

Abstract
The membrane-associated flavoprotein Ero1p promotes disulfide bond formation in the endoplasmic reticulum (ER) by selectively oxidizing the soluble oxidoreductase protein disulfide isomerase (Pdi1p), which in turn can directly oxidize secretory proteins. Two redox-active disulfide bonds are essential for Ero1p oxidase activity: Cys100-Cys105 and Cys352-Cys355. Genetic and structural data indicate a disulfide bond is transferred from Cys100-Cys105 directly to Pdi1p, whereas a Cys352-Cys355 disulfide bond is used to reoxidize the reduced Cys100-Cys105 pair through an internal thiol-transfer reaction. Electron transfer from Cys352-Cys355 to molecular oxygen, by way of a flavin cofactor, maintains Cys352-Cys355 in an oxidized form. Herein, we identify a mixed disulfide species that confirms the Ero1p intercysteine thiol-transfer relay in vivo and identify Cys105 and Cys352 as the cysteines that mediate thiol-disulfide exchange. Moreover, we describe Ero1p mutants that have the surprising ability to oxidize substrates in the absence of Cys100-Cys105. We show the oxidase activity of these mutants results from structural changes in Ero1p that allow substrates increased access to Cys352-Cys355, which are normally buried beneath the protein surface. The altered activity of these Ero1p mutants toward selected substrates leads us to propose the catalytic mechanism involving transfer between cysteine pairs evolved to impart substrate specificity to Ero1p.
INTRODUCTION
Disulfide bonds play an essential role in the folding and assembly of secretory proteins and the extracellular domains of membrane proteins. Disulfide bonds are formed in vivo by the cooperative activity of two groups of proteins: soluble oxidoreductases of the thioredoxin family, and membrane associated oxidases that couple protein oxidation with small molecule redox reactions. Both groups of proteins use a thioldisulfide exchange mechanism to transfer disulfide bonds from themselves to substrate proteins (for recent reviews, see Fassio and Sitia, 2002; Sevier and Kaiser, 2002; Kadokura et al., 2003; Jessop et al., 2004; Nakamoto and Bardwell, 2004; Tan and Bardwell, 2004; Tu and Weissman, 2004).
The major disulfide bond formation pathway in eukaryotes is located in the endoplasmic reticulum (ER) and is comprised of the soluble thioredoxin-like protein Pdi1p and the membrane associated flavoprotein Ero1p (Frand and Kaiser, 1998; Pollard et al., 1998; Frand and Kaiser, 1999). A disulfide bond formed between the active site cysteines of Pdi1p is transferred directly to folding secretory proteins (Frand and Kaiser, 1999). Ero1p reoxidizes reduced Pdi1p, whereas reduced Ero1p, in turn, becomes reoxidized by molecular oxygen via its flavin cofactor (Frand and Kaiser, 1999; Tu et al., 2000; Tu and Weissman, 2002). An alternative, nonessential oxidation pathway uses the membrane-associated flavoprotein Erv2p to drive protein disulfide bond formation via the transfer of oxidizing equivalents to Pdi1p (Gerber et al., 2001; Sevier et al., 2001). Like Ero1p, Erv2p is maintained in an oxidized state by transferring electrons to its flavin cofactor, which donates electrons to molecular oxygen (Sevier et al., 2001). The Erv2p-driven protein oxidation pathway is unique to fungi and may facilitate oxidation of a group of nonessential disulfide-bonded proteins. Alternatively, mammalian cells contain an extracellular protein not present in fungi that contains an Erv2p-like domain fused with an amino-terminal thioredoxin domain (Hoober et al., 1999; Hoober and Thorpe, 2002; Thorpe et al., 2002). An attractive hypothesis is that these secreted fusion proteins, alternatively referred to as SOX or Quiescin, facilitate extracellular disulfide bond formation.
Despite a lack of sequence similarity between the disulfide carrier proteins Ero1p and Erv2p, their catalytic domains have fundamental structural features in common (Gross et al., 2002; Gross et al., 2004). The core domain of both proteins is comprised of a bundle of four antiparallel α-helices that hold the flavin cofactor close to a conserved cysteine pair in a Cys-x-x-Cys motif (Cys352-Cys355 in Ero1p and Cys121-Cys124 in Erv2p) that is positioned on a tight turn between two of the bundled helices. Ero1p and Erv2p also contain a second cysteine pair located in a flexible domain that is devoid of significant α-helical or β-sheet content, which comes in proximity to the first cysteine pair. In Ero1p, the second pair of cysteines (Cys100-Cys105) is situated in a polypeptide loop that rests on the surface of the protein, whereas in Erv2p the second cysteine pair (Cys176-Cys178) is positioned at the protein C terminus in a flexible tail. In Ero1p and Erv2p, both conserved cysteine pairs are essential for oxidative activity (Frand and Kaiser, 2000; Gross et al., 2002).
Structural and biochemical data suggest both Ero1p and Erv2p use a catalytic mechanism that involves a direct disulfide bond transfer between two cysteine pairs in each protein. Flexible domains containing the second cysteine pair can adopt at least two different conformations that have been observed in the crystal forms of both proteins (Gross et al., 2002; Gross et al., 2004). In one conformation, the cysteine pair in the flexible domain is adjacent to the FAD-proximal cysteine pair, suggestive of a direct interaction between the two cysteine pairs, whereas in the other conformation the flexible region is displaced from the protein core where the cysteines may be more accessible to soluble Pdi1p. Capture of a disulfide bond between the two essential Erv2p cysteine pairs supports the disulfide relay mechanism suggested by the structural data (Gross et al., 2002). Mutagenesis of the Ero1p cysteines has demonstrated a role for the loop cysteines in interaction with Pdi1p, and a role for Cys352-Cys355 in reoxidation of the loop cysteine pair (Frand and Kaiser, 2000; Bertoli et al., 2004). However, a direct interaction between the two cysteine pairs in Ero1p has never been observed.
Here, we consider the mechanistic details of the interdisulfide shuttle between the two essential cysteine pairs in Ero1p. We provide direct evidence of thiol-disulfide exchange between the two Ero1p cysteine pairs and identify Cys105 and Cys352 as the cysteines mediating the thioldisulfide transfer event. Our mutational studies show that the Cys352-Cys355 disulfide is sufficient for catalysis of disulfide bond formation by Ero1p. However, access to this active site disulfide bond is controlled by the domain containing Cys100-Cys105, which serves as a substrate selectivity filter by not only relaying disulfides to Pdi1p but also by physically blocking the access of other cellular proteins and small molecules to Cys352-Cys355. We suggest the use of a catalytic mechanism involving transfer between cysteine pairs by Ero1p evolved to impart substrate specificity to the major cellular protein disulfide bond formation pathway.
MATERIALS AND METHODS
Strains and Growth Conditions
Saccharomyces cerevisiae strains were grown and genetically manipulated using standard techniques (Adams et al., 1998). YPD is rich medium with 2% glucose. SMM and SMM Gal are minimal medium supplemented with amino acids containing 2% glucose or 2% galactose, respectively.
Isolation and characterization of the ero1-1 strain (CKY598) has been described previously (Frand and Kaiser, 1998). The ero1-1 ire1Δ::KanMX (CKY932) strain was made by one-step gene replacement of the coding region of IRE1 with KanMX in CKY598. To generate a chromosomal ERO1 deletion, the ERO1 coding sequence was replaced with KanMX in a homozygous GAL2 ura3-52 leu2-3112 diploid background. The resultant heterozygous diploid was transformed with pAF82, and a viable MATa Ura+ KanMX+ segregant was recovered after sporulation (CKY933). The viability of CKY933 depends on the episomal ERO1 allele, as demonstrated by the failure of this strain to grow on medium containing 5-fluoroorotic acid (5-FOA; Toronto Research Laboratories, Downsview, Ontario, Canada).
Plasmid Construction
Table 1 lists the plasmids used in this study. To create pCS253, a thrombin site (DNA sequence GGAGGCTTAGTACCTAGAGGATCCGGT) was made after residue 325 in ERO1 by two-step fusion PCR. C90A and C349A mutations were amplified during the first fusion PCR step from previously constructed ERO1 plasmids (Frand and Kaiser, 2000). A triple myc tag was added to the C terminus of the ERO1 coding sequence as described in Frand and Kaiser (1998). An amino-terminal hemagglutinin (HA) epitope in ERO1 was created by ligation of a triple HA epitope fragment into an XhoI site created after residue 27 by site-directed mutagenesis with the QuikChange kit (Stratagene, La Jolla, CA). To create pCS75, the C-terminal triple myc tag was replaced with a triple HA tag in a plasmid containing ero1-C100A-C105A. To make plasmids pCS42 and pCS55, the triple myc epitope was inserted into a NotI site, followed by a stop codon, created after residue 181 of ERO1 by site-directed mutagenesis. To create the remaining yeast ERO1 plasmids, additional amino acid replacements were made by site-directed mutagenesis or by subcloning mutations from previously constructed ERO1 plasmids using standard molecular biology techniques.
Table 1.
Plasmids
| Name | Description | Markers | Source |
|---|---|---|---|
| pAF82 | ERO1-myc | CEN URA3 | Frand and Kaiser (1998) |
| pAF85 | ERO1-myc | CEN LEU2 | Frand and Kaiser (1998) |
| pAF89 | ERO1-myc | 2μ LEU2 | Frand and Kaiser (1998) |
| pAF125 | pAF89 with C100A | 2μ LEU2 | Frand and Kaiser (1998) |
| pAF126 | pAF89 with C105A | 2μ LEU2 | Frand and Kaiser (1998) |
| pCS214 | pAF89 with C100A, C105A | 2μ LEU2 | This study |
| pAF129 | pAF89 with C352A | 2μ LEU2 | Frand and Kaiser (1998) |
| pAF130 | pAF89 with C355A | 2μ LEU2 | Frand and Kaiser (1998) |
| pCS261 | pAF89 with C352A, C355A | 2μ LEU2 | This study |
| pCS253 | ero1-HA-C90A-C349A-myc with thrombin site (ERO1-t) | 2μ LEU2 | This study |
| pCS257 | pCS253 with C100A | 2μ LEU2 | This study |
| pCS258 | pCS253 with C105A | 2μ LEU2 | This study |
| pCS254 | pCS253 with C352A | 2μ LEU2 | This study |
| pCS255 | pCS253 with C355A | 2μ LEU2 | This study |
| pCS259 | pCS253 with C100A, C105A | 2μ LEU2 | This study |
| pCS256 | pCS253 with C352A, C355A | 2μ LEU2 | This study |
| pCS347 | pCS253 with I66N | 2μ LEU2 | This study |
| pCS349 | pCS253 with L70P | 2μ LEU2 | This study |
| pCS348 | pCS253 with L70R | 2μ LEU2 | This study |
| pCS306 | pCS253 with L84P | 2μ LEU2 | This study |
| pCS307 | pCS253 with L86S | 2μ LEU2 | This study |
| pCS262 | pCS253 with W120R | 2μ LEU2 | This study |
| pCS274 | ero1-C90A-C349A-myc with thrombin site (ERO1-t-myc) | 2μ URA3 | This study |
| pCS275 | ero1-HA-C90A-C349 with thrombin site (ERO1-t-HA) | 2μ LEU2 | This study |
| pCS42 | PGAL-ero1-1-181-myc | CEN LEU2 | This study |
| pCS55 | PGAL ero1-1-181-C100A-C105A-myc | CEN LEU2 | This study |
| pCS341 | pCS42 with I66N | CEN LEU2 | This study |
| pCS342 | pCS42 with L70P | CEN LEU2 | This study |
| pCS321 | pCS42 with L70R | CEN LEU2 | This study |
| pCS318 | pCS42 with L84P | CEN LEU2 | This study |
| pCS319 | pCS42 with L86S | CEN LEU2 | This study |
| pCS320 | pCS42 with W120R | CEN LEU2 | This study |
| pCS75 | ero1-C100A-C105A-HA | CEN URA3 | This study |
| pCS169 | ero1-C100A-C105A-myc | CEN LEU2 | This study |
| pCS196 | pCS169 with I66N | CEN LEU2 | This study |
| pCS194 | pCS169 with L70P | CEN LEU2 | This study |
| pCS235 | pCS169 with L70R | CEN LEU2 | This study |
| pCS233 | pCS169 with L84P | CEN LEU2 | This study |
| pCS230 | pCS169 with L86S | CEN LEU2 | This study |
| pCS184 | pCS169 with W120R | CEN LEU2 | This study |
| pCS343 | pCS261 with I66N | 2μ LEU2 | This study |
| pCS344 | pCS261 with L70P | 2μ LEU2 | This study |
| pCS325 | pCS261 with L70R | 2μ LEU2 | This study |
| pCS322 | pCS261 with L84P | 2μ LEU2 | This study |
| pCS323 | pCS261 with L86S | 2μ LEU2 | This study |
| pCS324 | pCS261 with W120R | 2μ LEU2 | This study |
| ECVS WT | GST-His6-ero1-56-424 (ERO1-c) | AMP | Gross et al. (2004) |
| pCS302 | ECVS WT with C100A-C105A | AMP | This study |
| pCS304 | ECVS WT with L84P-C100A-C105A | AMP | This study |
| pCS305 | ECVS WT with C100A-C105A-W120R | AMP | This study |
| pET Trx1 | Trx1-His6 | KAN | Gross et al. (2005) |
A plasmid encoding glutathione S-transferase (GST) fused to a His6-tagged Ero1p (residues 56–424) was generously provided by E. Gross and D. Fass (Department of Structural Biology, The Weizmann Institute of Science, Rehovot, Israel) (Gross et al., 2004). ERO1 mutants were subcloned into this bacterial expression plasmid after PCR amplification from the yeast expression plasmids.
Intragenic Suppressor Screen
A vector containing ero1-C100A-C105A was used as a template to generate random intragenic ERO1 mutants by PCR. A 2687-base pair fragment including the entire ERO1 coding sequence plus 560 base pairs of the 5′ untranslated region (UTR) and 309 base pairs of the 3′ UTR was amplified using AmpliTaq Gold (PerkinElmer Life and Analytical Sciences, Boston, MA) in a 100-μl reaction containing 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 7 mM MgCl2, 0.3 mM MnCl2, primers at 0.5 μM each, 1 mM dCTP and dTTP each, and 0.2 mM dATP and dGTP each. PCR conditions included a hot start at 94°C for 12 min, 35 cycles of 94°C for 1.5 min, 50°C for 2 min, 72°C for 3 min, followed by a 10-min extension at 72°C. To prevent overrepresentation of a single mutant, eight separate 100-μl reactions were run and pooled after completion of the PCR run. PCR products were cloned by homologous recombination in CKY932 into a gapped CEN LEU2 vector that contained the noncoding 5′ and 3′ ERO1 sequence in addition to the first 124 base pairs of the ERO1 open reading frame. Transformants were selected at 24°C on SMM plates lacking leucine and subsequently replica plated onto SMM-selective plates and incubated at 24 or 37°C. Transformants able to grow at 37°C were isolated at a frequency of ∼5 per every 100 replica-plated colonies. Plasmids recovered from colonies that suppressed the growth defect of CKY932 cells at 37°C initially and after retransformation, 15 in total, were isolated and sequenced. Of the sequenced plasmids, eight plasmids contained ERO1 that had recombined to restore Cys100 and Cys105 to wild type, and seven plasmids contained ERO1 coding sequence with between two and five nonsilent coding changes in addition to the C100A and C105A mutations. Mutations were separated by standard molecular biology techniques, and six individual mutations were determined to be responsible for growth of plasmid transformed CKY932 at 37°C.
Radiolabeling and Immunoprecipitations
For Ero1p immunoprecipitation experiments, CKY598 or CKY932 containing ERO1-t plasmids were grown to exponential phase. Cells, 5 OD600 units, were collected by centrifugation, resuspended in 1 ml of SMM lacking methionine, incubated at 30 or 38°C for 15 min, and labeled with [35S]methionine and [35S]cysteine (EXPRESS; PerkinElmer Life and Analytical Sciences) at 30 or 38°C for 30 min. Extracts were collected by centrifugation and suspended in 10% (wt/vol) trichloroactetic acid (TCA). Cell membranes were disrupted by agitation with glass beads, and proteins were collected by centrifugation at 4°C. Protein pellets were resuspended in 50 μl of 80 mM Tris-HCl, pH 7.4, 10% glycerol, 2% SDS, 0.1% bromcresol purple, 50 mM N-ethylmaleimide (NEM), and 1 mM phenylmethylsulfonyl fluoride (PMSF), and the pH of the samples was adjusted by gradual addition of 1 M Tris-HCl, pH 8.0, until samples turned purple. After vortex-mixing for 15 min at 4°C and a 15-min incubation at room temperature, samples were diluted 1:20 in immunoprecipitation (IP) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, and 0.2% SDS) containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and immunoprecipitated with monoclonal anti-myc (9E10) or anti-HA (12CA5). Immunoprecipitated proteins were collected by protein A-Sepharose (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) and were washed twice with IP buffer, once with detergent-free IP buffer, and once with phosphate-buffered saline (PBS) (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4). Immunoprecipitated proteins were treated with 0.1 U of thrombin protease (GE Healthcare) per microliter of PBS at 30°C for 18 h at which time the reaction was stopped with an equal volume of 2× sample buffer (160 mM Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, and 0.2% bromphenol blue) with or without 0.2 M dithiothreitol (DTT). When desired, samples were diluted 1:10 in IP buffer without SDS for reimmunoprecipitation with anti-myc or anti-HA. Samples were resolved by SDS-PAGE (10% gel) and analyzed with a 445si PhosphorImager (GE Healthcare).
For carboxypeptiase Y (CPY) immunoprecipitations, CKY598 or CKY932 containing the indicated plasmids were grown to exponential phase at which time cells were harvested and resuspended at 5 OD600 units/ml in SMM lacking methionine. Cells were shifted to 30 or 38°C for 30 min and radiolabeled at 30 or 38°C for 30 min. Cells, 2 OD600 equivalents, were collected in 20 mM NaN3 and lysed by agitation with glass beads in 30 μl of sample buffer containing 1 mM PMSF. Extracts were diluted to 1 ml in IP buffer with 0.1% SDS, and proteins were immunoprecipitated with polyclonal anti-CPY. Immunoprecipitates were collected with protein A-Sepharose, washed twice in IP buffer, once in detergent-free IP buffer, and solubilized in sample buffer with 0.1 M DTT. Samples were resolved by SDS-PAGE (8% gel) and analyzed with a 445si PhosphorImager.
Expression and Purification of Ero1p and Trx1
Plasmid encoding wild-type or mutant Ero1p-c was transformed into the Escherichia coli strain Origami B (DE3) pLysS (Novagen, Madison, WI). Transformants were grown at 37°C in LB medium containing 100 μg/ml ampicillin, 34 μg/ml chloramphenicol, 12.5 μg/ml tetracycline, and 15 μg/ml kanamycin to an OD600 of 0.5–0.7 at which time cultures were shifted to 24°C, and isopropyl β-d-thiogalactoside and FAD were added to a final concentration of 1.0 mM and 10 μM, respectively. After 16–18 h of induction, cells were harvested by centrifugation, solubilized in 10 ml of PBS plus protease inhibitors per 1 liter of cell culture, and lysed by treatment with lysozyme followed by sonication. Insoluble material was removed by centrifugation, and supernatant was applied to a GSTrap HP column (GE Healthcare) equilibrated with PBS. After washing the column with PBS, the column was filled by syringe with 5 ml of thrombin solution (10 U/ml in PBS) and rotated at room temperature for 2 h. After incubation, cleaved protein was eluted with PBS and loaded onto a HiTrap Chelating HP column (GE Healthcare) that had been charged with NiSO4 and equilibrated with PBS. The column was washed with wash buffer (50 mM NaH2PO4, pH 8, 300 mM NaCl, and 20 mM imidazole), and protein was eluted with a gradient of 20 to 250 mM imidazole in 50 mM NaH2PO4, pH 8, and 300 mM NaCl. Fractions containing Ero1p were pooled and dialyzed against 50 mM Tris-HCl, pH 8, 150 mM NaCl. Samples were aliquoted and stored at –80°C in 8% glycerol. The concentration of purified Ero1p was determined spectroscopically using an extinction coefficient of 12.5 mM–1 cm–1 at 454 nm.
Plasmid encoding His6-tagged E. coli thioredoxin I (Trx1) was kindly provided by E. Gross and D. Fass (Gross et al., 2005). His6-tagged Trx1 was expressed and purified essentially as described for His6-Pdi1p in Sevier et al. (2001). Proteins were eluted from the Ni-column with a gradient of 20 to 250 mM imidazole in 50 mM NaH2PO4, pH 8, and 300 mM NaCl, and fractions containing Trx1 were pooled and dialyzed against 10 mM Tris-HCl, pH 8, and 25 mM NaCl.
FAD Reduction Assay
Thiol substrate stock solutions were prepared in 50 mM Tris-HCl, pH 8, and 150 mM NaCl buffer and standardized with Ellman's reagent using an extinction coefficient of 13.6 mM–1 cm–1 at 412 nm. When necessary, NaOH was added to raise thiol substrate stock solutions to pH 8. Reduced Trx1 was prepared by incubation with 0.1 M DTT for 1 h at room temperature. Reduced Trx1 solution (2.5 ml) was applied to a PD-10 column equilibrated with 50 mM Tris-HCl, pH 8, and 150 mM NaCl, 0.5-ml fractions were collected, and free thiols were detected with Ellman's reagent. Fractions containing reduced Trx1 were pooled, and the concentration of reduced Trx1 was calculated with Ellman's reagent.
Recombinant Ero1p-c was diluted to a final concentration of 5 μM in 50 mM Tris-HCl, pH 8, and 150 mM NaCl with the indicated concentration of thiol substrate and reduction of the Ero1p-c FAD cofactor was monitored at 454 nm. The time lag between the moment of Ero1p-c addition and reduction of the bound FAD corresponds to the time required for the oxidase activity to consume a significant quantity of oxygen in the spectrophotometer cuvette. Using a Clark oxygen electrode, we observed the rate of oxygen consumption by Ero1p-c in the presence of DTT decreased when ∼80% of the oxygen in the chamber was consumed (our unpublished data). Therefore, we estimated a similar amount of oxygen (45 nmol in our 200-μl cuvette) had been consumed when the Ero1p-c FAD began to bleach. To control for the range of slopes observed during Ero1p-c flavin reduction, we used the time point at which half of the total change in flavin absorbance was achieved to represent the time for bound flavin reduction. Average oxygen consumption values, with standard deviations, were calculated from a minimum of three independent reactions.
RESULTS
Capture of a Disulfide Transfer Intermediate between Cys100-Cys105 and Cys352-Cys355
To test for the catalytic transfer of oxidizing equivalents between Ero1p Cys100-Cys105 and Cys352-Cys355 in vivo, we modified the Ero1 protein to facilitate detection of a disulfide bond connecting the two cysteine pairs, which is a predicted transient intermediate of the thiol-disulfide exchange process. Modified Ero1p (hereafter referred to as Ero1p-t) includes a thrombin cleavage site in an exposed loop after residue 325 as well as epitope tags in the amino- and carboxy-terminal regions to permit the independent detection of either domain after proteolysis (Figure 1A). Ero1p-t also contains Ala residues in place of Cys90 and Cys349 to prevent the formation of the nonessential Cys90-Cys349 bond, which spans the thrombin protease site (Frand and Kaiser, 2000; Gross et al., 2004). We confirmed Ero1p-t is active by demonstrating its ability to restore growth (Figure 1B) and CPY processing (Figure 1D) to an inviable ero1Δ strain as well as its ability to rescue the disulfide bond formation defect of the ero1-1 strain under restrictive conditions, which cause CPY to accumulate in the ER in the p1 form (Figure 1D). Processing of CPY was slightly delayed in an ero1-1 strain expressing Ero1p-t, relative to Ero1p (Figure 1D), suggesting Ero1p-t function may be slightly compromised relative to Ero1p. However in other assays Ero1p and Ero1p-t demonstrated similar function, including a comparable oxidizing capacity based on growth rates in the presence of the reducing agent DTT (Figure 1C). We further showed Cys105, Cys352, and Cys355 are essential for Ero1p-t function as is the case for wild-type Ero1p (Figure 1; Frand and Kaiser, 2000), and Cys100 is required for oxidative activity of Ero1p-t (Figure 1C), although induction of the unfolded protein response permits a C100A mutant to support viability (Figure 1B) (Frand and Kaiser, 2000).
Figure 1.

Functionality of Ero1p-t. (A) Ero1p amino acid sequence depicting insertion of two epitope tags (HA and myc), replacement of Cys90 and Cys349 with Ala, and addition of a thrombin protease recognition site. Lines denote cysteine connectivity found in the Ero1p-c crystal structure. (B) Complementation of an ero1Δ mutant by ERO1-t alleles. Transformants of CKY933 (ero1Δ [pAF82]) containing the following plasmids were selected on SMM minus leucine and spotted onto SMM plates containing 5-FOA or SMM plates lacking leucine: pCS253, pCS257, pCS258, pCS259, pCS254, pCS255, pCS256, pAF89, pAF125, pAF126, pCS214, pAF129, pAF130, pCS261, or an empty vector. (C) Transformants of CKY598 (ero1-1) or ero1Δ cells containing the ERO1-t plasmids in B were grown overnight in SMM minus leucine and spotted onto YPD plates containing the indicated amounts of DTT. Strains were grown for 2 d at 30°C. (D) Processing of newly synthesized CPY in CKY598 (ero1-1) or ero1Δ cells containing the ERO1-t plasmids in B. Cells were radiolabeled at 30 or 38°C for 30 min, and CPY was immunoprecipitated and resolved by SDS-PAGE. ER (p1) and vacuolar mature (m) forms of CPY are indicated.
To trap any mixed disulfide intermediates, ero1Δ cells overproducing Ero1p-t were labeled with [35S]methionine and subsequently treated with TCA to inhibit thiol-disulfide reactivity and precipitate cellular proteins. Free thiols were then blocked with the thiol-alkylation agent NEM, and proteins were immunoprecipitated with anti-myc under nonreducing but denaturing conditions. After cleavage with thrombin, immunoprecipitates were resolved by SDS-PAGE under reducing or nonreducing conditions (Figure 2A). Efficient cleavage of Ero1p-t into two domains by thrombin proteolysis was evident from the disappearance of a band corresponding to full-length Ero1p and the presence of two smaller fragments on the reducing gel (Figure 2A). Identity of the small fragments was confirmed by reimmunoprecipitation with anti-HA or anti-myc antibodies that recognize the N- or C-terminal Ero1p fragments, respectively (our unpublished data).
Figure 2.
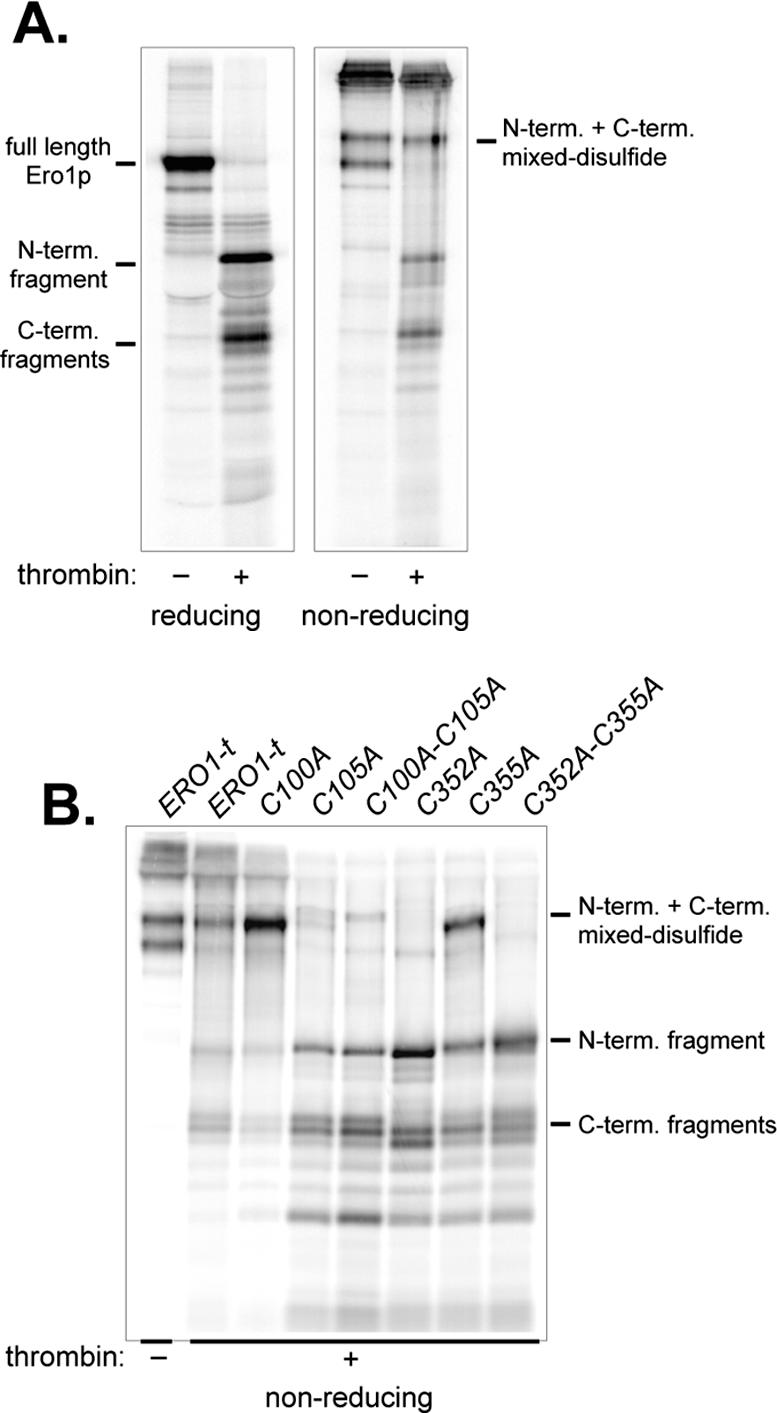
A transient disulfide bond forms between Ero1p Cys105 and Cys352. Cells with a chromosomal deletion of ERO1 containing pCS253 (A) or transformants of CKY598 (ero1-1) containing pCS253, pCS257, pCS258, pCS259, pCS254, pCS255, or pCS256 (B) were radiolabeled for 30 min at 30°C (A) or 38°C (B), lysed in conditions to block disulfide exchange, and Ero1p was immunoprecipitated under denaturing, nonreducing conditions with monoclonal anti-myc. Immunoprecipitates were cleaved with thrombin and samples were resolved by SDS-PAGE under reducing or nonreducing conditions.
When samples were run under nonreducing conditions, most of the thrombin treated Ero1p-t was observed as N-terminal and C-terminal fragments connected by a disulfide bond (Figure 2A). The disulfide-linked complex had an electrophoretic mobility similar to full-length uncleaved Ero1p under nonreducing conditions and resolved into N-terminal and C-terminal fragments on a reducing gel. Unexpectedly, under nonreducing conditions untreated Ero1p-t ran as a doublet of relatively high molecular weight (Figure 2A). We are uncertain what disulfide bond connectivity differences yield the two different oxidized Ero1p forms; however, the migration differences may reflect the presence or absence of a transient disulfide bond connecting Cys100-Cys105 and Cys352-Cy355, as suggested by the resolution of one of the oxidized species into N- and C-terminal fragments after thrombin treatment (Figure 2A). The slow migration of both oxidized species relative to reduced Ero1p most likely reflects the presence of at least two disulfide bonds (Cys143-Cys166 and Cys150-Cys295; Figure 1).
To determine which cysteines participate in the interdomain disulfide bond observed after thrombin cleavage, we evaluated the ability of cysteine mutants to form an interdomain mixed disulfide bond. For these experiments, Ero1p-t cysteine mutants were overexpressed in ero1-1 cells that were shifted to 38°C during radiolabeling, to inactivate the chromosomal ero1-1 allele. An interdomain disulfide bond was not observed when either Cys105 or Cys352 were replaced with Ala, indicating these two cysteines are normally responsible for the interdomain connection (Figure 2B). In agreement with Cys105 and Cys352 forming a transient interdomain disulfide bridge, Cys105 and Cys352 are the cysteines from each essential cysteine pair that come in proximity within the crystallized forms of Ero1p (Gross et al., 2004). Interestingly, substitution of Cys100 with Ala enhanced the recovery of an interdomain mixed disulfide relative to wild type, which almost certainly reflects the contribution of Cys100 in resolving the transient Cys105-Cys352 disulfide bond; when Cys100 is absent, the intersubunit disulfide seems to be longer lived (Figure 2B). Recovery of a Cys105-Cys352 disulfide in a Cys355 mutant was unexpected; formation of the mixed disulfide is an anticipated product of attack of the Cys352-Cys355 disulfide (absent in a Cys355 mutant) by a Cys105 thiolate (see Discussion; Figure 8). Formation of the Cys105-Cy352 interdomain disulfide could be a product of the reverse reaction, wherein the Cys352 thiolate (formed in a Cys355 mutant) attacks the Cys100-Cys105 disulfide.
Figure 8.

Proposed catalytic mechanism for reoxidation of Pdi1p by Ero1p. Thin black arrows reflect electron movement, whereas thin gray arrows denote movement of the loop domain.
Intragenic Complementation of Mutations in Different Domains of ERO1
Transfer of a disulfide bond between Ero1p domains suggested that the N- and C-terminal regions may function autonomously, allowing intragenic complementation between ERO1 alleles in different domains. To test this idea, we created ERO1 alleles that each encoded a different functional domain of Ero1p with one essential cysteine pair: an amino-terminal segment of Ero1p (Ero1p-1-181), which contains the Cys100-Cys105 pair; and Ero1p with alanine substitutions for both Cys100 and Cys105 (Ero1p-C100A-C105A), which only contains the Cys352-Cys355 pair. We established coexpression of two nonfunctional forms of Ero1p can reconstitute activity as demonstrated by ability of the coexpressed proteins to complement the ero1-1 allele lethality at 37°C and to partially restore oxidative protein folding to the ER (Figure 3). Complementation of the ero1-1 allele required expression of both polypeptides and was dependent upon Cys100-Cys105 in the amino-terminal Ero1p-1-181 fragment (Figure 3).
Figure 3.

Intragenic complementation of ERO1 mutations. (A) Diagram of the ERO1 alleles used in the complementation assays. Constructs 1–4 correspond to plasmids pAF82, pCS75, pCS42, and pCS55, respectively. (B) Transformants of CKY598 (ero1-1) containing empty vector (–) or the constructs described in A were plated on SMM (Glu) or SMM Gal (Gal) plates, and incubated for 2 d at 24 or 37°C. (C) The strains described in B were radiolabeled for 30 min at 38°C. CPY was immunoprecipitated and resolved by SDS-PAGE. ER (p1) and mature (m) forms of CPY are indicated.
Intermolecular Disulfide Transfer between Ero1 Proteins
The crystallized form of Ero1p is a monomeric protein with Cys100-Cys105 on a flexible loop approaching a four-helix bundle containing Cys352-Cys355 (Gross et al., 2004). However, our intragenic complementation studies indicated it was possible for redox-active cysteine pairs from separate Ero1 polypeptides to form an active enzyme inside the cell, implying the possibility of an active dimeric or multimeric Ero1p form. By co-overexpressing Ero1p-t with an HA epitope near the amino terminus (Ero1p-t-HA), and Ero1p-t with a myc tag at the carboxy terminus (Ero1p-t-myc), we were able to detect a disulfide bond linking the two differentially tagged proteins thereby establishing intermolecular exchange in vivo. Immunoprecipitation of Ero1p-t-HA under nonreducing denaturing conditions resulted in coprecipitation of Ero1p-t-myc, as shown by the presence of a myc-tagged carboxy-terminal fragment that could be reimmunoprecipitated from the original anti-HA lysate after thrombin cleavage (Figure 4, lanes 2 and 4). When the primary immunoprecipitation with anti-HA antibody was done under reducing denaturing conditions, no coprecipitating fragments derived from Ero1p-t-myc (a myc-tagged carboxy-terminal and an untagged amino-terminal fragment) were observed, confirming that Ero1p-t-HA and Ero1p-t-myc were linked via a disulfide bond (Figure 4, lane 1). Ero1p-t-myc immunoprecipitation produced comparable results to those observed with Ero1p-t-HA (Figure 4, lanes 6 and 8). Ability to capture a significant portion of Ero1p-t forming interdomain disulfide bonds suggests that intermolecular disulfide exchange between Ero1p molecules accounts for a significant portion of the Ero1p cycle. However, these experiments do not exclude the possibility that some Ero1p molecules engage in intramolecular dithiol-disulfide transfer events as suggested by the crystal structure.
Figure 4.

Thiol-disulfide exchange occurs between Ero1p proteins. Diagram of two differentially epitope-tagged ERO1-t alleles (A) and outline of experimental protocol used to trap mixed disulfides between different Ero1 polypeptides (B). Note lanes 1 and 5 are controls in which disulfide bonds are reduced before the first immunoprecipitation. (C) Reducing SDS-polyacrylamide gel of Ero1p immunoprecipitates. Black circles indicate Ero1p fragments coprecipitating under nonreducing conditions. The loading in lanes 4 and 8 is 4 times that of lanes 1–3 and 5–7.
Ero1p Loop Mutations Bypass the Requirement for Cys100 and Cys105
The structure of Ero1p as well as the inability of a Cys100-Cys105 mutant to facilitate protein oxidation in vivo suggests protein substrates, such as Pdi1p, may be too large to gain access the Cys352-Cys355 pair directly, which is found below the protein surface partially buried underneath the flexible loop domain (Frand and Kaiser, 2000; Gross et al., 2004). To test the idea that Cys100-Cys105 might be dispensable if there was another way for substrate thiols to gain access to the active site disulfide pair, we developed a genetic suppression approach to isolate second-site mutations that restored activity to an Ero1p-C100A-C105A mutant.
A plasmid borne copy of ero1-C100A-C105A was mutagenized and screened for intragenic mutations that could complement the ero1-1 ire1Δ strain growth defect at the restrictive temperature. Use of an ire1Δ mutant prevented induction of the unfolded protein response, which can partially compensate for certain ERO1 alleles (Frand and Kaiser, 2000). Six suppressing mutations were isolated and all mapped to a 55-amino acid region of Ero1p that included the ends of the flexible loop domain (L84P, L86S, and W120R) as well as the α-helix directly preceding the loop domain (I66N, L70P, and L70R) (Figure 5). Growth in the presence of the reducing agent DTT (Figure 5A) and proper CPY processing (Figure 5C) in an ero1-1 ire1Δ strain covered by the suppressor alleles confirmed the intragenic suppressors restored oxidative protein folding to the ER. Presence of the suppressor alleles in the ero1-1 ire1Δ strain was not sufficient to allow growth in the presence of 10 mM DTT, suggesting the suppressor alleles possess less in vivo oxidative activity than wild-type Ero1p (Figure 5A). None of the second site mutations were able to suppress loss of Cys352-Cys355, illustrating that suppression required the core catalytic activity of Ero1p (Figure 5B). Location of the suppressor mutations in the loop region suggests the different suppressor alleles may cause the loop to be displaced from the catalytic core, allowing substrates direct access to Cys352-Cys355.
Figure 5.

Ero1p loop mutations can suppress loss of Cys100 and Cys105. (A) Transformants of CKY932 (ero1-1 ire1Δ) or CKY933 (ero1Δ [pAF82]) containing empty vector, pAF85, pCS169, pCS196, pCS194, pCS235, pCS233, pCS230, or pCS184 were plated on (from left to right) SMM plates lacking leucine and incubated for 3 d at 37 or 24°C, or on YPD plates with the indicated concentration of DTT, and incubated for 3 d at 24°C (B) CKY598 (ero1-1) containing empty vector, pAF89, pCS261, pCS343, pCS344, pCS325, pCS322, pCS323, or pCS324 were plated on SMM plates lacking leucine and incubated for 3 d at 24 or 37°C. (C) The ero1-1 ire1Δ strains described in A were radiolabeled for 30 min at 38°C. CPY was immunoprecipitated and resolved by SDS-PAGE. ER (p1) and mature (m) forms of CPY are indicated. (D) Residues that when mutated can suppress loss of Cys100 and Cys105 are represented as red sticks in the Ero1p-c structure. The two essential cysteine pairs, Cys100-Cys105 and Cys352-Cys355, and flavin cofactor are drawn as orange and yellow sticks respectively. Residues 55–136, which include the loop domain and α-helix preceding the loop, are highlighted in blue. The four helices forming the catalytic core domain of Ero1p are represented as brown cylinders.
To probe the position of the loop in different suppressor mutants, we took advantage of the thrombin cleavage assay described previously. We introduced amino acid changes isolated in the intragenic suppressor screen into the ERO1-t plasmid (wild-type Cys100 and Cys105) and determined whether the mutants were capable of forming interdomain disulfide bonds. If the suppressor mutations displace the loop from the active site cysteines, a corresponding decrease in recovery of the Cys105-Cys352 disulfide should be observed after thrombin cleavage. Indeed, no interdomain mixed disulfide complex was recovered from most of the suppressor mutants, whereas two mutants caused a severe decrease in complex recovery (Figure 6). Interestingly, the two suppressor mutants capable of forming some mixed disulfide complexes were also the only two mutants that could complement an ero1Δ strain (our unpublished data). Complete cleavage of all the suppressor mutants was confirmed by resolving the thrombin treated fragments under reducing conditions (our unpublished data). Results from a comparable experiment suggest the suppressor mutants also exhibit decreased formation of the Cys90-Cys349 bond (our unpublished data).
Figure 6.
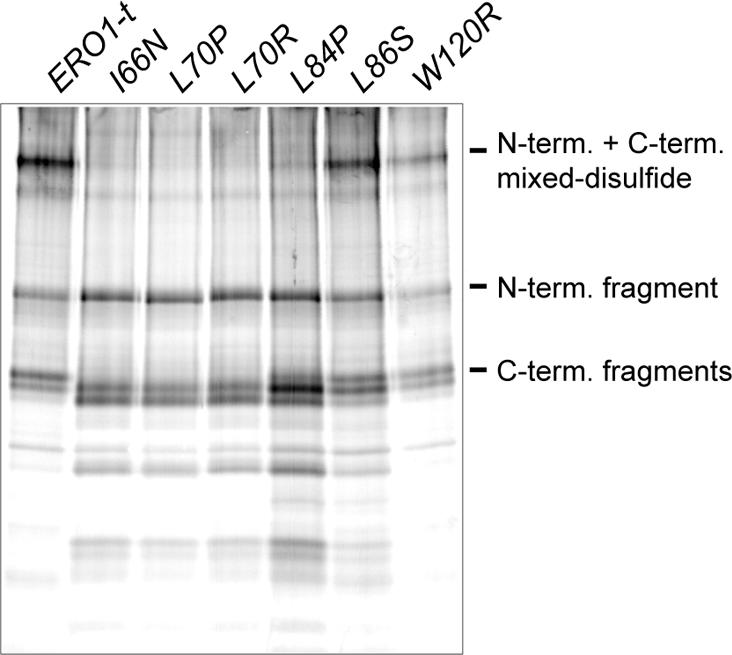
Substitution of Ero1p loop residues decreases recovery of an interdomain Cys105-Cys352 disulfide bond. Transformants of CKY598 (ero1-1) containing pCS253, pCS347, pCS349, pCS348, pCS306, pCS307, or pCS262 were radiolabeled at 38°C for 30 min, lysed in conditions to block disulfide exchange, and Ero1p was immunoprecipitated with anti-myc antibody under denaturing, nonreducing conditions. Immunoprecipitates were cleaved with thrombin and samples were resolved by SDS-PAGE under nonreducing conditions.
Ero1p Loop Mutations Prevent Intragenic Complementation between Ero1 Polypeptides
If the suppressor mutations disrupt the interaction between the loop domain and the enzyme core, when the mutations are added to the amino-terminal Ero1p-1-181 fragment they should block the ability of this fragment to complement mutations in full-length Ero1p. Coexpression of Ero1p-C100A-C105A and an Ero1p-1-181 fragment each containing one of five different suppressor mutations prevented intragenic complementation (Figure 7). An exception was the Trp120 mutation that when added to the amino-terminal fragment could still suppress the ero1-1 growth defect when coexpressed with the Ero1p-C100A-C105A protein (Figure 7). The Trp120 mutation also had the least effect on the recovery of the Cys105-Cys352 disulfide shown previously (Figure 6), suggesting this mutation has the least impact of all the suppressor mutants on the loop position. Expression of the Ero1-1-181 suppressor fragments in galactose media was confirmed by Western analysis (our unpublished data).
Figure 7.

Mutations in the Ero1p loop can prevent intragenic complementation. (A) Diagram of constructs used in B. ERO1 alleles correspond to products of plasmids pCS75 (1), pCS42 (2), pCS45 (3), pCS341 (4-I66N), pCS342 (4-L70P), pCS321 (4-L70R), pCS318 (4-L84P), pCS319 (4-L86S), and pCS320 (4-W120R). (B) Transformants of CKY598 (ero1-1) containing constructs described in A were plated on SMM (Glu) or SMM Gal (Gal) plates and incubated at 24 or 37°C for 2 d.
Suppressor Mutations in the Loop Give Increased Access to Cys352-Cys355
To test the idea that displacement of the Ero1p loop by the suppressor mutations allows substrates direct access to Cys352-Cys355, we assayed the ability of wild-type and mutant Ero1p to oxidize a range of thiol-containing substrates in vitro. Recombinant Ero1p (residues 56–424, referred to as Ero1p-c; Gross et al., 2004), Ero1p-c with C100A-C105A mutations, and Ero1p-c with C100A-C105A and either an L86S or W120R substitution were purified from E. coli. Ero1p-c proteins containing other suppressor mutations (I66N, L70P/R, or L84P) were insoluble when expressed in bacteria and could not be purified in sufficient quantities for biochemical analysis (our unpublished data). The correlation between insolubility and inability to form the Cys105-Cys352 bond (Figure 6), suggests gross perturbations in the loop region of the suppressor mutants led to aggregation of the bacterially expressed proteins. Large conformational changes in the loop domain may also affect the ability of these mutants to form stable structures in yeast cells completely deficient in the normal oxidative protein-folding pathway, which could account for the inability of these same mutants to complement a strain lacking ERO1.
Analyses of recombinant wild-type and mutant Ero1 proteins for their ability to oxidize a range of thiol-containing substrates in vitro revealed that Cys100 and Cys105 are important for oxidation of relatively large thiol-containing substrates, including reduced cysteine, glutathione, and thioredoxin, but not for oxidation of small substrates such as DTT and β-mercaptoethanol (Table 2). Small molecules such as DTT and β-mercaptoethanol may have direct access to the buried Cys352-Cys355 pair, bypassing the requirement for Cys100-Cys105. Ero1p-c-C100A-C105A did have some capacity to oxidize larger molecules, suggesting these substrates had limited access the active site. Combining the suppressor mutations L84P or W120R to Ero1p-c with the double C100A-C105A substitution, increased the rates of all substrate oxidation relative to Ero1p-c-C100A-C105A, suggesting Cys352-Cys355 in the suppressor mutants is more accessible than in wild-type Ero1p-c (Table 2). Ability of Cys352-Cys355 to oxidize a variety of substrates when exposed by the suppressor mutants, shows the Ero1p loop, displaced in the suppressor mutants, normally plays a role in concealing the active site from potential substrates.
Table 2.
Analysis of thiol oxidation by Ero1p-C100A-C105A intragenic suppressors
| Activity (nmol O2 min-1/nmol Ero1p-c)a
|
|||||
|---|---|---|---|---|---|
| Molecular weightb | Wild-type | C100A C105A | L86S C100A C105A | C100A C105A W120R | |
| 5 mM Dithiothreitol | 152 | 16.1 ± 1.9 (129) | 12.5 ± 2.0 (100) | 23.7 ± 4.4 (190) | 64.3 ± 10.7 (514) |
| 50 mM β-Mercaptoethanol | 154 | 3.6 ± 0.2 (109) | 3.3 ± 0.8 (100) | 4.8 ± 0.3 (145) | 6.3 ± 0.3 (191) |
| 50 mM Cysteine | 240 | 5.8 ± 0.8 (187) | 3.1 ± 0.7 (100) | 5.8 ± 0.2 (187) | 8.2 ± 0.5 (265) |
| 50 mM Glutathione | 613 | 1.7 ± 0.1 (189) | 0.9 ± 0.0 (100) | 1.7 ± 0.2 (189) | 3.8 ± 0.5 (422) |
| 0.5 mM Thioredoxin | 12,626 | 75.0 ± 15.0 (2083) | 3.6 ± 0.1 (100) | 8.5 ± 0.7 (236) | 12.5 ± 1.1 (347) |
Activity values were extrapolated from the rate of FAD reduction as described in Materials and Methods. Percentage of activity relative to Ero1p-C100A-C105A is noted in parentheses.
Molecular weight of the oxidized form.
Thioredoxin and Pdi1p are structural homologues, sharing a conserved structural fold and Cys-x-x-Cys active site; the small size and large yield of recombinant thioredoxin led us to use thioredoxin in place of Pdi1p for these in vitro studies. The two- to threefold increase in activity of the suppressor mutants toward thioredoxin relative to Ero1p-C100A-C105A shows the increased accessibility of the active site cysteine in the loop mutants (Table 2). The 10-fold increase in activity of Ero1p-c toward thioredoxin, beyond the two- and threefold increase activity of the suppressor mutants toward thioredoxin, suggests the loop cysteines facilitate a specific association with thioredoxin (Table 2). It seems likely the Ero1p loop domain has evolved to interact specifically with proteins of the thioredoxin structural family.
DISCUSSION
Mutational and structural studies led to the idea that disulfide bond transfer from Ero1p to Pdi1p involves a disulfide shuttling mechanism between the two essential Ero1p cysteine pairs (Frand and Kaiser, 2000; Gross et al., 2004). In this article, we provide the first direct evidence of a thiol-disulfide transfer event between the Ero1p cysteines and identify Cys105 and Cys352 as the cysteines directly involved in the exchange reaction. We suggest the relay of oxidizing equivalents between the two Ero1p cysteine pairs serves to direct the flow of oxidizing equivalents from the buried flavinproximal Cys352-Cys355 pair to the substrate protein Pdi1p.
Thiol-Disulfide Exchange between Ero1p Cysteine Pairs
Combining the data from our studies with previous work on Ero1p allows us to propose a detailed picture of the electron transfer events in the oxidation of Pdi1p by Ero1p outlined in Figure 8. The cycle begins when a thiolate from reduced Pdi1p attacks the Ero1p Cys100-Cys105 disulfide, resulting in formation of a transient mixed disulfide intermediate between Ero1p-Cys100 and Pdi1p (Figure 8, stages 1–2; Frand and Kaiser, 2000). This intermediate is resolved upon transfer of electrons from a second cysteine in Pdi1p to Ero1p, which releases oxidized Pdi1p (Figure 8, stages 2–3). The reduced loop domain then moves closer to the catalytic core of Ero1p where the Cys105 thiolate attacks the Cys352-Cys355 disulfide bond, forming a mixed disulfide complex between Cys105 and Cys352 (Figure 8, stages 3–5). This intermediate is resolved upon attack of the Cys105-Cys352 disulfide by the Cys100 thiolate and transfer of electrons to Cys352-Cys355, which results in an oxidized Cys100-Cys105 pair (Figure 8, stages 5–6). The Ero1p catalytic cycle is complete when Cys352-Cys355 is reoxidized by transfer of electrons from the Cys355 thiol to molecular oxygen, via the flavin cofactor (Figure 8, stages 6–7; Tu et al., 2000; Tu and Weissman, 2002; Gross et al., 2004). It will require further study to determine whether the transfer reactions outlined in Figure 8 take place in distinct steps or are coupled to formation of transient ternary complexes, as outlined in a catalytic scheme for the prokaryotic functional homologue of Ero1p, DsbB (Kadokura and Beckwith, 2002).
Studies with the human Ero1 proteins Ero1-Lα and Ero1-Lβ (Cabibbo et al., 2000; Pagani et al., 2000) suggest the mechanisms of electron transfer outlined in Figure 8 are conserved among the eukaryotic Ero1 proteins. Ero1-Lα contains two essential cysteine pairs, Cys94-Cys99 and Cys394-Cys397 (Benham et al., 2000; Cabibbo et al., 2000; Mezghrani et al., 2001; Bertoli et al., 2004). Recently, an Ero1-Lα-C94S mutant was shown to be defective in its association with protein disulfide isomerase (PDI), suggesting that Cys94 (like Ero1p Cys100) normally undergoes thiol-disulfide exchange with PDI (Bertoli et al., 2004). Our model implies an internal electron exchange between Cys99 and Cys394 is also part of the human Ero1-L catalytic mechanism.
Consistent with our proposed catalytic model, we observed an enhanced trapping of the interdomain mixed disulfide intermediate in an Ero1p-C100A mutant because of poor resolution of this intermediate in the absence of nucleophilic attack by Cys100 (Figure 8, stage 5). Ability of Ero1p-C100A to suppress a complete deletion of ERO1 suggests Pdi1p may be able to attack and receive a disulfide bond directly from the Cys105-Cys352 interdomain disulfide. Lack of a captured interdomain mixed disulfide in an Ero1p-C105A mutant demonstrates that Cys100 cannot substitute for Cys105, which may be the result of structural constraints imposed by the relative positions of Cys100 and Cys105 in the loop domain.
It remains puzzling that it was possible to recover a mixed domain disulfide bond in a Cys355 mutant, which lacks the Cys352-Cys355 disulfide bond normally attacked by the Cys105 thiolate (Figure 8, stage 4). Formation of the Cys105-Cys352 disulfide bond in the Cys355 mutant may reflect a reverse reaction wherein the Cys352 thiolate attacks Cys100-Cys105. Interestingly, a similar internal mixed disulfide intermediate has been captured with analogous cysteine mutants in Erv2p and DsbB, proteins that have been proposed to use a catalytic mechanism similar to the one outlined in Figure 8 (Gross et al., 2002; Kadokura and Beckwith, 2002).
An important mechanistic question is whether the interdomain thiol-disulfide exchange reaction in Ero1p occurs by transfer between domains of an individual polypeptide, as suggested by the crystal structure (Gross et al., 2004), or by transfer between different Ero1p polypeptides, as suggested by our ability to capture interchain disulfide bonds. It seems likely that in the cell both intra- and interchain transfer events can occur. Based on the structural similarity between the catalytic core of Ero1p and Erv2p, it seems possible that a conformational rearrangement to create an Erv2p-like antiparallel dimer with two catalytic centers, wherein each protomer contributes a single cysteine pair, could provide for an intermolecular exchange event. The catalytic cycle of an Erv2p-like, dimeric Ero1p would involve relay of oxidizing equivalents from the buried active site Cys352-Cys355 of one protomer to Pdi1p via the loop Cys100-Cys105 pair from a second Ero1p protomer. Recent studies with Ero1-Lβ suggest interchain transfer events may also be a catalytic feature of human Ero1 proteins (Dias-Gunasekara et al., 2005).
Creation of a functional Ero1p dimer would require a significant change in the loop position, from the arbitrarily designated re face to the si face, to allow for an interaction between Cys100-Cys105 and Cys352-Cys355 in the associated protomer. Redox-dependent transitions between monomeric and dimeric structures have been described for several proteins, including peroxiredoxin 5 and the intracellular chloride ion channel protein CLIC1 (Declercq et al., 2001; Evrard et al., 2004; Littler et al., 2004). However, further structural and biochemical data will be required to elucidate whether, and if so, how the extensive rearrangement necessary for Ero1p dimerization is possible.
Internal Disulfide Transfer and Specificity
Inability of an Ero1p-C100A-C105A mutant to promote protein oxidation in vivo (Frand and Kaiser, 2000) suggests the requirement for Cys100-Cys105 stems from the inability of Pdi1p to directly interact with Cys352-Cys355. This interpretation is supported by our observation that in vitro Cys100-Cys105 facilitates oxidation of bulky thiol-containing substrates, which have limited access to Cys352-Cys355. The physical isolation of Cys352-Cys355 from large substrates is evident in the Ero1p structure which shows Cys352-Cys355 situated below the protein surface, underneath the flexible loop domain (Gross et al., 2004).
Using a genetic suppression approach, we isolated six intragenic mutations that could bypass the requirement for Cys100-Cys105. Biochemical and genetic tests showed the suppressor mutations dislodge the loop domain from the catalytic core, which allows substrates direct access to the normally inaccessible Cys352-Cys355 pair. These mutants suggest the loop domain not only facilitates oxidation of Pdi1p but also prevents thiols direct access to Cys352 and Cys355.
We suggest the two different functions of the loop domain, to prevent the active site pair from nonspecifically oxidizing small molecules or proteins and to transfer disulfides from the buried active site cysteines directly to Pdi1p, impart substrate selectivity to Ero1p. By physically blocking access to Cys352-Cys355, the loop domain prevents nonspecific oxidation of proteins or molecules in the ER. Use of a second pair of cysteines in the loop domain with properties designed to facilitate the specific oxidation of Pdi1p guides substrate selection by Ero1p and thereby may prevent oxidation of undesired in vivo substrates such as glutathione. Mutants recently isolated in the yeast ER oxidase Erv2p indicate the intercysteine relay used by Erv2p confers a similar specificity to the Erv2p catalytic cycle (Vala et al., 2005). Structural changes in Erv2p that allow substrates direct access to the active site, bypassing the requirement for the secondary tail cysteine pair, cause an altered activity toward selected substrates similar to that observed with our Ero1p suppressor mutants (Vala et al., 2005).
Ability of Ero1p-C100A-C105A suppressor mutants to oxidize many of the thiol compounds we studied at a rate similar to or exceeding wild-type Ero1p suggests the relay of oxidizing equivalents thru the cysteines in the loop domain diminishes the oxidizing capacity from Cys352-Cys355. However, this loss in oxidizing potential seems to be an acceptable trade-off for the increased specificity toward thioredoxin-like substrates provided by the loop domain. It is an attractive possibility that the unstructured nature of the flexible loop domain drives the specific association of Cys100-Cys105 with Pdi1p or thioredoxin by mimicking an unfolded nascent polypeptide chain, which is a known substrate for Pdi1p.
Coincident operation of various protein oxidation and reduction pathways within a single cellular compartment, such as the ER, requires that mechanisms exist to ensure transfer of disulfides to appropriate substrates. Inadvertent transfer of oxidizing or reducing equivalents to the wrong substrate could result in undesired substrate activation or incapacitation. In addition, unnecessary cellular damage may be caused by excess reactive oxygen species generated from unregulated catalytic cycles of Ero1p or Erv2p (Harding et al., 2003; Tu and Weissman, 2004). We suggest one way to control oxidation of thiol-containing substrates is to use an intercysteine relay mechanism wherein the second cysteine pair drives certain thiol transfer events. Many different thiol oxidoreductases, including Ero1p, use an intraprotein disulfide transfer event to affect substrate oxidation or reduction, signifying the value of this type of mechanism (Niimura and Massey, 1996; Katzen and Beckwith, 2000; Gross et al., 2002; Kadokura and Beckwith, 2002; Raje and Thorpe, 2003; Argyrou and Blanchard, 2004; Messens et al., 2004). An improved understanding of the specific recognition and oxidation of Pdi1p by Ero1p will likely rely upon defining the association between Pdi1p and the flexible loop domain of Ero1p containing Cys100 and Cys105 at a biochemical and structural level.
Acknowledgments
We thank Hongjing Qu for invaluable assistance with protein purification and plasmid construction. This work was supported by National Institutes of Health Grant GM-46941 (to C.A.K.).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–05–0417) on February 22, 2006.
References
- Adams, A., Gottschling, D. E., Kaiser, C. A., and Stearns, T. (1998). Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Argyrou, A., and Blanchard, J. S. (2004). Flavoprotein disulfide reductases: advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol. 78, 89–142. [DOI] [PubMed] [Google Scholar]
- Benham, A. M., Cabibbo, A., Fassio, A., Bulleid, N., Sitia, R., and Braakman, I. (2000). The CXXCXXC motif determines the folding, structure and stability of human Ero1-Lalpha. EMBO J. 19, 4493–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli, G., Simmen, T., Anelli, T., Molteni, S. N., Fesce, R., and Sitia, R. (2004). Two conserved cysteine triads in human Ero1alpha cooperate for efficient disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 279, 30047–30052. [DOI] [PubMed] [Google Scholar]
- Cabibbo, A., Pagani, M., Fabbri, M., Rocchi, M., Farmery, M. R., Bulleid, N. J., and Sitia, R. (2000). ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275, 4827–4833. [DOI] [PubMed] [Google Scholar]
- Declercq, J. P., Evrard, C., Clippe, A., Stricht, D. V., Bernard, A., and Knoops, B. (2001). Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 A resolution. J. Mol. Biol. 311, 751–759. [DOI] [PubMed] [Google Scholar]
- Dias-Gunasekara, S., Gubbens, J., van Lith, M., Dunne, C., Williams, J. A., Kataky, R., Scoones, D., Lapthorn, A., Bulleid, N. J., and Benham, A. M. (2005). Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1β. J. Biol. Chem. 280, 33066–33075. [DOI] [PubMed] [Google Scholar]
- Evrard, C., Capron, A., Marchand, C., Clippe, A., Wattiez, R., Soumillion, P., Knoops, B., and Declercq, J. P. (2004). Crystal structure of a dimeric oxidized form of human peroxiredoxin 5. J. Mol. Biol. 337, 1079–1090. [DOI] [PubMed] [Google Scholar]
- Fassio, A., and Sitia, R. (2002). Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochem. Cell Biol. 117, 151–157. [DOI] [PubMed] [Google Scholar]
- Frand, A. R., and Kaiser, C. A. (1998). The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1, 161–170. [DOI] [PubMed] [Google Scholar]
- Frand, A. R., and Kaiser, C. A. (1999). Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 4, 469–477. [DOI] [PubMed] [Google Scholar]
- Frand, A. R., and Kaiser, C. A. (2000). Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol. Biol. Cell 11, 2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, J., Muhlenhoff, U., Hofhaus, G., Lill, R., and Lisowsky, T. (2001). Yeast ERV2p is the first microsomal FAD-linked sulfhydryl oxidase of the Erv1p/Alrp protein family. J. Biol. Chem. 276, 23486–23491. [DOI] [PubMed] [Google Scholar]
- Gross, E., Kastner, D. B., Kaiser, C. A., and Fass, D. (2004). Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117, 601–610. [DOI] [PubMed] [Google Scholar]
- Gross, E., Sevier, C. S., Heldman, N., Vitu, E., Bentzur, M., Kaiser, C. A., Thorpe, C., and Fass, D. (2005). Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 103, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, E., Sevier, C. S., Vala, A., Kaiser, C. A., and Fass, D. (2002). A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat. Struct. Biol. 9, 61–67. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hoober, K. L., Glynn, N. M., Burnside, J., Coppock, D. L., and Thorpe, C. (1999). Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J. Biol. Chem. 274, 31759–31762. [DOI] [PubMed] [Google Scholar]
- Hoober, K. L., and Thorpe, C. (2002). Flavin-dependent sulfhydryl oxidases in protein disulfide bond formation. Methods Enzymol. 348, 30–34. [DOI] [PubMed] [Google Scholar]
- Jessop, C. E., Chakravarthi, S., Watkins, R. H., and Bulleid, N. J. (2004). Oxidative protein folding in the mammalian endoplasmic reticulum. Biochem. Soc. Trans. 32, 655–658. [DOI] [PubMed] [Google Scholar]
- Kadokura, H., and Beckwith, J. (2002). Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 21, 2354–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura, H., Katzen, F., and Beckwith, J. (2003). Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135. [DOI] [PubMed] [Google Scholar]
- Katzen, F., and Beckwith, J. (2000). Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell 103, 769–779. [DOI] [PubMed] [Google Scholar]
- Littler, D. R., et al. (2004). The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J. Biol. Chem. 279, 9298–9305. [DOI] [PubMed] [Google Scholar]
- Messens, J., Van Molle, I., Vanhaesebrouck, P., Limbourg, M., Van Belle, K., Wahni, K., Martins, J. C., Loris, R., and Wyns, L. (2004). How thioredoxin can reduce a buried disulphide bond. J. Mol. Biol. 339, 527–537. [DOI] [PubMed] [Google Scholar]
- Mezghrani, A., Fassio, A., Benham, A., Simmen, T., Braakman, I., and Sitia, R. (2001). Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 20, 6288–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto, H., and Bardwell, J. C. (2004). Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim. Biophys. Acta 1694, 111–119. [DOI] [PubMed] [Google Scholar]
- Niimura, Y., and Massey, V. (1996). Reaction mechanism of Amphibacillus xylanus NADH oxidase/alkyl hydroperoxide reductase flavoprotein. J. Biol. Chem. 271, 30459–30464. [DOI] [PubMed] [Google Scholar]
- Pagani, M., Fabbri, M., Benedetti, C., Fassio, A., Pilati, S., Bulleid, N. J., Cabibbo, A., and Sitia, R. (2000). Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275, 23685–23692. [DOI] [PubMed] [Google Scholar]
- Pollard, M. G., Travers, K. J., and Weissman, J. S. (1998). Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1, 171–182. [DOI] [PubMed] [Google Scholar]
- Raje, S., and Thorpe, C. (2003). Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation. Biochemistry 42, 4560–4568. [DOI] [PubMed] [Google Scholar]
- Sevier, C. S., Cuozzo, J. W., Vala, A., Aslund, F., and Kaiser, C. A. (2001). A flavoprotein oxidase defines a new endoplasmic reticulum pathway for biosynthetic disulphide bond formation. Nat. Cell Biol. 3, 874–882. [DOI] [PubMed] [Google Scholar]
- Sevier, C. S., and Kaiser, C. A. (2002). Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell. Biol. 3, 836–847. [DOI] [PubMed] [Google Scholar]
- Tan, J. T., and Bardwell, J. C. (2004). Key players involved in bacterial disulfide-bond formation. Chembiochem 5, 1479–1487. [DOI] [PubMed] [Google Scholar]
- Thorpe, C., Hoober, K. L., Raje, S., Glynn, N. M., Burnside, J., Turi, G. K., and Coppock, D. L. (2002). Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch. Biochem. Biophys. 405, 1–12. [DOI] [PubMed] [Google Scholar]
- Tu, B. P., Ho-Schleyer, S. C., Travers, K. J., and Weissman, J. S. (2000). Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290, 1571–1574. [DOI] [PubMed] [Google Scholar]
- Tu, B. P., and Weissman, J. S. (2002). The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 10, 983–994. [DOI] [PubMed] [Google Scholar]
- Tu, B. P., and Weissman, J. S. (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vala, A., Sevier, C. S., and Kaiser, C. A. (2005). Structural determinants of substrate access to the disulfide oxidase Erv2p. J. Mol. Biol. 354, 952–966. [DOI] [PubMed] [Google Scholar]