblasts could potentially result from delayed COPI release. However, our finding that βCOP and the trans-Golgi network adaptor protein AP-1 were released at the same rate in control and patient cells (Figure 2) suggests an alternate mechanism. Because BFA-induced tubulation has been shown to occur along microtubules (Lippincott-Schwartz et al., 1990; Sciaky et al., 1997; Duran et al., 2003), the COG complex may facilitate Golgi-to-ER transport by tethering retrograde-bound tubules to the cytoskeleton. If this is the mechanism of action, it is notable that the COG complex would be acting independently of COPI. Because recent studies have shown that BFA-induced tubules fuse with the ER at ER exit sites, loss of Cog7p could result in failure of tubules to fuse properly with the ER at these sites (Mardones et al., 2006). Although COG may play a direct role in the tethering and fusion of BFA-induced tubules, secondary loss of other proteins may contribute to this effect. Finally, a defect in ER-Golgi recycling, as evidenced by the abnormal localization of multiple recycling proteins such as ERGIC-53, may lead to changes in the lipid composition of the Golgi that are unfavorable for tubule formation.
After prolonged BFA treatment of patient fibroblasts, several of the Golgi proteins examined accumulated in nocodazole-sensitive solitary clusters that colocalized with the MTOC. These clusters have a different appearance from the previously noted BFA-induced Golgi-like “remnants,” which have a scattered, peripheral punctate distribution and are now considered to represent ER exit sites (Miles et al., 2001; Ward et al., 2001; Puri and Linstedt, 2003). The MTOC-associated clusters were not detected in the COG7-deficient fibroblasts in the absence of BFA treatment, so it is unclear whether Golgi proteins accumulate at the MTOC at steady state in the mutant cells or whether these structures are a specific consequence of BFA-induced overloading of the ER export system with Golgi proteins in cells impaired in vesicular trafficking. Coincident with its role in positioning the Golgi complex, the microtubule-organizing center serves to organize pre-Golgi intermediates before their transport along microtubules to the Golgi (Lippincott-Schwartz, 1998; Rios and Bornens, 2003). Thus, the presence of Golgi proteins at the MTOC under pressure of BFA may represent the failure of pre-Golgi membranes to efficiently move away from the centrosome in the absence of a functional COG complex. This would be consistent with the known interaction between COPI and the COG complex because the transport of vesicles from the ERGIC to the Golgi has been shown to involve COPI (Aridor et al., 1995; Scales et al., 1997; Stephens et al., 2000). Indeed, the accumulation of ERGIC-53 in an atypical ERGIC compartment in patient fibroblasts at steady state might also reflect a block in ERGIC-to-Golgi transport.
The striking loss of GS15 in patient fibroblasts and the dependence of this reduction on COG complex subunits are consistent with recent observations in COG-deficient CHO cells (Oka et al., 2004). In this study by Krieger and colleagues, a reduction in the level of additional Golgi proteins (termed GEARs) was noted. Although decreases in some GEAR proteins such as GS28 and GPP130 were observed in P1 fibroblasts, this reduction was highly variable and was not seen in P2 fibroblasts. The observation by Lupashin and coworkers that GS15 is mislocalized in CCD vesicles upon depletion of COG3 in HeLa cells reinforces the hypothesis that loss of COG results in mislocalization of Golgi proteins and their eventual degradation (Zolov and Lupashin, 2005). Because this degradation might occur via either ER-associated proteasomal or lysosomal pathways, we sought to determine the degradative pathway responsible for the GS15 reduction. Surprisingly, inhibition of the proteasome or lysosomal proteases failed to restore GS15 in patient fibroblasts. The mechanism underlying the depletion of GS15 in patient fibroblasts is still unclear.
Although the alterations in protein trafficking between the ER and Golgi may be a direct consequence of impaired COG complex integrity, these abnormalities could also be due to the secondary loss of recycling factors (i.e., SNAREs) required for this transport. In support of this notion, small interfering RNA (siRNA)-mediated knockdown of COG3 (Zolov and Lupashin, 2005) or GS15 (Xu et al., 2002) in HeLa cells produced a similar phenotype, namely, the redistribution of multiple Golgi markers, including βGalT, to dispersed, punctate vesicular structures. Because CDG-IIe fibroblasts are deficient in several COG subunits as well as GS15, it is surprising that more prominent differences in Golgi protein localization were not observed. Because siRNA knockdown experiments result in acute posttranscriptional depletion of the target protein, the lack of Golgi protein redistribution in the patient fibroblasts may reflect compensation for the genetic deficiency of COG7 (i.e., up-regulation or down-regulation of other trafficking proteins). Because the COG subunits 1-4 (comprising lobe A) are present at normal steady-state levels and localized to the Golgi in COG7-deficient fibroblasts (Wu et al., 2004, Oka et al., 2005), it is also possible that these subunits may form a partially functional COG subcomplex.
We previously demonstrated decreased sialylation of N- and O-linked oligosaccharides, reduced activity of multiple glycosyltransferases and nucleotide-sugar transporters and abnormal trafficking of a GFP-sialyltransferase chimera in COG7-deficient fibroblasts. Here, the observation that βGalT exhibits altered trafficking in patient fibroblasts provides additional support for the hypothesis that altered recycling of glycosylation enzymes between the ER and Golgi may lead to altered localization, activity, or both. The lack of any detectable defects in galactosylation in COG7-deficient fibroblasts despite the modest reduction in steady-state levels of βGalT is not surprising because fibroblasts from a βGalT-deficient CDG-IId patient, exhibited only minor changes in galactosylation of N-linked oligosaccharides despite a 95% reduction in enzyme activity (Hansske et al., 2002). The COG complex has been proposed to facilitate COPI-dependent intra-Golgi retrieval of resident proteins and enzymes (Suvorova et al., 2002; Ungar et al., 2002; Zolov and Lupashin, 2005). Therefore, it is possible that mislocalization of such enzymes to post-Golgi compartments such as endosomes or lysosomes may contribute to decreased activity or stability as has been recently shown in COG-deficient yeast cells (Bruinsma et al., 2004). Because efficient glycosylation requires the transport of nucleotide-sugars into the Golgi lumen via nucleotide-sugar transporters in addition to the action of glycosyltransferases, the mislocalization and/or degradation of the nucleotide-sugar transporters may be sufficient to disrupt glycosylation reactions in the absence of substantial effects on glycosyltransferase stability. A more precise determination of the mechanisms responsible for the observed glycosylation defects in P1 fibroblasts (i.e., decreased sialylation) will require the generation of appropriate antisera.
These results have several implications regarding the pathological consequences of COG deficiency. The multiple developmental abnormalities seen in the CDG-IIe patients stress the vital role of the COG complex in human development. Because efficient glycosylation is required for the proper development and function of many systems, altered modification of oligosaccharides due to decreased activity of glycosylation enzymes may account for many of the clinical features noted in these patients. In addition to the impact of abnormal glycosylation, loss of proper protein recycling in COG-deficient cells could have other relevant outcomes. Golgi disassembly and reassembly are necessary steps during mitosis (Misteli, 1996). The inability to efficiently transport proteins or link vesicles to the cytoskeleton upon Golgi reassembly in COG-deficient cells may result in defective growth of rapidly dividing cells or impaired ability to migrate, especially in light of the high demand for this process during development. The impact of COG deficiency on spermatogenesis, a sensitized developmental system involving cytokinesis and the assembly on specialized Golgi architecture, has been previously observed in the COG5-deficient Drosophila mutant four-way stop (fws) (Farkas et al., 2003). Finally, the abnormal localization of ERGIC-53 could impact the levels of several blood coagulation proteins in the CDG-IIe patients. Because ERGIC-53 has been shown to be necessary for the sorting and transport of Factor V and Factor VIII, its inability to recycle efficiently between the ER and cis-Golgi may lead to a failure in the proper sorting and export of these coagulation factors (Nichols et al., 1998).
Many patients with the diagnosis of CDG have unknown defects, and it is likely that a subset of them will have abnormalities in components of their protein trafficking machinery. Determination of the molecular defect via analysis of patient fibroblasts is often confounded by the lack of observable phenotypes (i.e., changes in glycan structure). Although COG7-deficient patient fibroblasts exhibit only subtle changes in their glycosylation profile, clear differences can be observed in their response to BFA. Thus, our results show the value of nonequilibrium conditions (i.e., BFA treatment) to bring out defects that may not be evident under steady-state conditions. We suggest that this simple procedure become a routine component of the analysis of fibroblasts from these patients.
Supplementary Material
Acknowledgments
We are indebted to Dr. Heather Flanagan-Steet for expert assistance in confocal microscopy. We acknowledge Drs. Eric Berger and Hans-Peter Hauri for the generous gift of antisera. We also thank Drs. Daniel Ory and Alexander Ungewickell for critical reading of the manuscript. This work is supported by National Institutes of Health Grant CA08759 (to S. K.) and National Institutes of Health Training Grant 5T32-HL07088-30 and American Heart Association Scientist Development Grant 0535026N (to R. S.).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-08-0822) on March 1, 2006.
Abbreviations used: βGalT, β1,4-galactosyltransferase; BFA, brefeldin A; CDG, congenital disorders of glycosylation; COG, conserved oligomeric Golgi; ER, endoplasmic reticulum; ERGIC, ER-Golgi intermediate compartment; MTOC, microtubule-organizing center.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Altan-Bonnet, N., Sougrat, R., and Lippincott-Schwartz, J. (2004). Molecular basis for Golgi maintenance and biogenesis. Curr. Opin. Cell Biol. 16, 364-372. [DOI] [PubMed] [Google Scholar]
- Aridor, M., Bannykh, S. I., Rowe, T., and Balch, W. E. (1995). Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131, 875-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma, P., Spelbrink, R. G., and Nothwehr, S. F. (2004). Retrograde transport of the mannosyltransferase Och1p to the early Golgi requires a component of the COG transport complex. J. Biol. Chem. 279, 39814-39823. [DOI] [PubMed] [Google Scholar]
- Donaldson, J. G., Lippincott-Schwartz, J., Bloom, G. S., Kreis, T. E., and Klausner, R. D. (1990). Dissociation of a 110-kD peripheral membrane protein from the Golgi apparatus is an early event in brefeldin A action. J. Cell Biol. 111, 2295-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran, J. M., Valderrama, F., Castel, S., Magdalena, J., Tomas, M., Hosoya, H., Renau-Piqueras, J., Malhotra, V., and Egea, G. (2003). Myosin motors and not actin comets are mediators of the actin-based Golgi-to-endoplasmic reticulum protein transport. Mol. Biol. Cell 14, 445-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas, R. M., Giansanti, M. G., Gatti, M., and Fuller, M. T. (2003). The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell 14, 190-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze, H. H. (2001). Update and perspectives on congenital disorders of glycosylation. Glycobiology 11, 129R-143R. [PubMed] [Google Scholar]
- Ghosh, P., Griffith, J., Geuze, H. J., and Kornfeld, S. (2003). Mammalian GGAs act together to sort mannose 6-phosphate receptors. J. Cell Biol. 163, 755-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansske, B., et al. (2002). Deficiency of UDP-galactose:N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J. Clin. Investig. 109, 725-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin, C., Foguet, M., Kappeler, F., Klumperman, J., and Hauri, H. P. (1995). Recycling of the endoplasmic reticulum/Golgi intermediate compartment protein ERGIC-53 in the secretory pathway. Biochem. Soc. Trans. 23, 541-544. [DOI] [PubMed] [Google Scholar]
- Klumperman, J., Schweizer, A., Clausen, H., Tang, B. L., Hong, W., Oorschot, V., and Hauri, H. P. (1998). The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J. Cell Sci. 111, 3411-3425. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J. (1993). Membrane cycling between the ER and Golgi apparatus and its role in biosynthetic transport. Subcell. Biochem. 21, 95-119. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J. (1998). Cytoskeletal proteins and Golgi dynamics. Curr. Opin. Cell Biol. 10, 52-59. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Donaldson, J. G., Schweizer, A., Berger, E. G., Hauri, H. P., Yuan, L. C., and Klausner, R. D. (1990). Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60, 821-836. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Yuan, L. C., Bonifacino, J. S., and Klausner, R. D. (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56, 801-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., and Zaal, K. J. (2000). Cell cycle maintenance and biogenesis of the Golgi complex. Histochem. Cell Biol. 114, 93-103. [DOI] [PubMed] [Google Scholar]
- Mardones, G. A., Snyder, C. M., and Howell, K. E. (2006). Cis-Golgi matrix proteins move directly to endoplasmic reticulum exit sites by association with tubules. Mol. Biol. Cell 17, 525-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt, T., and Denecke, J. (2003). Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 162, 359-379. [DOI] [PubMed] [Google Scholar]
- Miles, S., McManus, H., Forsten, K. E., and Storrie, B. (2001). Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155, 543-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T. (1996). Molecular mechanisms in the disassembly and reassembly of the mammalian Golgi apparatus during M-phase. FEBS Lett. 389, 66-69. [DOI] [PubMed] [Google Scholar]
- Morsomme, P., Prescianotto-Baschong, C., and Riezman, H. (2003). The ER v-SNAREs are required for GPI-anchored protein sorting from other secretory proteins upon exit from the ER. J. Cell Biol. 162, 403-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsomme, P., and Riezman, H. (2002). The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev. Cell 2, 307-317. [DOI] [PubMed] [Google Scholar]
- Nichols, W. C., et al. (1998). Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell 93, 61-70. [DOI] [PubMed] [Google Scholar]
- Oakley, B. R., and Akkari, Y. N. (1999). Gamma-tubulin at ten: progress and prospects. Cell Struct. Funct. 24, 365-372. [DOI] [PubMed] [Google Scholar]
- Oka, T., and Krieger, M. (2005). Multi-component protein complexes and Golgi membrane trafficking. J. Biochem. 137, 109-114. [DOI] [PubMed] [Google Scholar]
- Oka, T., Ungar, D., Hughson, F. M., and Krieger, M. (2004). The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 15, 2423-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka, T., Vasile, E., Penman, M., Novina, C. D., Dykxhoorn, D. M., Ungar, D., Hughson, F. M., and Krieger, M. (2005). Genetic analysis of the subunit organization and function of the COG complex: studies of Cog5 and Cog7 deficient mammalian cells. J. Biol. Chem. [DOI] [PubMed]
- Puri, S., and Linstedt, A. D. (2003). Capacity of the Golgi apparatus for biogenesis from the endoplasmic reticulum. Mol. Biol. Cell 14, 5011-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios, R. M., and Bornens, M. (2003). The Golgi apparatus at the cell centre. Curr. Opin. Cell Biol. 15, 60-66. [DOI] [PubMed] [Google Scholar]
- Rudd, P. M., Morgan, B. P., Wormald, M. R., Harvey, D. J., van den Berg, C. W., Davis, S. J., Ferguson, M. A., and Dwek, R. A. (1997). The glycosylation of the complement regulatory protein, human erythrocyte CD59. J. Biol. Chem. 272, 7229-7244. [DOI] [PubMed] [Google Scholar]
- Scales, S. J., Pepperkok, R., and Kreis, T. E. (1997). Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell 90, 1137-1148. [DOI] [PubMed] [Google Scholar]
- Sciaky, N., Presley, J., Smith, C., Zaal, K. J., Cole, N., Moreira, J. E., Terasaki, M., Siggia, E., and Lippincott-Schwartz, J. (1997). Golgi tubule traffic and the effects of brefeldin A visualized in living cells. J. Cell Biol. 139, 1137-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, D. J., Lin-Marq, N., Pagano, A., Pepperkok, R., and Paccaud, J. P. (2000). COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 113, 2177-2185. [DOI] [PubMed] [Google Scholar]
- Suvorova, E. S., Duden, R., and Lupashin, V. V. (2002). The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 157, 631-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztul, E., and Lupashin, V. (2006). Role of tethering factors in secretory membrane traffic. Am. J. Physiol. 290, C11-C26. [DOI] [PubMed] [Google Scholar]
- Ungar, D., Oka, T., Brittle, E. E., Vasile, E., Lupashin, V. V., Chatterton, J. E., Heuser, J. E., Krieger, M., and Waters, M. G. (2002). Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 157, 405-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S. M., Cao, X., Lupashin, V. V., Barlowe, C., and Waters, M. G. (1998). Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J. Cell Biol. 141, 1107-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S. M., Cao, X., Sapperstein, S. K., Chiang, E. C., Lupashin, V. V., Barlowe, C., and Waters, M. G. (1999). Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J. Cell Biol. 147, 729-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, T. H., Polishchuk, R. S., Caplan, S., Hirschberg, K., and Lippincott-Schwartz, J. (2001). Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155, 557-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Steet, R. A., Bohorov, O., Bakker, J., Newell, J., Krieger, M., Spaapen, L., Kornfeld, S., and Freeze, H. H. (2004). Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 10, 518-523. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Martin, S., James, D. E., and Hong, W. (2002). GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol. Biol. Cell 13, 3493-3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolov, S. N., and Lupashin, V. V. (2005). Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 168, 747-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.