

Abstract
Mating yeast cells provide a genetically accessible system for the study of cell fusion. The dynamics of fusion pores between yeast cells were analyzed by following the exchange of fluorescent markers between fusion partners. Upon plasma membrane fusion, cytoplasmic GFP and DsRed diffuse between cells at rates proportional to the size of the fusion pore. GFP permeance measurements reveal that a typical fusion pore opens with a burst and then gradually expands. In some mating pairs, a sudden increase in GFP permeance was found, consistent with the opening of a second pore. In contrast, other fusion pores closed after permitting a limited amount of cytoplasmic exchange. Deletion of FUS1 from both mating partners caused a >10-fold reduction in the initial permeance and expansion rate of the fusion pore. Although fus1 mating pairs also have a defect in degrading the cell wall that separates mating partners before plasma membrane fusion, other cell fusion mutants with cell wall remodeling defects had more modest effects on fusion pore permeance. Karyogamy is delayed by >1 h in fus1 mating pairs, possibly as a consequence of retarded fusion pore expansion.
INTRODUCTION
In cell fusion, the plasma membranes of two cells fuse to yield a single larger cell. Cell fusion is essential for human development and occurs in diverse contexts including the fusion of a sperm with an egg to produce a fertilized zygote, the fusion of myocytes to produce skeletal muscle fibers, the fusion of trophoblasts in the placenta, the fusion of macrophage/monocyte derived cells to produce osteoclasts for bone remodeling and multinucleated giant cells to scavenge large foreign bodies, and the fusion of fiber cells to form the lens of the eye (Kuszak et al., 1989; Mi et al., 2000; Vignery, 2000; Primakoff and Myles, 2002; Taylor, 2002; Chen and Olson, 2005). Cell-cell fusion may also occur in cancer, leading to the production of hybrid cells with multidrug resistance and/or enhanced metastatic potential (Duelli and Lazebnik, 2003). Similarly, stem cells can fuse with cells within damaged tissues to acquire differentiation markers (Alvarez-Dolado et al., 2003). In a related process, the plasma membranes of two cells can fuse and exchange membrane proteins without forming a hybrid cell, as occurs at immunological synapses and during the formation of a tunneling nanotube (Stinchcombe et al., 2001; Rustom et al., 2004). Despite the fundamental importance of cell-cell fusion, the mechanism for fusing the plasma membranes of two cells is poorly understood. The cell fusion event of yeast mating provides a genetically accessible system to elucidate the mechanism of cell fusion.
Yeast has two haploid mating types, MATa and MATα (Marsh and Rose, 1997). Mating initiates when pheromones secreted by haploid yeast bind to receptors expressed on cells of the opposite mating type. A signaling pathway involving G-proteins and MAP kinases is activated in both haploid cells, resulting in arrest of the cell cycle before DNA synthesis, a shift in the cellular growth axis toward the pheromone secreting partner, and transcriptional induction of genes involved in the mating process. Haploid cells of opposite mating type bind to each other and then remodel their cell walls to allow their plasma membranes to contact each other and fuse. Membrane fusion allows the exchange of cellular contents including cytoplasmic proteins, nuclei, mitochondria, secretory organelles, and vacuoles. Finally, a diploid daughter cell buds from the conjugation bridge connecting the two parents.
Cell fusion mutants have mating defects at a stage after haploid cells of opposite mating type have adhered to each other, but before they have fused (White and Rose, 2001). fus1 was the first cell fusion mutant to be discovered (McCaffrey et al., 1987; Trueheart et al., 1987). Arrested fus1 mating pairs have cell walls separating the two plasma membranes, indicating a defect in cell wall remodeling. A similar phenotype is found in many other cell fusion mutants, including fus2, rvs161, spa2, pea2, bni1, and fig1 (White and Rose, 2001). In contrast, fusion of prm1 mutant mating pairs often arrests at a stage after cell wall remodeling, but before fusion (Heiman and Walter, 2000). In addition, we recently reported that some prm1 mating pairs lyse immediately after plasma membrane contact, supporting a direct role for Prm1 in the membrane fusion process (Jin et al., 2004). Both Fus1 and Prm1 are membrane proteins that are expressed in response to mating pheromones and targeted to sites of cell-cell contact in mating pairs (Trueheart et al., 1987; Trueheart and Fink, 1989; Heiman and Walter, 2000). However, the molecular functions of these proteins are largely unknown, and the fusion proteins that actually merge the two plasma membranes during yeast mating have not been identified. Furthermore, all known cell fusion mutants are leaky, suggesting that yeast has multiple pathways leading to cell fusion.
The membrane fusion events associated with infection by enveloped viruses and intracellular membrane trafficking have been investigated more extensively than plasma membrane fusion during mating. Viral fusion proteins such as the gp41 protein of HIV and the hemagglutinin (HA) protein of influenza virus are transmembrane glycoproteins that are activated by binding to receptors on the host cell and/or by internalization to an acidic endosome (Bentz, 1993). Upon activation, they undergo a conformational shift that involves insertion of a hydrophobic fusion peptide into the target membrane and the formation of a helical bundle that pulls the transmembrane domain and fusion peptide together to mediate membrane merger (Carr and Kim, 1993; Bullough et al., 1994). SNARE proteins are thought to mediate intracellular membrane fusion by a similar mechanism (Weber et al., 1998). SNAREs from the two membranes assemble into an α-helical bundle that pulls the transmembrane domains and associated membranes into close proximity before fusion.
The initial aqueous connection between two membrane-bound compartments is known as a fusion pore. A typical viral or exocytic fusion pore opens with a burst and then gradually expands (Spruce et al., 1990, 1991). However, other modes of fusion have been described. Transient connections (flickers) from failed fusion attempts are often observed before a successful fusion (Spruce et al., 1990). Small pores that permit release of low-molecular-weight signaling molecules but not larger proteins can precede complete fusion (Albillos et al., 1997). In “kiss and run” exocytosis, secretory vesicle contents can be released through a transient fusion pore, which then closes to allow the vesicle membrane to recycle without ever merging completely with the plasma membrane (Aravanis et al., 2003). Changes in the dynamics of exocytic fusion pore opening and expansion may regulate the efficacy of intercellular signaling (Richards et al., 2005).
Fusion pore dynamics can be manipulated experimentally by interfering with the function of fusion proteins. Mutations in the fusion peptide, transmembrane domain, or cytoplasmic regions of viral fusion proteins can inhibit fusion pore dilation (Schoch and Blumenthal, 1993; Melikyan et al., 1999, 2000; Kozerski et al., 2000; Dutch and Lamb, 2001). Pore dilation can also be inhibited by a peptide that interferes with α-helical bundle formation, indicating that bundle formation may drive expansion as well as formation of a fusion pore (Markosyan et al., 2003). In exocytosis, mutations in the transmembrane domain of the t-SNARE syntaxin altered the conductance of fusion pores, suggesting that these domains may line the fusion pore (Han et al., 2004). Furthermore, the dynamics of exocytic fusion pores can also be manipulated by altering the expression of SNARE-binding proteins. Synaptotagmins I and IV regulate the relative frequency of kiss and run versus full fusion via interactions of their C2B domains with Ca2+ (Wang et al., 2003). A Munc18 mutation that promotes dissociation from syntaxin accelerated fusion pore expansion (Fisher et al., 2001). Complexin II overproduction promoted transient fusion by closing fusion pores (Archer et al., 2002), and cysteine string protein overproduction interfered with fusion pore expansion (Graham and Burgoyne, 2000). These results suggest that mutations that alter the dynamics of fusion pores between mating yeast may further our understanding of the regulation of cell fusion and lead to the discovery of a fusion protein.
This article reports the first study of fusion pore dynamics during a naturally occurring fusion event between two cells. GFP permeance measurements were used to show that fusion pores between mating yeast typically open with a burst and then gradually expand. A selection of cell fusion mutants was surveyed, leading to the discovery that the fusion pores of fus1 mating pairs have exceptionally low initial permeance and a slow expansion rate. One likely consequence of this fusion pore defect is a delay before karyogamy.
MATERIALS AND METHODS
Strains and Plasmids
Yeast strains were all derived from strains produced by the yeast knockout consortium in the BY4741/BY4742 strain background. Plasmids are listed in Table 1. The yeast integrating plasmids used in this study are derivatives of pRH475, an HMG1-eGFP expression plasmid with a GPD promoter and PGK1 terminator (Hampton et al., 1996). The 3′ UTR and 3′ end of the SSO1 gene were inserted at a KpnI site to permit directed integration at the SSO1 locus without altering normal SSO1 expression or function. Plasmids pEG311 for cytoplasmic eGFP expression and pEG223 for cytoplasmic DsRed expression have been described previously (Jin et al., 2004). The Gag-GFP fusion protein was constructed by inserting the coding sequence for eGFP flanked by two 5X glycine flexible linkers between T166 and A167 of the L-a virus GAG-POL gene in pJR63 (Ribas and Wickner, 1998). The resulting GAG-POL-GFP fusion gene was subcloned by PCR into the BamHI and SalI sites of pEG311 (replacing eGFP) to create pEG440. A PCR-amplified mCherry open reading frame was inserted between the BamHI and SalI sites of pEG311 to construct pEG463. An eGFP-SSO2 fusion protein was constructed by PCR and inserted between the BamHI and SalI sites of pEG311 to construct pEG361. Full-length SSO1 was inserted into the KpnI site of pRH475 to construct the Hmg1-GFP expression plasmid pEG218.
Table 1.
Plasmids
| pRH475 | HMG1-eGFP URA3 |
| pEG311 | eGFP URA3 SSO1(CT) |
| pEG223 | DsRed URA3 SSO1 |
| pEG361 | eGFP-SSO2 URA3 SSO1(CT) |
| pEG463 | mCherry URA3 SSO1(CT) |
| pEG218 | HMG1-eGFP URA3 SSO1 |
| pJR63 | GAG URA3 |
| pEG440 | GAG-eGFP URA3 SSO1(CT) |
Imaging
Mating yeast were prepared for time-lapse microscopy as previously described (Jin et al., 2004). MATa and MATα cells were grown to log phase, mixed, and then collected on 2.5-cm nitrocellulose filters. The filters were incubated on nutrient agar plates at 30°C for 45 min. Mating pairs were washed off the filters into liquid medium, concentrated by centrifugation for 10 s, and then applied to a 1-mm-thick pad of nutrient agar on a microscope slide. A coverslip was applied and sealed with VALAP (a 1:1:1 mixture of vasoline, lanolin, and paraffin), and cell fusion was imaged during the interval from 1 to 2.5 h (or later for fus1) after the initiation of mating. An acute shift to hyperosmotic medium prevented mating, so the measurements described in Figure 5A were made with cells that were both grown and mated in medium with 1 M sorbitol.
Figure 5.

Osmotic regulation of pore permeance. MATa GFP cells and MATα cells were grown and then mated in media supplemented with 1 M sorbitol (n = 20) or under standard conditions (n = 19). Mean initial permeances and rates of permeance increase were calculated. Error bars, SEM. p values are from a Student's t test.
Microscopy was performed with an Axioplan 2 imaging microscope (Zeiss, Thornwood, NY) outfitted with a mercury arc lamp, bandpass filters (Chroma, Brattleboro, VT), DIC optics, and an Orca ER digital camera (Hamamatsu, Bridgewater, NJ). Images were collected with 63×, NA 1.4, or 100×, NA 1.4, Plan Apochromat objective lenses. For time-lapse microscopy, the objective lens and microscope stage were heated to 30°C. Automated image acquisition, fluorescence quantification, contrast enhancement, colorization, and quicktime movie production were performed using Openlab software (Improvision, Lexington, MA). 2× binning was used to reduce exposure times and minimize photobleaching. CellTracker Blue and FM4-64 were purchased from Molecular Probes (Eugene, OR).
Diffusion Constant Measurements
FRAP experiments were performed on a Zeiss LSM510 confocal microscope using a 63×, 1.4 NA. Planapochromat objective with the pinhole fully opened to collect fluorescence from the entire cell. After collecting five prebleach images, a rectangular region overlying one-half of the cell was rapidly photobleached using unattenuated laser light. Immediately (3-4 ms) after photobleaching, successive images were collected to monitor the fluorescence redistribution. Images were collected with no delay between images in most cases, and the total time between successive images ranged from 70 to 120 ms. Two control image series were then collected from the same cell. The first control series had identical parameters except that no laser light was provided during the bleach time. In the second control series, the entire cell was photobleached. These controls were used to assess the extent of changes in intensity due to processes other than diffusion, including bleaching during monitoring and reversible bleaching resulting from a light-induced state transition (Dickson et al., 1997; Weber et al., 1999). In these experiments, the extent of reversible GFP and dsRed bleaching was negligible in the time of the diffusional recovery. For each time series an average background value determined from a cell-free area was first subtracted from each image, and intensity values were then averaged perpendicular to a line drawn down the long axis of the cell, generating a one-dimensional representation of fluorescence intensity. The decay in the bleaching induced asymmetry in fluorescence distribution was then used to calculate the diffusion coefficient (D) and mobile fraction (R) using a previously described modification of the normal mode analysis (Koppel, 1985; Cowan et al., 2003, 2004).
Permeance Calculations
GFP is the preferred fluorescent protein for monitoring fusion pores between mating yeast because a uniform level of GFP fluorescence was measured in all GFP-expressing cells. The level of DsRed fluorescence varied between cells, perhaps because of its slow maturation rate (Baird et al., 2000). mCherry had a uniform expression level, but it was not as bright as GFP.
A permeance equation describing the diffusion of GFP through a fusion pore between two cells was derived from Fick's law by integration (Atkinson and Sheridan, 1988).
We use Fick's Equation:
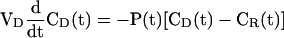 |
where VD is the donor volume, CD(t) is the concentration in the donor volume, CR(t) is the concentration in the receptor volume, and P(t) is the permeance. We define MT as a total weight concentration:
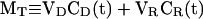 |
and write CR(t) in terms of CD(t):
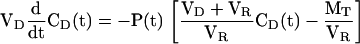 |
This gives
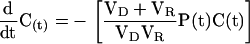 |
where we have defined a C(t), equal to the concentration difference, CD(t) - CR(t):
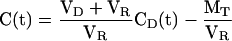 |
The general solution of this differential equation is
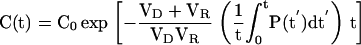 |
We can then express the permeance in terms of the derivative with time of the natural log of C(t):
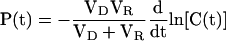 |
The integrated intensity (I) in each cell is proportional to the mass of GFP. Normalizing for the difference in volumes of the two cells, CT ∝ ID - IR(VD/VR). Thus, our permeance equation can be restated in terms of parameters that can be quantified by microscopy:
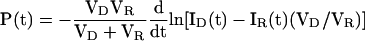 |
To measure permeance, we begin with the volume constant VDVR/(VD + VR). Cytoplasmic volumes are estimated using the simplifying assumption that mating yeast have a spheroid shape. For each cell in a mating pair, length (l) along the mating axis and width (w) perpendicular to the axis are measured at the time of mating, and the corresponding volume (πlww/6) is calculated. This volume is then corrected to account for the fact that GFP is excluded from intracellular organelles. The correction factor was determined by using confocal microscopy to measure the percentage of the cell that is occupied by vacuoles, which are by far the largest organelles that exclude GFP, which diffuses freely through nuclear pores. Cells for confocal microscopy were prepared by the same procedure used to observe cell fusion, because vacuolar morphology is regulated in response to growth conditions (Weisman, 2003). The lumen of vacuoles within MATα DsRed cells was prelabeled with Cell-tracker Blue (Molecular Probes), and the cellular and vacuolar boundaries on a Z-series of fluorescent images were delineated at the point of half-maximal DsRed and Cell-tracker Blue intensities. Results from 40 cells revealed that 24 ± 11% of the cytoplasmic volume is occupied by vacuoles. Because of the wide range of observed vacuole sizes, we use GFP and DsRed images to estimate whether a mating cell has small (15%), medium (25%), or large (35%) vacuoles, resulting in correction factors of 0.85 (100% - 15%), 0.75, and 0.65.
The VD/VR ratio in the logarithmic expression is measured from the ratio of GFP intensities between the two cells at a time after fusion when GFP has diffused to equilibrium. At this time, the GFP concentrations (C = I/V) in the two cells are equal, so VD/VR = ID/IR as t → ∞. If this ratio is used instead of the less accurate ratio derived from length, width and vacuole volume measurements, the value of the expression ID - IR(VD/VR) approaches 0 as expected when the fluorescent proteins reach equilibrium.
Next, regions of interest were drawn around each cell. Mean GFP intensities within these regions were measured as a function of time, corrected for background fluorescence and photobleaching, and then multiplied by the area of the defined region to yield an integrated intensity (Figure 2Ab). After the initiation of fusion, GFP intensity declined in the MATa cell and increased in the MATα cell, but the total intensity in the cell pair remained constant as expected if fluorescence intensity is directly proportional to the number of GFP proteins and GFP does not leak into the medium during fusion. This data is used to calculate the natural log of the volume-adjusted difference in intensities between cells (Figure 2, C and D). The slope (first differential) of the logarithmic curve is proportional to the permeance of the pore. Time points after GFP has diffused to within 5-10% of equilibrium are excluded because the data becomes noisy when we calculate a small difference between two relatively large numbers. For most pores, the data can be accurately fit (R2 > 0.995) with a second-order polynomial equation, yielding a final permeance versus time equation with an initial permeance defined by the first time point at which GFP can be detected in the MATα cell and a linear rate of permeance increase (Figure 2, E and F). All calculations and curve fittings were executed using Microsoft Excel. The logarithmic data were multiplied by 1000 to avoid truncation of significant figures.
Figure 2.

Permeance calculation for a typical fusion pore. The permeance equation, P(t) = [VDVR/(VD + VR)]dln[ID(t) - IR(t)(VD/VR)]/dt, was derived from Fick's law of diffusion. (A) Fluorescence intensity measurements. A series of fluorescent images of a field of mating MATa GFP and MATα cells was collected at 2.5-s intervals. Boundaries were drawn around the two cells of a mating pair and the mean fluorescence intensity in each cell was measured for each image. To control for photobleaching and other extrinsic factors, fluorescence intensity was also measured for a set of adjacent MATa GFP cells that did not fuse and for a background region. (B) The raw fluorescence intensity data were corrected for background fluorescence and photobleaching and then multiplied by the area of the cell. (C) The volume adjusted intensity difference between the cells approaches 0 as GFP diffuses to equilibrium. (D) The natural log of the volume adjusted intensity difference was calculated to fit the form of the permeance equation shown above. (E) A second-order polynomial equation was fit to the logarithmic data for the interval corresponding to the start of GFP movement until GFP approached equilibrium. (F) The fitted curve was multiplied by a volume constant to yield permeance in μm3/s.
RESULTS
Permeance of Wild-Type Fusion Pores
To detect cell fusion during yeast mating, MATa cells expressing GFP as a soluble cytoplasmic protein were mated to MATα cells expressing cytoplasmic DsRed. When the plasma membranes of two cells fused, GFP and DsRed diffused through the resulting fusion pore (Figure 1A, Supplementary Movie 1). GFP diffused to equilibrium more rapidly than DsRed because GFP is a 27-kDa monomer whereas DsRed is a tetramer of 27-kDa subunits (Baird et al., 2000). The two fluorescent proteins rapidly filled the entire cytoplasmic volume after transferring through the fusion pore, indicating that the pore provides the only significant barrier to diffusion.
Figure 1.

Cell fusion detected by the exchange of fluorescent markers. (A) Cytoplasmic mixing. MATa cells expressing cytoplasmic GFP were mated with MATα cells expressing cytoplasmic DsRed and monitored by time-lapse microscopy (Supplementary Movie 1). (B) Slow transfer of a plasma membrane protein. MATa SSO2-GFP cells were mated to MATα DsRed cells and monitored by time-lapse microscopy. Sso2-GFP diffused slowly into the MATα cell and was absent from emerging buds (arrowheads).
We expected that cell fusion would also lead to rapid mixing of plasma membrane proteins. However, the plasma membrane t-SNARE Sso2 transferred very slowly between mating partners. Thirty-four minutes after complete equilibration of DsRed, GFP-Sso2 remained concentrated in the lobe of the zygote that originated from the MATa cell, with only a small amount visible in the MATα lobe near the junction (Figure 1B). Even after diploid buds emerged, GFP-Sso2 largely remained in one lobe of the zygote. The fact that such a small amount of fluorescent GFP-Sso2 was found in the MATα lobe suggested the possibility of a barrier preventing GFP-Sso2 diffusion across the expanded fusion pore. In this situation, the MATα lobe might contain only newly synthesized GFP-Sso2 delivered by the secretory pathway. However, newly synthesized GFP-Sso2 is delivered via the secretory pathway to emerging buds, which are not fluorescent. The lack of bud fluorescence can be explained by slow maturation of the GFP fluorophore (Heim et al., 1995). The GFP hybrid of a related protein, GFP-Sso1, has an unusually slow diffusion constant, 0.0025 μm2/s, in the yeast plasma membrane (Valdez-Taubas and Pelham, 2003). Because GFP-Sso2 exhibited a similar diffusion constant, the expected time constant for transfer of GFP-Sso2 between two 6-μm-diameter cells separated by a large 1.5-μm fusion pore is greater than 80 min, using a method developed for calculating lateral diffusion on fused cell doublets (Koppel, 1984). Thus, plasma membrane proteins are exchanged very slowly after cell fusion of mating yeast.
To examine the suitability of cytoplasmic GFP and DsRed as diffusion probes, fluorescence in one-half of a cell was photobleached and the rate and extent of fluorescence recovery by diffusion from the unbleached half of the cell was measured (Table 2). The diffusion constant for GFP in yeast cytoplasm is sevenfold less than the diffusion constant in CHO cell cytoplasm, but 50% greater than in Escherichia coli, indicating that yeast cytoplasm has an intermediate effective viscosity (Swaminathan et al., 1997; Elowitz et al., 1999). The ratio of the GFP and DsRed diffusion constants is consistent with DsRed behaving as a tetramer with an effective radius 41/3 = 1.59 times that of GFP. Free diffusion of >90% of both GFP and DsRed in yeast cytoplasm validates their use as diffusion probes.
Table 2.
Photobleaching results
| D (μm2/s) | Mobile fraction | |
|---|---|---|
| GFP (n = 12) | 12.4 ± 2.6 | 0.94 ± 0.06 |
| DsRed (n = 13) | 8.11 ± 1.33 | 0.91 ± 0.06 |
| GFP (1 M sorbitol) (n = 14) | 9.0 ± 2.0 | 0.91 ± 0.05 |
| Gag-GFP (n = 13) | 1.12 ± 0.6 | 0.54 ± 0.07 |
Fusion pore permeances were measured using Fick's law of diffusion, which states that the rate of GFP transfer between donor (D) and recipient (R) cells is equal to the permeance (P) of the pore times the difference between the GFP concentrations of the two cells:
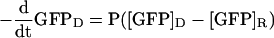 |
Because cellular dimensions and the fluorescence intensity of GFP can be conveniently measured, but the absolute GFP concentration cannot, Fick's equation was integrated and solved for permeance as a function of intracellular volume and GFP intensity:
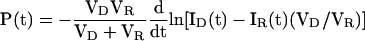 |
The integrated equation shows that fusion pore permeance is proportional to the slope of a dln[ID - IR(VD/VR)]/dt vs. time curve. As seen in an example calculation (Figure 2), data are well fit by a second-order polynomial equation. Differentiating this equation to yield a permeance versus time curve reveals that the initial permeance of this typical pore is large relative to the rate of permeance increase, indicating abrupt opening followed by more gradual expansion.
Wild-type fusion pores had a wide distribution of permeances, and there was no clear correlation between the initial permeance and rate of permeance increase for individual pores (Supplementary Figure S1). Forty percent of the pores had an essentially stable permeance, with a dln[ID - IR(VD/VR)]/dt curve that could be fit (R2 > 0.99) with a straight line. These nonexpanding pores had initial permeances that ranged from 0.5 to 8.5 μm3/s. In seven independent experiments, the mean initial permeance of wild-type mating pairs under standard conditions was 1.53 ± 1.63 μm3/s and the mean rate of permeance increase was 0.064 ± 0.082 μm3/s2 (n = 109, SD).
The permeance traces of some fusion pores did not fit the typical pattern of abrupt opening followed by gradual expansion. In up to 15% of the fusions between wild-type yeast, permeance abruptly increased or decreased after the initial opening phase (Figure 3). A sudden increase in permeance could indicate anomalous expansion of the existing pore, but we favor the more conservative possibility that a second pore has opened between the two cells. An abrupt decrease in permeance indicates that the pore has either closed or been blocked by a cytoplasmic occlusion. GFP flux usually resumed shortly after pore closure, indicating either opening of a new pore or removal of the occlusion. In two examples, however, GFP flux did not resume in the 10-15-min interval between pore closure and the end of the time-laspe recording. Furthermore, a scan of many microscopic fields 3 h after the initiation of mating revealed a small number of mating pairs where the two partner cells had different concentrations of GFP. At this late time point, most mating pairs have already produced a diploid daughter cell, and no new fusion pores can be detected. This observation indicates that a fusion pore can open transiently and then close permanently.
Figure 3.

Abrupt expansion (A), and transient opening (B) of fusion pores between wild-type mating yeast. Top, GFP and DsRed fluorescence intensity measurements for the two cells of a mating pair. Bottom, the logarithms of the volume adjusted intensity differences were calculated as shown in Figure 2D and then fit with second-order polynomial curves. The accuracy (R2 value) of each fit is displayed. In A, permeance abruptly expands after 169 s. In B, a small pore opens at 16 s and closes at 234 s. A larger permeance flux initiates at 477 s. Note that DsRed transfers later and more slowly than GFP because it is a 108-kDa tetramer.
Although permeance is the only property of the fusion pore that can actually be measured by following transfer of fluorescent proteins between cells, we can estimate the actual dimensions of a fusion pore. In Fick's law, permeance (P) is equal to the diffusion constant (D) in the pore times the area (A) divided by the length (L) of the pore:
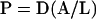 |
Assuming that the diffusion constant of GFP within the pore is equal to the measured diffusion constant of GFP in the cytoplasm (12.4 μm2/s), and that a fusion pore is a regular cylinder with a length of 17 nm, corresponding to the thickness of two 4.5-nm membranes plus the 8-nm distance observed between plasma membrane bilayers in arrested prm1 mating pairs, a theoretical curve can be drawn expressing the relationship between the permeance and the radius of a pore (Figure 4). According to this curve, the median wild-type fusion pore has an initial radius of 25 nm and expands to 40 nm 30 s later. However, if the initial pore is not a perfect cylinder, it will restrict the transfer of objects with a radius <25 nm.
Figure 4.

Theoretical relationship between GFP permeance and the fusion pore radius.
To evaluate the accuracy of this theoretical relationship between permeance and pore size, we examined small fusion pores that initially permitted flux of GFP, but not DsRed. GFP has a β-barrel structure with a diameter of 3.2 nm and a height of 5 nm, and DsRed is a tetramer of β-barrels with overall dimensions of 5.3 × 7.3 × 7.5 nm (Ormo et al., 1996; Yang et al., 1996; Yarbrough et al., 2001). DsRed-restricted pores (n = 7) all had a GFP permeance of <0.2 μm3/s. This permeance corresponds to a theoretical radius of 9 nm, which is not grossly larger than the size of DsRed considering that anomalous diffusion would be expected in cases where the pore is small relative to the size of the probe (Wei et al., 2000; Verkman, 2002). As a control, when MATa GFP cells were mated to MATα cells expressing the monomeric red fluorescent protein mCherry (Shaner et al., 2004), the GFP and mCherry permeances were identical.
Various conditions were examined to determine if fusion pore dynamics can be regulated by environmental conditions. When yeast were mated in medium supplemented with 1 M sorbitol, the average initial fusion pore permeance was reduced by 72%, and the average rate of permeance increase was reduced by 73% after correcting for a small reduction in the GFP diffusion constant (Figure 5, Table 2). Notably, although there was no correlation between the initial permeance and rate of permeance increase of individual pores measured in any single condition, there was a good correlation between the average values for these parameters in comparisons between different mating conditions and mutant strains.
Pore Permeance in Cell Fusion Mutants
Mutations in fusion proteins or regulators of fusion proteins can alter the dynamics of viral and exocytic fusion pores. We therefore asked whether any cell fusion mutants would alter the permeance of fusion pores between mating yeast. In these experiments, the same gene was deleted from both mating partners because cell fusion phenotypes are often stronger in “bilateral” matings (Trueheart et al., 1987; Brizzio et al., 1998).
Initially, the prm1 mutant was of greatest interest because Prm1 has been implicated at the plasma membrane fusion stage of mating (Heiman and Walter, 2000; Jin et al., 2004). Unfortunately, it was challenging to measure pore permeance in prm1 mating pairs because under the conditions used for live cell imaging these mating pairs either arrested or lysed upon plasma membrane contact and few fusion events could be recorded (Jin et al., 2004). After varying several parameters, it was found that prm1 mating pairs fuse more frequently after a hypoosmotic shift. Under these conditions, prm1 mutant pores had a slightly reduced initial permeance and rate of permeance increase compared with a PRM1 control, but the differences were not statistically significant (Figure 6A).
Figure 6.

Fusion pore permeance in cell fusion mutants. (A) prm1. Fusion pore permeance was measured after a hypoosmotic shift for wild-type (n = 19) and prm1 (n = 18) mating pairs. (B) Cell wall remodeling mutants. Fusion pore permeance was measured under standard conditions for wild-type (n = 32), fus1 (n = 31), fus2 (n = 37), rvs161 (n = 41), and spa2 (n = 47) mating pairs.
In contrast to the modest effect of the prm1 mutation, deleting the FUS1 gene had a compelling effect on fusion pore permeance. fus1 pores had significantly less permeance than wild-type pores (t test, p < 0.001), with initial permeances and rates of permeance increase that were reduced by >10-fold compared with wild-type pores (Figure 6B). A defect in cell wall remodeling prevents plasma membrane contact and fusion in >50% of fus1 mating pairs (McCaffrey et al., 1987; Trueheart et al., 1987; Gammie et al., 1998). Furthermore, cell wall remnants remain after cytoplasmic mixing in the fus1 mating pairs that do fuse (Gammie et al., 1998). These cell wall remnants might act as a collar preventing fusion pore expansion. This possible mechanism was addressed by measuring fusion pore permeance in mating pairs with mutations in other cell fusion mutants known to have cell wall remodeling defects. Like fus1, the fus2, rvs161, and spa2 proteins are all required for proper remodeling of the cell wall before the membrane fusion stage of mating (Trueheart et al., 1987; Valtz and Herskowitz, 1996; Gammie et al., 1998). Cytoplasmic mixing was inhibited to a comparable degree in all five cell wall remodeling mutants, and the mutant mating pairs all had visible cell wall remnants after mating for 1.5 h (Supplementary Figure S2). Nevertheless, fusion pore permeance and expansion were reduced by <2-fold in the fus2, rvs161, and spa2 mutants, compared with the 10-fold reduction in fus1 (Figure 6B). Thus, the mere presence of cell wall remnants does not fully explain the reduced permeance of fus1 fusion pores. A fus1 suppression experiment provides additional support for the conclusion that FUS1, but not FUS2 regulates pore permeance. The cell fusion defect of fus1 mating pairs can be partially suppressed by FUS2 overproduction (Trueheart et al., 1987). FUS2 on a CEN plasmid rescued 70% of the mating defect of fus1 mating pairs, but increased pore permeance and expansion by only 30%. Thus, pore permeance regulation is a specific function of FUS1.
If Fus1 regulates fusion pore dynamics by acting on fusion proteins or other unique constituents of the initial fusion pore, the pore expansion rate in fus1 mutant mating pairs might increase once the pore expands to the point where these initial components are diluted out by an influx of additional membrane. A larger probe was required to examine later stages of pore expansion after monomeric GFPs diffused to equilibrium. For this purpose, GFP was inserted into an external loop of the L-a virus Gag protein using the crystal structure of this 39-nm icosahedral virus as a guide (Naitow et al., 2001). The L-a virus is native to many yeast strains and is known to be transmitted by cell fusion (Wickner, 1992). In wild-type mating pairs, Gag-GFP flux between mating partners was rarely detected before mCherry had diffused to equilibrium, indicating that a typical fusion pore takes minutes to expand to a size sufficient to permit Gag-GFP transfer (Figure 7A). Unfortunately, permeance measurements were impractical with Gag-GFP since 46% of the protein was immobile in a 30-s FRAP experiment (Table 2). We therefore measured the interval between the initiation times for mCherry and Gag-GFP flux to gauge the initial rate of pore expansion and the time required for Gag-GFP to diffuse to 33% of its equilibrium value as an indicator of the rate of expansion at times after the pore had exceeded 39 nm. As expected, there was a longer delay before the initiation of Gag-GFP flux in the fus1 mating pairs (Figure 7B). Furthermore, once Gag-GFP started to transfer between mating partners, it transferred more slowly in the fus1 mating pairs, indicating a persistently slow fusion pore expansion rate (Figure 7C). This result implies that pore expansion is not regulated exclusively by components of the original fusion pore.
Figure 7.

Persistently slow expansion of fus1 fusion pores. (A) Monitoring late stages of fusion pore expansion with Gag-GFP. GFP was fused to the structural protein of the L-a virus. MATa cells expressing Gag-GFP were mated to MATα mCherry cells. Cytoplasmic mixing was followed by time-lapse microscopy. (B) Delayed initiation of Gag-GFP transfer in fus1 mating pairs. The time interval between the initiation of plasma membrane fusion marked by mCherry transfer and the time point when the pore had expanded sufficiently to detect Gag-GFP transfer was compared for wild-type (n = 22) and fus1 (n = 21) fusion pores. (C) Slow Gag-GFP transfer in fus1 mating pairs. After the initiation of Gag-GFP transfer, the time required for 33% of the mobile fraction of Gag-GFP to diffuse across the fusion pore was compared for wild-type (n = 22) and fus1 (n = 19) fusion pores. Gag-GFP did not reach equilibrium before the end of the recorded interval in 2 fus1 mating pairs.
Karyogamy and Vacuole Mixing Are Delayed in fus1 Mating Pairs
Mutations that interfere with plasma membrane fusion could indirectly inhibit downstream events in the mating pathway, which include fusion between the nuclei of the two parent cells, vacuolar fusion, and the emergence of a diploid bud (Weisman and Wickner, 1988; Marsh and Rose, 1997; Weisman, 2003). We therefore developed assays to compare the kinetics of plasma membrane fusion, karyogamy, and vacuole mixing.
Karyogamy and plasma membrane fusion were monitored in the same mating pairs by mating MATa cells expressing the fluorescent ER/nuclear envelope protein Hmg1-GFP to MATα cells expressing cytoplasmic DsRed. DNA was stained with DAPI after arresting mating at various times. Transfer of DsRed to the MATa cell indicated plasma membrane fusion. Karyogamy was detected by congression of DAPI-stained nuclear DNA into a single structure surrounded by an Hmg1-GFP labeled nuclear envelope. Interestingly, Hmg1-GFP often appeared in the nuclear envelope and endoplasmic reticulum of the MATα cell in zygotes with two distinct nuclei, suggesting that ER derived vesicles transfer between mating partners and fuse with the ER and/or nuclear envelope before karyogamy (Figure 8A).
Figure 8.

Late mating events are delayed in fus1 mating pairs. (A) Detecting karyogamy and vesicle transfer. MATa cells expressing Hmg1-GFP were mated to MATα DsRed cells. At various time points, mating was arrested by transferring the yeast to azide buffer at 4°C. Mating pairs were then stained with DAPI and observed by fluorescence microscopy. Cytoplasmic DsRed transferred after plasma membrane fusion. Hmg1-GFP transferred slowly between cells in ER derived transport vesicles and then rapidly diffused throughout the combined nuclear envelope after karyogamy. (B) Images from a time-lapse series documenting plasma membrane and vacuole fusion (Supplementary Movie 2). MATa GFP cells were pulse-labeled with FM4-64 to mark vacuole membranes and then mated to MATα cells containing CellTracker Blue CMAC-labeled vacuoles. Vacuoles were retained within their original parent until a diploid bud emerged 50 min after plasma membrane fusion. (C-F) Mating pairs that had completed plasma membrane fusion as evidenced by GFP or DsRed transfer were scored for Hmg1-GFP vesicle transfer (C), karyogamy (D), zytogic bud emergence (E), and vacuole fusion (F). The apparent reduction in zygotic budding at the 4-h time point in wild-type mating pairs results from separation of the first zygotic bud from the fused mating pair.
To follow vacuoles after cell fusion, the vacuolar membrane of MATa GFP cells was prelabeled with FM4-64 by a pulse-chase procedure, and the vacuolar content of the MATα cells was prelabeled with CellTracker Blue. The two populations of vacuoles remained within their respective halves of the zygote for an extended period after plasma membrane fusion marked by GFP transfer. During this time, <15% of the total FM4-64 and CellTracker Blue transferred to vacuoles in the other lobe of the zygote. Shortly after bud emergence, vacuoles from each parent cell simultaneously streamed toward the bud, where they fused. Ultimately, vacuoles in both parental lobes and the emerging bud were marked with both FM4-64 and CellTracker Blue (Figure 8B, Supplementary Movie 2).
To examine the functional consequences of small fusion pores on later mating events, fus1 mating pairs that had already initiated plasma membrane fusion and exchanged cytoplasmic GFP or DsRed were compared with wild-type controls. As previously noted, cell wall remnants visible by DIC microscopy often separated the two parent cells of a fused fus1 mating pair (Gammie et al., 1998). Cell wall remnants could also be detected shortly after fusion in wild-type mating pairs, but the fus1 remnants were more stable. After cytoplasmic mixing, the fus1 mating pairs had a slight reduction in the transfer and fusion of ER-derived Hmg1-GFP vesicles, but a delay of >1 h before karyogamy (Figure 8, C and D). Karyogamy appears to be inhibited at the nuclear congression step, because the two nuclei in many fus1 mating pairs were not closely associated with the site of cell fusion. The karyogamy delay could result from an inability of astral microtubules to form a network through the restricted fusion pore (Maddox et al., 1999). Despite this delay, >80% of the fus1 mating pairs eventually completed karyogamy. Thus, even the smallest fus1 fusion pores eventually expanded sufficiently to permit fusion of the two nuclei, which have a diameter of 2 μm.
Later mating events were delayed to a lesser extent by the fus1 mutation. In wild-type mating pairs, there is a 1-h interval between plasma membrane fusion and zygotic bud emergence. This interval was modestly longer in the fus1 mating pairs (Figure 8E). Shortly after bud emergence, the vacuoles streamed toward each other and fused. Pairs of unfused vacuoles straddling the site of cell fusion were never detected, indicating that both wild-type and fus1 pores had expanded to a size that does not impede vacuole fusion at the time of bud emergence (Figure 8F).
DISCUSSION
GFP flux was used to measure the size and expansion rate of fusion pores between mating yeast. Most fusion pores opened abruptly and then gradually expanded. Viral and exocytic fusion pores also open abruptly and gradually expand, suggesting that fundamental aspects of the mechanism of membrane fusion are conserved. Yeast plasma membrane fusion pores have a wide distribution of initial permeances and expansion rates. Nevertheless, fusion pore size can be regulated by increasing the osmolarity of the media. Sorbitol, which mimics growth on a sun-dried grape, reduced the size and expansion rate of the pores. Hyperosmotic media causes a reduction in the lateral tension of the plasma membrane. Because fusion pore expansion relieves membrane tension, reduced tension will reduce the force available to drive pore expansion. Activation of intracellular signaling pathways might also contribute to the reduced rate of fusion pore expansion. However, the osmotic stress activated HOG1 MAP kinase pathway is unlikely to be involved because Hog1 is transiently activated by a shift to hyperosmotic conditions (Ferrigno et al., 1998), whereas pore permeance was reduced when cells were continuously maintained in hyperosmotic media.
Although fusion pore permeance increased at a linear rate in most mating pairs, exceptional pairs displayed an abrupt increase or decrease in permeance. We interpret abrupt permeance increases as an indication that a second pore has opened in the same cell pair. Because a second pore cannot be detected if it opens after GFP (or DsRed) has diffused to equilibrium through the first pore, formation of a second pore may occur more frequently than it can be detected. On the other hand, if the first pore expands rapidly, the entire zone of plasma membrane contact may be depleted before a second pore can open. Multiple fusion pores have previously been observed between myoblasts in Drosophila, which have a much larger zone of plasma membrane apposition (Doberstein et al., 1997).
Some fusion pores between mating yeast cells close before GFP has diffused to equilibrium. Viral and exocytic fusion pores often flicker open and closed, but the open state of these flickering pore is typically smaller than the 3-nm diameter of a GFP protein. For example, the kiss-and-run fusion pores of hippocampal synaptic vesicles have an estimated size of 1 nm (Klyachko and Jackson, 2002; Richards et al., 2005). Larger fusion pores can open and then close during exocytosis from dense core secretory vesicles and lysosomes in order to facilitate recycling of vesicle membranes (Klyachko and Jackson, 2002; Jaiswal et al., 2004). Dynamin, the GTPase required for pinching off endocytic vesicles, is also involved in the closure of at least some of these larger fusion pores, indicating that large fusion pores do not close by a simple reversal of the fusion process (Graham et al., 2002; Holroyd et al., 2002). In the same way that exocytosis and endocytosis are opposing processes that regulate the size and composition of the plasma membrane, the opposing process of cell fusion is cytokinesis. By analogy to the role of dynamin in closing large exocytic fusion pores, the chiton synthetase and contractile ring that drive cytokinesis might participate in the closure of fusion pores between mating yeast.
As a first step toward identifying components of the fusion pore for yeast mating, we measured fusion pore permeance in known cell fusion mutants. Attention was initially drawn to PRM1, because Prm1 is the only protein reported to act at the plasma membrane fusion stage of mating. prm1 mutant fusion pores had modestly reduced initial permeances and rates of permeance increase, but the most dramatic effect of deleting PRM1 was the lysis that usually occurred immediately after plasma membrane contact (Jin et al., 2004). Although >20% of prm1 mating pairs fuse when mated on a nitrocellulose filter layered over a nutrient agar plate, the fusion frequency was reduced to <1% when these same prm1 strains were mated on a nutrient agar pad under a coverslip. A hypoosmotic shift increased the frequency of successful fusion on microscope slides. This treatment increases lateral tension on the plasma membrane, which could directly promote membrane fusion or act indirectly by activating the cell wall integrity signaling pathway (Hohmann, 2002). Prm1 is likely to facilitate the initial pore opening of plasma membrane fusion, because cells lyse before the fusion pore permits significant GFP transfer. A smaller fluorescent probe would allow a higher-resolution study of this stage of fusion and might provide further insight into the function of Prm1.
Deletion of the FUS1 gene dramatically reduced pore permeance and expansion. In principle, fusion pore expansion could be restrained from within the plasma membrane by components of the fusion pore, from outside of the membrane by the cell wall, or from the cytoplasmic surface of the membrane by binding to cytoskeletal components. Although the mechanism remains unknown, we have been able to exclude two likely possibilities.
Experiments with Gag-GFP, which assembles into large viral particles, revealed that pore expansion continued at a slow rate for longer than 10 min and that pores continued to expand slowly after dilating to a diameter of >40 nm. At this stage, protein components of the initial fusion pore would be diluted out by an influx of membrane lipids and other proteins. Thus, membrane proteins are unlikely to be sufficient to restrict the rate of pore expansion in fus1 mating pairs.
FUS1 was previously shown to regulate the cell wall remodeling stage of mating that precedes plasma membrane fusion. Cell wall remodeling and pore permeance could be linked if cell wall remnants in the fused fus1 mating pairs act as a collar restraining the fusion pore. Arguing against this idea, the cell wall remnants of fus2, rvs161, and spa2 mating pairs have little if any effect on the initial fusion pore permeance or expansion rate. An alternative approach to examine the role of the cell wall in restricting fusion pore expansion would be to mutate a glucanase. Yeast express multiple glucanases with potentially overlapping functions, but the glucanase(s) that execute cell wall remodeling during mating have not been conclusively identified. Mating defects were observed in the scw4/10 and exg1 mutants (Cappellaro et al., 1998; Nolan and Grote, unpublished observation). Neither of these mutations had a significant effect on fusion pore permeance, supporting the proposal that the cell wall does not inhibit fusion pore expansion. There is little hope of directly visualizing an interaction between the cell wall and a nascent fusion pore. Cell wall remnants in fused mating pairs are visible by light microscopy, but neither light nor electron microscopy have sufficient resolution to detect the gap in the cell wall where fusion occurs during the time interval of GFP flux immediately after the initiation of plasma membrane fusion. Thus, it remains possible that the fus1 cell wall remnants are more restrictive for fusion pore expansion than those of fus2, rvs161, and spa2. On the other hand, one clue favoring an extensive zone of plasma membrane contact in fus1 mating pairs before membrane fusion comes from the kinetics of GFP transfer. An abrupt increase in GFP flux suggesting the opening of a second fusion pore was frequently observed during the unusually long interval when GFP flux could be measured for these small pores. Thus, despite the known role of Fus1 in cell wall remodeling, the available data suggest that cell wall remnants do not restrain fusion pore expansion.
Previous work on Fus1 has provided some clues concerning its molecular function. Fus1 is a 58-kDa type 1 membrane protein whose expression is strongly induced by mating pheromones. It is targeted to the surface of mating projections in α-factor treated MATa cells and to sites of cell-cell contact in mating pairs (Trueheart et al., 1987). Fus1 targeting depends upon expression of the chitosome transport regulator Chs5, o-glycosylation of an N-terminal signal by Pmt4, and a plasma membrane enriched in ergosterol and sphingolipids (Bagnat and Simons, 2002; Santos and Snyder, 2003; Proszynski et al., 2004). Fus1 might function as a scaffold to recruit other proteins to fusion sites. In a two-hybrid assay, Fus1 interacted with itself, Fus2, Sho1, Fus3, Kss1, Bni1, Bnr1, Chs5, Cdc42, Pea2, and Ste5 (Nelson et al., 2004). A C-terminal SH3 domain and an adjacent proline-rich domain mediate some of these interactions. However, these C-terminal domains are not required to complement the Δfus1 mating and pore permeance defects in our strain background, whereas the remainder of Fus1 has no obviously homologous to other known proteins. Mapping of additional functional domains in Fus1 and the identification of relevant binding partners will be essential for a mechanistic understanding of Fus1 function and its role in fusion pore expansion.
Supplementary Material
Acknowledgments
Laurence Pelletier, Judsen Sheridan, and David Sheff provided invaluable assistance during the preliminary stages of this study, which was initiated in the laboratory of Peter Novick. We thank Rajani Rao, Lois Weisman, Richard Cone, Benjamin Podbilowicz, and Ed Chapman for helpful discussions; Reed Wickner, Mark Rose, and Yunri Du for contributing plasmids; and Elizabeth Chen for comments on the manuscript. The microscopy facilities at the Center for Cell Analysis and Modeling are funded in part by National Institutes of Health Grant RR13186. This work was partly supported by a Research Scholar Award (RSG-05-205-01-MBC) from the American Cancer Society.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-11-1015) on February 22, 2006.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Albillos, A., Dernick, G., Horstmann, H., Almers, W., Alvarez de Toledo, G., and Lindau, M. (1997). The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389, 509-512. [DOI] [PubMed] [Google Scholar]
- Alvarez-Dolado, M., Pardal, R., Garcia-Verdugo, J. M., Fike, J. R., Lee, H. O., Pfeffer, K., Lois, C., Morrison, S. J., and Alvarez-Buylla, A. (2003). Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 425, 968-973. [DOI] [PubMed] [Google Scholar]
- Aravanis, A. M., Pyle, J. L., and Tsien, R. W. (2003). Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423, 643-647. [DOI] [PubMed] [Google Scholar]
- Archer, D. A., Graham, M. E., and Burgoyne, R. D. (2002). Complexin regulates the closure of the fusion pore during regulated vesicle exocytosis. J. Biol. Chem. 277, 18249-18252. [DOI] [PubMed] [Google Scholar]
- Atkinson, M. M., and Sheridan, J. D. (1988). Altered junctional permeability between cells transformed by v-ras, v-mos, or v-src. Am. J. Physiol. 255, C674-C683. [DOI] [PubMed] [Google Scholar]
- Bagnat, M., and Simons, K. (2002). Cell surface polarization during yeast mating. Proc. Natl. Acad. Sci. USA 99, 14183-14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2000). Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 97, 11984-11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. (1993). Viral fusion mechanisms, CRC Press: Boca Raton, FL.
- Brizzio, V., Gammie, A. E., and Rose, M. D. (1998). Rvs161p interacts with Fus2p to promote cell fusion in Saccharomyces cerevisiae. J. Cell Biol. 141, 567-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough, P. A., Hughson, F. M., Skehel, J. J., and Wiley, D. C. (1994). Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37-43. [DOI] [PubMed] [Google Scholar]
- Cappellaro, C., Mrsa, V., and Tanner, W. (1998). New potential cell wall glucanases of Saccharomyces cerevisiae and their involvement in mating. J. Bacteriol. 180, 5030-5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, C. M., and Kim, P. S. (1993). A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823-832. [DOI] [PubMed] [Google Scholar]
- Chen, E. H., and Olson, E. N. (2005). Unveiling the mechanisms of cell-cell fusion. Science 308, 369-373. [DOI] [PubMed] [Google Scholar]
- Cowan, A. E., Koppel, D. E., Setlow, B., and Setlow, P. (2003). A soluble protein is immobile in dormant spores of Bacillus subtilis but is mobile in germinated spores: implications for spore dormancy. Proc. Natl. Acad. Sci. USA 100, 4209-4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan, A. E., Olivastro, E. M., Koppel, D. E., Loshon, C. A., Setlow, B., and Setlow, P. (2004). Lipids in the inner membrane of dormant spores of Bacillus species are largely immobile. Proc. Natl. Acad. Sci. USA 101, 7733-7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson, R. M., Cubitt, A. B., Tsien, R. Y., and Moerner, W. E. (1997). On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388, 355-358. [DOI] [PubMed] [Google Scholar]
- Doberstein, S. K., Fetter, R. D., Mehta, A. Y., and Goodman, C. S. (1997). Genetic analysis of myoblast fusion: blown fuse is required for progression beyond the prefusion complex. J. Cell Biol. 136, 1249-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli, D., and Lazebnik, Y. (2003). Cell fusion: a hidden enemy? Cancer Cell 3, 445-448. [DOI] [PubMed] [Google Scholar]
- Dutch, R. E., and Lamb, R. A. (2001). Deletion of the cytoplasmic tail of the fusion protein of the paramyxovirus simian virus 5 affects fusion pore enlargement. J. Virol. 75, 5363-5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz, M. B., Surette, M. G., Wolf, P. E., Stock, J. B., and Leibler, S. (1999). Protein mobility in the cytoplasm of Escherichia coli. J. Bacteriol. 181, 197-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno, P., Posas, F., Koepp, D., Saito, H., and Silver, P. A. (1998). Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J. 17, 5606-5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. J., Pevsner, J., and Burgoyne, R. D. (2001). Control of fusion pore dynamics during exocytosis by Munc18. Science 291, 875-878. [DOI] [PubMed] [Google Scholar]
- Gammie, A. E., Brizzio, V., and Rose, M. D. (1998). Distinct morphological phenotypes of cell fusion mutants. Mol. Biol. Cell 9, 1395-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, M. E., and Burgoyne, R. D. (2000). Comparison of cysteine string protein (Csp) and mutant alpha-SNAP overexpression reveals a role for csp in late steps of membrane fusion in dense-core granule exocytosis in adrenal chromaffin cells. J. Neurosci. 20, 1281-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, M. E., O'Callaghan, D. W., McMahon, H. T., and Burgoyne, R. D. (2002). Dynamin-dependent and dynamin-independent processes contribute to the regulation of single vesicle release kinetics and quantal size. Proc. Natl. Acad. Sci. USA 99, 7124-7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, R. Y., Koning, A., Wright, R., and Rine, J. (1996). In vivo examination of membrane protein localization and degradation with green fluorescent protein. Proc. Natl. Acad. Sci. USA 93, 828-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X., Wang, C. T., Bai, J., Chapman, E. R., and Jackson, M. B. (2004). Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289-292. [DOI] [PubMed] [Google Scholar]
- Heim, R., Cubitt, A. B., and Tsien, R. Y. (1995). Improved green fluorescence. Nature 373, 663-664. [DOI] [PubMed] [Google Scholar]
- Heiman, M. G., and Walter, P. (2000). Prm1p, a pheromone-regulated multispanning membrane protein, facilitates plasma membrane fusion during yeast mating. J. Cell Biol. 151, 719-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann, S. (2002). Osmotic adaptation in yeast-control of the yeast osmolyte system. Int. Rev. Cytol. 215, 149-187. [DOI] [PubMed] [Google Scholar]
- Holroyd, P., Lang, T., Wenzel, D., De Camilli, P., and Jahn, R. (2002). Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc. Natl. Acad. Sci. USA 99, 16806-16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, J. K., Chakrabarti, S., Andrews, N. W., and Simon, S. M. (2004). Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS. Biol. 2, E233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, H., Carlile, C., Nolan, S., and Grote, E. (2004). Prm1 prevents contact-dependent lysis of yeast mating pairs. Eukaryot. Cell 3, 1664-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko, V. A., and Jackson, M. B. (2002). Capacitance steps and fusion pores of small and large-dense-core vesicles in nerve terminals. Nature 418, 89-92. [DOI] [PubMed] [Google Scholar]
- Koppel, D. E. (1984). Lateral diffusion on fused cell doublets. Biophys. J. 46, 837-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel, D. E. (1985). Normal-mode analysis of lateral diffusion on a bounded membrane surface. Biophys. J. 47, 337-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozerski, C., Ponimaskin, E., Schroth-Diez, B., Schmidt, M. F., and Herrmann, A. (2000). Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74, 7529-7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszak, J. R., Ennesser, C. A., Bertram, B. A., Imherr-McMannis, S., Jones-Rufer, L. S., and Weinstein, R. S. (1989). The contribution of cell-to-cell fusion to the ordered structure of the crystalline lens. Lens Eye Toxic Res. 6, 639-673. [PubMed] [Google Scholar]
- Maddox, P., Chin, E., Mallavarapu, A., Yeh, E., Salmon, E. D., and Bloom, K. (1999). Microtubule dynamics from mating through the first zygotic division in the budding yeast Saccharomyces cerevisiae. J. Cell Biol. 144, 977-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan, R. M., Cohen, F. S., and Melikyan, G. B. (2003). HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 14, 926-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, L., and Rose, M. D. (1997). The pathway of cell and nuclear fusion during mating in S. cerevisiae. In: The Molecular and Cellular Biology of the Yeast Saccharomyces, Vol. 3, ed. J. R. Pringle, J. R. Broach, and E. W. Jones, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 827-888. [Google Scholar]
- McCaffrey, G., Clay, F. J., Kelsay, K., and Sprague, G. F., Jr. (1987). Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. Mol. Cell Biol. 7, 2680-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., Lin, S., Roth, M. G., and Cohen, F. S. (1999). Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell 10, 1821-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., Markosyan, R. M., Roth, M. G., and Cohen, F. S. (2000). A point mutation in the transmembrane domain of the hemagglutinin of influenza virus stabilizes a hemifusion intermediate that can transit to fusion. Mol. Biol. Cell 11, 3765-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi, S. et al. (2000). Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785-789. [DOI] [PubMed] [Google Scholar]
- Naitow, H., Canady, M. A., Lin, T., Wickner, R. B., and Johnson, J. E. (2001). Purification, crystallization, and preliminary X-ray analysis of L-A: a dsRNA yeast virus. J. Struct. Biol. 135, 1-7. [DOI] [PubMed] [Google Scholar]
- Nelson, B., Parsons, A. B., Evangelista, M., Schaefer, K., Kennedy, K., Ritchie, S., Petryshen, T. L., and Boone, C. (2004). Fus1p interacts with components of the HOG1p mitogen-activated protein kinase and Cdc42p morphogenesis signaling pathways to control cell fusion during yeast mating. Genetics 166, 67-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormo, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R. Y., and Remington, S. J. (1996). Crystal structure of the Aequorea victoria green fluorescent protein. Science 273, 1392-1395. [DOI] [PubMed] [Google Scholar]
- Primakoff, P., and Myles, D. G. (2002). Penetration, adhesion, and fusion in mammalian sperm-egg interaction. Science 296, 2183-2185. [DOI] [PubMed] [Google Scholar]
- Proszynski, T. J., Simons, K., and Bagnat, M. (2004). O-glycosylation as a sorting determinant for cell surface delivery in yeast. Mol. Biol. Cell 15, 1533-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, J. C., and Wickner, R. B. (1998). The Gag domain of the Gag-Pol fusion protein directs incorporation into the L-A double-stranded RNA viral particles in Saccharomyces cerevisiae. J. Biol. Chem. 273, 9306-9311. [DOI] [PubMed] [Google Scholar]
- Richards, D. A., Bai, J., and Chapman, E. R. (2005). Two modes of exocytosis at hippocampal synapses revealed by rate of FM1-43 efflux from individual vesicles. J. Cell Biol. 168, 929-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom, A., Saffrich, R., Markovic, I., Walther, P., and Gerdes, H. H. (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007-1010. [DOI] [PubMed] [Google Scholar]
- Santos, B., and Snyder, M. (2003). Specific protein targeting during cell differentiation: polarized localization of Fus1p during mating depends on Chs5p in Saccharomyces cerevisiae. Eukaryot. Cell 2, 821-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch, C., and Blumenthal, R. (1993). Role of the fusion peptide sequence in initial stages of influenza hemagglutinin-induced cell fusion. J. Biol. Chem. 268, 9267-9274. [PubMed] [Google Scholar]
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N., Palmer, A. E., and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572. [DOI] [PubMed] [Google Scholar]
- Spruce, A. E., Breckenridge, L. J., Lee, A. K., and Almers, W. (1990). Properties of the fusion pore that forms during exocytosis of a mast cell secretory vesicle. Neuron 4, 643-654. [DOI] [PubMed] [Google Scholar]
- Spruce, A. E., Iwata, A., and Almers, W. (1991). The first milliseconds of the pore formed by a fusogenic viral envelope protein during membrane fusion. Proc. Natl. Acad. Sci. USA 88, 3623-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe, J. C., Bossi, G., Booth, S., and Griffiths, G. M. (2001). The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15, 751-761. [DOI] [PubMed] [Google Scholar]
- Swaminathan, R., Hoang, C. P., and Verkman, A. S. (1997). Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72, 1900-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, M. V. (2002). Muscle differentiation: how two cells become one. Curr. Biol. 12, R224-R228. [DOI] [PubMed] [Google Scholar]
- Trueheart, J., Boeke, J. D., and Fink, G. R. (1987). Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol. Cell. Biol. 7, 2316-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart, J., and Fink, G. R. (1989). The yeast cell fusion protein FUS1 is O-glycosylated and spans the plasma membrane. Proc. Natl. Acad. Sci. USA 86, 9916-9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Taubas, J., and Pelham, H. R. (2003). Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr. Biol. 13, 1636-1640. [DOI] [PubMed] [Google Scholar]
- Valtz, N., and Herskowitz, I. (1996). Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J. Cell Biol. 135, 725-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman, A. S. (2002). Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27, 27-33. [DOI] [PubMed] [Google Scholar]
- Vignery, A. (2000). Osteoclasts and giant cells: macrophage-macrophage fusion mechanism. Int. J. Exp. Pathol. 81, 291-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. T., Lu, J. C., Bai, J., Chang, P. Y., Martin, T. F., Chapman, E. R., and Jackson, M. B. (2003). Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature 424, 943-947. [DOI] [PubMed] [Google Scholar]
- Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., Sollner, T. H., and Rothman, J. E. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759-772. [DOI] [PubMed] [Google Scholar]
- Weber, W., Helms, V., McCammon, J. A., and Langhoff, P. W. (1999). Shedding light on the dark and weakly fluorescent states of green fluorescent proteins. Proc. Natl. Acad. Sci. USA 96, 6177-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Q., Bechinger, C., and Leiderer, P. (2000). Single-file diffusion of colloids in one-dimensional channels. Science 287, 625-627. [DOI] [PubMed] [Google Scholar]
- Weisman, L. S. (2003). Yeast vacuole inheritance and dynamics. Annu. Rev. Genet. 37, 435-460. [DOI] [PubMed] [Google Scholar]
- Weisman, L. S., and Wickner, W. (1988). Intervacuole exchange in the yeast zygote: a new pathway in organelle communication. Science 241, 589-591. [DOI] [PubMed] [Google Scholar]
- White, J. M., and Rose, M. D. (2001). Yeast mating: getting close to membrane merger. Curr. Biol. 11, R16-R20. [DOI] [PubMed] [Google Scholar]
- Wickner, R. B. (1992). Double-stranded and single-stranded RNA viruses of Saccharomyces cerevisiae. Annu. Rev. Microbiol. 46, 347-375. [DOI] [PubMed] [Google Scholar]
- Yang, F., Moss, L. G., and Phillips, G. N., Jr. (1996). The molecular structure of green fluorescent protein. Nat. Biotechnol. 14, 1246-1251. [DOI] [PubMed] [Google Scholar]
- Yarbrough, D., Wachter, R. M., Kallio, K., Matz, M. V., and Remington, S. J. (2001). Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-A resolution. Proc. Natl. Acad. Sci. USA 98, 462-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.