

Abstract
Protein kinase C (PKC) ζ has been implicated in insulin-induced glucose uptake in skeletal muscle cell, although the underlying mechanism remains unknown. In this study, we investigated the effect of PKCζ on actin remodeling and glucose transport in differentiated rat L6 muscle cells expressing myc-tagged glucose transporter 4 (GLUT4). On insulin stimulation, PKCζ translocated from low-density microsomes to plasma membrane accompanied by increase in GLUT4 translocation and glucose uptake. Z-scan confocal microscopy revealed a spatial colocalization of relocated PKCζ with the small GTPase Rac-1, actin, and GLUT4 after insulin stimulation. The insulin-mediated colocalization, PKCζ distribution, GLUT4 translocation, and glucose uptake were inhibited by wortmannin and cell-permeable PKCζ pseudosubstrate peptide. In stable transfected cells, overexpression of PKCζ caused an insulin-like effect on actin remodeling accompanied by a 2.1-fold increase in GLUT4 translocation and 1.7-fold increase in glucose uptake in the absence of insulin. The effects of PKCζ overexpression were abolished by cell-permeable PKCζ pseudosubstrate peptide, but not wortmannin. Transient transfection of constitutively active Rac-1 recruited PKCζ to new structures resembling actin remodeling, whereas dominant negative Rac-1 prevented the insulin-mediated PKCζ translocation. Together, these results suggest that PKCζ mediates insulin effect on glucose transport through actin remodeling in muscle cells.
INTRODUCTION
Insulin stimulates glucose uptake into skeletal muscle tissue mainly through GLUT4 translocation from intracellular pools to the plasma membrane (Klip et al., 1993; Bryant et al., 2002; Saltiel and Pessin, 2002). Tyrosine phosphorylation of insulin receptor substrate-1 by insulin activates phosphatidylinositol 3-kinase (PI3-K) and induces activation of downstream signal molecules protein kinase B (PKB/Akt) (Kohn et al., 1996; Tanti et al., 1997; Hill et al., 1999; Wang et al., 1999) and atypical PKCs (aPKCs) ζ and λ/ι (Bandyopadhyay et al., 1997a,b, 1999; Standaert et al., 1997; Kotani et al., 1998). aPKCs have been implicated in insulin action in adipocytes and muscle tissues (Kotani et al., 1998; Kim et al., 1999; Sajan et al., 2004), although this notion is not supported consistently by some studies (Tsuru et al., 2002). Evidence indicates that activation of aPKCs by insulin in skeletal muscles is defective in type 2 diabetic patients, monkeys, and rodents, and this defect seems to contribute significantly to the diminution in insulin-stimulated glucose disposal and muscle-dependent insulin resistance seen in these diabetic states (Bandyopadhyay et al., 1997a, 1999; Standaert et al., 2002; Beeson et al., 2003; Kim et al., 2003).
We have previously shown that insulin causes a rapid and dynamic remodeling of actin into a cortical mesh (Khayat et al., 2000; Tong et al., 2001). Within the submembrane mesh, insulin-effective molecules such as glucose transporter (GLUT) isoform 4, vesicle-associated membrane protein (VAMP) 2, and phosphatidylinositol-3,4,5-trisphosphate have been enriched (Khayat et al., 2000; Tong et al., 2001). It is conceivable that the spatial and temporal change of actin structure provides a scaffold for the transmission of signals from insulin receptor to insulin responsive GLUT4 vesicles. The link between actin remodeling, insulin signaling molecule relocation, and GLUT4 translocation remains poorly understood. Experiments using both constitutively active and dominant negative mutants of Rho family members have shown that Rac causes actin ruffling and is necessary for formation of lamellipodia and cell movement (Ridley et al., 1992). In muscle cells, Rac-1 is the candidate small GTPase involved in insulin-stimulated actin reorganization that is necessary for GLUT4 translocation (JeBailey et al., 2004). On the other hand, PKCζ has been implicated in maintaining cell polarity in yeast and mammalian cells by forming quaternary complex with GTP-binding Rac/Cdc42 (Lin et al., 2000; Noda et al., 2001). Therefore, it is plausible that PKCζ may lie downstream of PI3-K in the insulin signaling cascade and link with Rac-1 and actin remodeling for GLUT4 translocation in muscle cells. In the present study, we provide evidence that PKCζ mediates insulin effect on glucose transport by interacting with Rac-1 and actin remodeling.
MATERIALS AND METHODS
Reagents
Tissue culture medium, serum, and other tissue culture regents were purchased from Invitrogen (Carlsbad, CA). Soluble insulin was purchased from Novo Nordisk (Bagsværd, Denmark). ProLong Antifade mounting solution, 4,6-diamidino-2-phenylindole (DAPI), Oregon Green-phalloidin, Alexa-phalloidin, and Alexa Fluor anti-rabbit or anti-mouse secondary antibodies were purchased from Invitrogen. Antibody against Thr410 PKCζ/λ was purchased from Cell Signaling Technology (Beverly, MA). Polyclonal antibody against PKCζ (C20), polyclonal anti-c-myc antibody, goat anti-rabbit, and goat anti-mouse secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Bio-Rad protein assay was purchased from Bio-Rad (Hercules, CA). Enhanced chemical luminescence (ECL) [γ-32P]ATP, 2-deoxy-[3H]d-glucose, and protein G beads were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom). Antibody against Rac-1, [159Ser]PKC-ε (AA153-164)-NH2 were purchased from Upstate Biotechnology (Lake Placid, NY). OptiPhase HiSafe 2 scintillation solution was purchased from Wallac Scintillation Product (Wallac Oy, Turku, Finland). Phenylmethylsulfonyl fluoride (PMSF) was purchased from Calbiochem (San Diego, CA). Dithiothreitol (DTT), orthovanadate, β-mercaptoethanol, bovine serum albumin (BSA), cell-permeable PKCζ pseudosubstrate (PS) myristoyl trifluoroacetate, O-phenylenediamine dihydrochloride (OPD), and phosphate-buffered saline (PBS) were obtained from Sigma-Aldrich (St. Louis, MO). Lipofectmine 2000, Opiti-MEM, and pEGFP-N1 vector were purchased from Invitrogen. G418 was purchased from Alexis Biochemicals (Lausen, Switzerland). PKCζ gene was kindly provided by Prof. R. V. Farese (University of South Florida College of Medicine, Tampa, FL) and was inserted into pEGFP-N1 vector through PCR.
Cell Culture and Transfection
L6 muscle cells expressing c-myc epitope tagged GLUT4 (L6myc cells) (Kanai et al., 1993; Ueyama et al., 1999) were maintained in myoblast monolayer culture in α-minimal essential medium containing 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) antibiotic-antimyocotic solution (100 U/ml penicillin G, 10 μg/ml streptomycin, and 25 mg/ml amphotericin B) in an atmosphere of 5% CO2 at 37°C. For differentiation into myotubes, myoblasts were plated in medium containing 2% (vol/vol) FBS at 104 cells/ml to allow spontaneous myoblast fusion. Medium was changed every 48 h, and myotubes were ready for experiment 6-8 d after plating. Transfection of L6 GLUT4myc myotubes was performed in six-well plates as described previously for L6 myoblast (Wang et al., 1999) with the following modifications: DNA was introduced to the cells at the start of the day 4 after seeding, and cells were maintained for another 72 h until experimentation. For stable transfection, 2 μg of DNA was introduced into the myoblasts at 104 cells/ml, and cells were maintained for another 72 h and then maintained in the normal culture medium contained 0.5 mg/ml G418. Proper clones were selected by immunofluorescence and subcultured with G418 all the time. The transfected cells were used within six generations. In this study, L6myc PKCζ cells specifically referred to the differentiated L6myc cells stably overexpressing transfected PKCζ.
Fluorescence and Confocal Laser Microscopy
L6 GLUT4myc muscle cells were grown to the stage of myotubes on 25-mm-diameter glass coverslips placed in six-well plates. Myotubes are characterized by multinucleation. Myotubes were deprived of serum for 3 h and treated with 100 nmol/l insulin for 5 min at 37°C. Myotubes were fixed with 3% (vol/vol) paraformaldehyde in PBS for 20 min and then washed with 0.1 mol/l glycine in PBS for 10 min, permeabilized with 0.1% (vol/vol) Triton X-100 in PBS for 3 min, and then washed with PBS. For labeling of actin filaments, fixed and permeabilized cells were incubated for 1 h at room temperature with Alexa-labeled phalloidin (0.01 U/coverslip). To assess autofluorescence, additional samples were treated for 1 h with PBS without labeled phalloidin. For immunostaining, fixed and permeabilized myotubes were first incubated for 1 h at room temperature with primary antibodies [dilution factors were myc, 1:100; PKCζ, 1:100; hemagglutinin (HA), and 1:100 in 0.1% (wt/vol) BSA/phosphate-buffered saline]. The cells were then washed with PBS and subsequently incubated with either Alexa-conjugated goat anti-rabbit or anti-mouse secondary antibodies, respectively, at a dilution of 1:250 for 1 h at room temperature. Cells were washed by PBS and labeled with 600 nmol/l DAPI in PBS for 5 min at room temperature. Cell monolayers were washed further with PBS and mounted in ProLong Antifade solution onto glass slides. For fluorescence and confocal laser microscopy, stained cells were examined with a Zeiss Axioplan 2 imaging microscope and a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
Measurement of PKCζ Enzyme Activity
After serum deprivation for 3 h, L6myc myotubes were left untreated or treated with 100 nmol/l insulin for 5 min at 37°C. Cells were washed three times with ice-cold PBS and collected. Then PKCζ enzyme activity was measured in specific immunoprecipitates as described previously by using [γ-32P]ATP (Bandyopadhyay et al., 1997a,b).
Densitometric Assay of Surface GLUT4myc
After serum deprivation for 3 h, L6 myotubes were left untreated or treated with 100 nM insulin for various times at 37°C. Cells were washed three times with ice-cold PBS, followed by blocking with 5% (vol/vol) goat serum in PBS for 10 min. Cells were incubated with anti-myc monoclonal antibody in HEPES-buffered RPMI 1640 medium containing 3% (vol/vol) goat serum for 60 min at 4°C before fixation with 3% (vol/vol) formaldehyde in PBS for 3 min. Cells were incubated with 100 mM glycine in PBS at 4°C for 10 min followed by horseradish peroxidase-conjugated goat anti-mouse IgG (1:1000) in PBS containing 3% goat serum for 60 min. To quantify the amount of bound antibody, OPD reagent was added at room temperature for up to 30 min, and the reaction was stopped by adding 3 M hydrochloric acid. An aliquot of the reaction was removed for measuring the absorbance at 492 nm (Wang et al., 1998).
2-Deoxy-[3H]deoxyglucose Uptake
After serum deprivation, L6 myotubes were left untreated or treated with 100 nmol/l insulin for different times at 37°C. After this period, cells were washed three times with glucose-free HEPES-buffered saline solution (140 mmol/l NaCl, 20 mmol/l Na-HEPES, pH 7.4, 2.5 mmol/l MgSO4, 5 mmol/l KCl, and 1 mmol/l, CaCl2). Glucose uptake was measured as described previously by using 2-deoxy-[3H]deoxyglucose (Klip et al., 1982). Each condition was assayed in triplicate.
Cell Fractionation
All steps subsequent to the incubation of the cell with or without insulin were performed at 4°C. At least four culture dishes (15 cm) of L6 myotubes were used as one group to get enough plasma membrane (PM), cytosol (CTS), and low-density microsome (LDM) for Western blotting. After treating the myotubes as indicated, the cells were washed with ice-cold PBS immediately and were scraped with a rubber policeman in ice-cold homogenization buffer (20 mmol/l HEPES, 1 mmol/l EDTA, pH 7.4, 20 μg/ml leupeptin, 10 μg/ml aprotinin, 2 μg/ml pepstain, 1 mmol/l PMSF, and 1 mmol/l DTT). Then, the cells were enriched in 1.5-ml centrifuge tubes by centrifugation at 1200 rpm. Cells were homogenized by 50 passes through a 27-gauge needle in 300 μl of HES buffer (20 mmol/l HEPES, 1 mmol/l EDTA, 250 mmol/l sucrose, pH 7.4, 1 mmol/l PMSF, and 1 mmol/l DTT). The suspension was centrifuged for 10 min at 1000 × g to remove the nucleus and complete cells. The supernatant was centrifuged at 17,300 × g for 20 min. The pellet was washed and layered onto a 1.12 mol/l sucrose cushion (20 mmol/l HEPES, 1 mmol/l EDTA, and 1.12 mol/l sucrose, pH 7.4) and centrifuged at 100,000 × g for 60 min. The membrane layer above the cushion contains highly enriched PM. The membrane layer was transferred to a new tube and enriched by centrifuging at 32,000 × g. The supernatant from the initial spin was subsequently centrifuged at 38,700 × g for 20 min to remove endoplasmic reticulum and smaller pieces of the plasma membrane. The resulting supernatant was centrifuged at 200,000 × g for 1 h to generate LDM fraction that is enriched in the intracellular GLUT4 storage vesicles and CTS. The layers of PM and LDM were resuspended in 50 μl of HES buffer contain protease inhibitors as described above. The protein concentration of these subcellular fractions was determined using the Bio-Rad protein assay.
Western Blot
Aliquots proteins were separated by SDS-PAGE (10% polyacrylamide). Thereafter, proteins were electrophoretically transferred to polyvinylidene difluoride membrane and block in 5% BSA and 0.05% Tween 20 in Tris-buffered saline (TBST) for 1.5 h at room temperature. Membranes were incubated overnight at 4°C with indicated first antibodies. Membranes were washed (3 times for 5 min each) in TBST and incubated with horseradish peroxidase-conjugated IgG for 0.5 h at room temperature, followed by additional washes (3 times for 15 min) in TBST. Proteins were visualized by ECL and quantified by densitometry.
Statistical Analysis
Data are expressed in mean ± SEM. For Western blot, x-ray films were quantified in the linear range by densitometry using Bio-Rad Image software. Differences between two means were analyzed by Student's t test. A two-tailed p value < 0.05 was considered to be significant.
RESULTS
Insulin-mediated PKCζ Phosphorylation, Expression, and Activity
In L6myc cells, treatment with insulin increased the PKCζ phosphorylation on Thr-410 by a 1.3-fold and expression by 1.2-fold of basal value. Moreover, PKCζ activity increased 1.6-fold after insulin (Figure 1). There is positive association of PKCζ phosphorylation on Thr-410 and kinase activity induced by insulin in L6myc cells.
Figure 1.
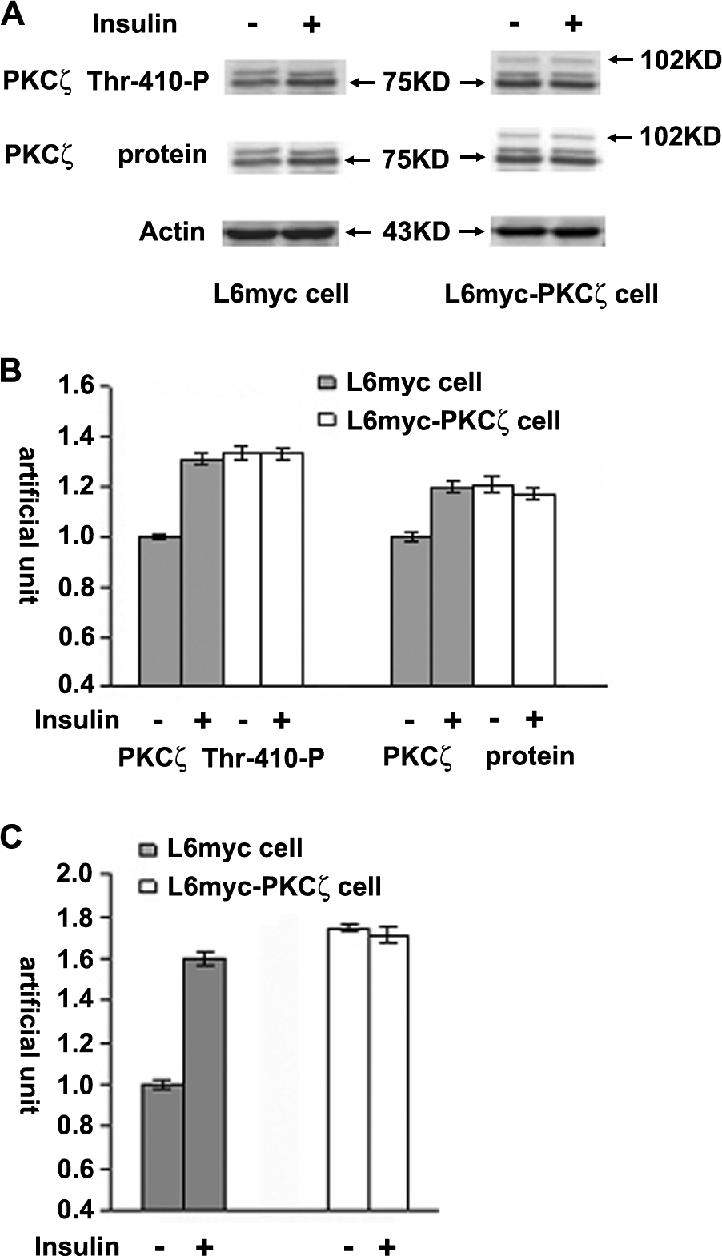
Insulin-mediated PKCζ phosphorylation, expression, and activity. L6myc and L6myc-PKCζ myotubes were treated with or without 100 nmol/l insulin for 5 min at 37°C, and then with 15 μg of whole cell lysates were subjected to 10% SDS-PAGE and immunoblotted with indicated antibodies (anti-phospho-PKCλ/ζ for PKCζ activity, anti-PKC λ/ζ for PKCζ protein, and anti-actin for actin protein). The same membranes were first blotted for phosphorylation followed by stripping and reblotting for protein. The EGFP tag ensured transfected PKCζ protein running ∼27 kDa higher than the endogenous PKCζ, ∼75 kDa indicated by arrowhead (A). The amounts of phosphorylated PKCζ and protein kinase Cζ protein were quantified by densitometric scanning and analyzed with Bio-Rad Molecular Analysis software. Pixel intensity was normalized, and a value of 1 was assigned to the basal condition (B). Insulin-induced PKCζ activity was measured with specific immunoprecipitates from the 30 μg of whole cell lysates, as described in Materials and Methods (C). The data are representative of three experiments.
Compared with normal L6myc cells, there was higher level of PKCζ activity (1.7-fold) in L6myc PKCζ cells in basal state, the changes of PKCζ phosphorylation, expression, and activity caused by insulin treatment were not significant (Figure 1). The expression of transfected PKCζ protein was evidenced by the presence of a 102-kDa band that represented the enhanced green fluorescent protein (EGFP)-tagged PKCζ (Figure 1). Treatment of insulin did not further increase the PKCζ phosphorylation, expression, and activity in L6myc PKCζ cells (Figure 1).
Insulin-mediated PKCζ Activation and Glucose Transport
Cell fractionation study showed an insulin-induced PKCζ activation in parallel with GLUT4 translocation and glucose uptake (Figure 2). In L6myc cells treated with insulin, PKCζ protein and phosphorylation increased 30% in PM fraction and decreased 20% in LDM fraction (Figure 2). Similarly, the GLUT4 protein increased 30% in PM fraction and decreased 20% in LDM fraction (Figure 2). In agreement with previous studies (Mitsumoto and Klip, 1992; Sumitani et al., 1997), insulin promotes GLUT4 translocation from LDM to PM.
Figure 2.
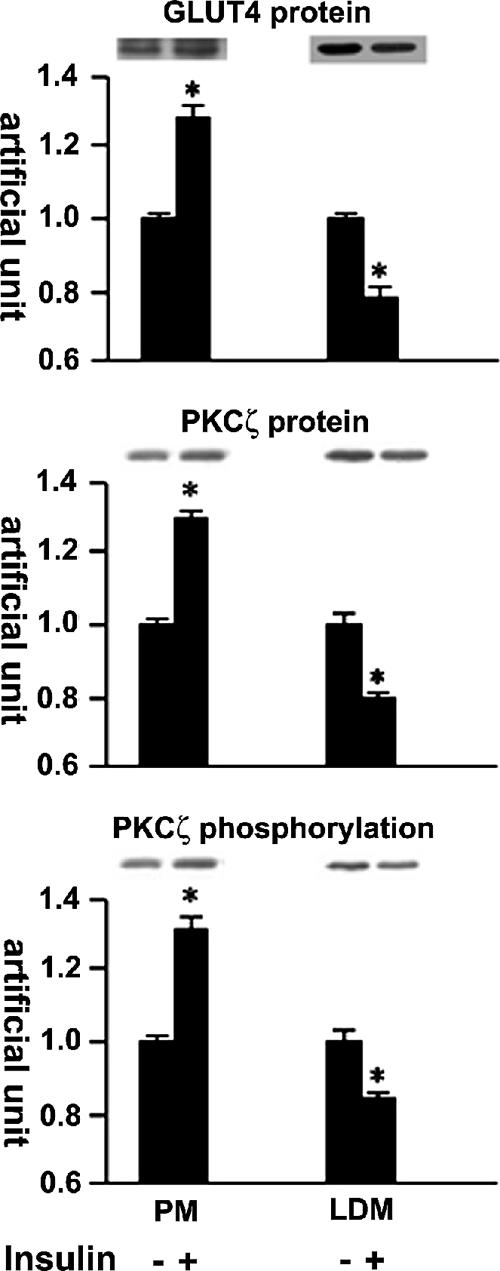
Insulin-mediated protein kinase Cζ and GLUT4 translocation shown by cell fractionation. Serum-depleted (3 h) L6myc cells were treated with or without 100 nmol/l insulin for 5 min. Subcellular fractionation was performed to purify the PM and LDM fractions as described in Materials and Methods. Equal amount proteins (20 μg) were subjected to 10% SDS-PAGE to immunoblot for GLUT4myc and PKCζ phosphorylation with corresponding antibodies first (anti-phospho-PKCλ/ζ for PKCζ phosphorylation, and anti-myc for GLUT4myc protein), after stripping, the same membrane (measured for protein kinase Cζ phosphorylation) was reblotted for PKCζ protein (anti-PKCλ/ζ for PKCζ protein) after stripping as described in Materials and Methods. The amount of phosphorylated PKCζ, PKCζ protein, and GLUT4myc were quantified by densitometric scanning and analysis with Bio-Rad Molecular Analysis software. Pixel intensity was normalized, and a value of 1 was assigned to the basal condition. The data are representative of three experiments as the mean ± SE and *p < 0.01 compared with basal state.
In L6myc PKCζ cells, overexpression of PKCζ enhanced the amount of GLUT4 protein in the PM, with a corresponding reduction of GLUT4 protein in the LDM. Insulin stimulation failed to further mobilize the movement of GLUT4 from LDM to PM. Similarly, both the expression and the phosphorylation of PKCζ in PM fraction significantly increased 20% in parallel to a 20% reduction in LDM fraction and treatment with insulin failed to cause significant changes in the PKCζ expression and activity in both PM and LDM fractions of L6myc PKCζ cells (our unpublished data).
Insulin-mediated Colocalization of PKCζ with Actin
We have previously reported insulin induces cortical actin remodeling and facilitates the association of PI3-K with GLUT4 vesicles leading to the recruitment of GLUT4-containing vesicles to the plasma membrane (Khayat et al., 2000; Tong et al., 2001). To investigate the participation of PKCζ in actin remodeling upon insulin stimulation, optical sections of 0.5 μm running from the ventral to the dorsal cell surface were examined using laser scanning confocal microscopy. After insulin stimulation, the staining pattern of filamentous actin (F-actin) reorganized into a meshlike structure that extended to the dorsal cell surface in L6myc cells. A portion of PKCζ colocalized with these new actin structures (Figure 3). Away from the dorsal surface, most actin filaments remained in the form of stress fibers, and the staining pattern of PKCζ was diffuse.
Figure 3.

Insulin-mediated colocalization of PKCζ with actin shown by laser confocal microscopy. L6myc cells were serum starved 3 h and then treated with 100 nmol/l insulin for 5 min at 37°C. Afterward, the cells were fixed, permeabilized, and double stained for PKCζ (anti-PKCζ antibody followed by Alexa 488 green-conjugated second antibody) and actin (Alexa red-phalloidin) as described in Materials and Methods. Serial optical sections of 0.5 μm thickness along the z-axis were generated as described in Materials and Methods. Optical slices taken from the dorsal to the ventral surface of myotubes show filamentous PKCζ (green) and actin (red) after 5 min of 100 nmol/l insulin treatment. The right line shows the merge of PKCζ and actin. The progression of images is linear from left to right and from top to bottom. Bar, 10 μm. The images are representative of five experiments.
Confocal results illustrated longitudinal stress fibers of filamentous actin and diffuse staining of PKCζ in L6myc cells in the absence of insulin (Figure 4, a-c). On insulin stimulation, the L6myc cells showed a spatial colocalization of reorganized PKCζ with actin (Figure 4, d-f). The insulin-mediated colocalization of PKCζ with actin in L6myc cells was abolished by preincubation with the PI3-K inhibitor wortmannin (WM; Figure 4, g-i) and the PKCζ PS (Figure 4, j-l).
Figure 4.

Inhibition effect of WM and PKCζ PS on insulin-mediated colocalization of PKCζ with actin remodeling in L6myc cells shown by laser confocal microscopy. Three hour-serum-starved L6myc cells were left untreated (a-c), or treated with 100 nmol/l insulin (d-f) for 5 min at 37°C, pretreated with 100 nmol/l WM (g-i) for 25 min, or 5 μmol/l PS (j-l) for 55 min at 37°C, followed by adding 100 nmol/l insulin and incubated at 37°C for 5 min. Then, the cells were fixed, permeabilized, stained, and scanned as described as Figure 3. Bar, 10 μm. The images are representative of five experiments.
PKCζ-mediated Insulin Effect on Actin Remodeling
PKCζ stimulated an insulinlike change of actin remodeling. In L6myc PKCζ cells, overexpression of PKCζ caused actin remodeling similar to that of insulin effect (Figure 5b). The stable overexpressed PKCζ (Figure 5a) colocalized with new actin structures in the absence of insulin (Figure 5c). In the presence of insulin, L6myc PKCζ cells showed a nonsignificant increase of actin remodeling and colocalization (Figure 5, d-f).
Figure 5.

PKCζ-mediated insulin effect on actin remodeling and the inhibition effect of WM and PKCζ PS in L6myc PKCζ cells shown by laser confocal microscopy. L6myc-protein kinase Cζ cells were serum starved 3 h and were left untreated (a-c), or treated with 100 nmol/l insulin for 5 min at 37°C (d-f), preincubated with 100 nmol/l WM (g-i) for 25 min, or 5 μmol/l PS (j-l) for 55 min at 37°C, followed by adding 100 nmol/l insulin and incubated at 37°C for 5 min. Then, the cells were fixed, permeabilized, stained, and scanned as described as Figure 3. Because the EGFP-tagged PKCζ gave the auto-green fluorescence in L6myc-PKCζ cells, Alexa red-phalloidin was used for illustrating the F-actin. Bar, 10 μm. The images are representative of five experiments.
In L6myc PKCζ cells, the insulin-mediated additional PKCζ redistribution, actin remodeling, and colocalization were largely abolished by preincubation with WM (Figure 5, g-i). In contrast, preincubation with PS abolished the insulinlike effects of PKCζ overexpression, actin remodeling, and colocalization in L6myc PKCζ cells (Figure 5, j-l). These data suggest that insulin effect on actin remodeling is mediated by PKCζ at the downstream of PI3 kinase in insulin signaling pathway.
PKCζ-mediated Insulin Effect on Glucose Transport
PKCζ stimulated insulinlike effects on GLUT4 translocation and glucose uptake. In L6myc PKCζ cells, overexpression of PKCζ caused 2.1-fold increase of surface GLUT4 (Figure 6A) and 1.7-fold increase of glucose uptake (Figure 6B). Neither surface GLUT4 (Figure 6A) nor glucose uptake (Figure 6B) increased significantly after treatment with insulin.
Figure 6.

PKCζ-mediated insulin effect on surface GLUT4 translocation and glucose uptake. L6myc cells and L6myc PKCζ cells were prepared on 24-well plates or 12-well plates for measurement of surface GLUT4 (A) or glucose uptake (B) as described in Materials and Methods. L6myc and L6myc-PKCζ cells were treated for the indicated times with 100 nmol/l insulin at 37°C. Cell surface density of GLUT4myc and 2-deoxyglucose uptake was measured in parallel culture plates. Data points are the mean ± SE of five to eight experiments performed in triplicate. Insulin-stimulated GLUT4myc translocation and glucose uptake were expressed as optical density (OD) and picomoles per minute per milligram of protein separately.
In L6myc cells, insulin stimulated twofold increase in both surface GLUT4 (Figures 6A and 7A) and glucose uptake (Figures 6B and 7B). The insulin-mediated increase in glucose transport echoes the effect of GLUT4 translocation from LDM to PM (Figure 2). To further investigate the contribution of PKCζ to GLUT4 translocation and glucose uptake, WM and PS were used to inhibit PI3-K and PKCζ activity, respectively. Although WM and PS had no effect on the basal surface GLUT4 and glucose uptake, both inhibitors significantly reduced the effects of insulin in L6myc cells (Figure 7, A and B).
Figure 7.

Inhibition effects of WM and PKCζ PS on GLUT4 translocation and glucose uptake in L6myc cells and L6myc PKCζ cells. L6myc cells (A and B) and L6myc PKCζ cells (C and D) were prepared on 24-well plates or 12-well plates for measurement of surface GLUT4 (A and C) and glucose uptake (B and D) as described in Materials and Methods. For insulin stimulation groups, cells were treated with 100 nmol/l insulin for 5 min at 37°C; for the inhibition groups, cells were first treated with 100 nmol/l wortmannin for 25 min or 5 μmol/l PS for 55 min, followed by adding 100 nmol/l insulin and incubated at 37°C for 5 min. Cell surface density of GLUT4myc (A and C) and 2-deoxyglucose uptake (B and D) were measured in parallel culture plates. Data points are the mean ± SE of four experiments performed in triplicate. Insulin-stimulated GLUT4myc translocation and glucose uptake are expressed as OD and picomoles per minute per milligram of protein. **p < 0.01.
In L6myc PKCζ cells, increases in the basal surface GLUT4 and glucose uptake were abolished by PS, whereas WM had no effect (Figure 7, C and D). In the presence of WM or PS, neither surface GLUT4 nor glucose uptake increased upon treatment of insulin, and PS showed significant inhibition effect (Figure 7, C and D). These data suggest that insulin effect on glucose transport is mediated by PKCζ at the downstream of PI3-K in insulin signaling pathway.
Association of PKCζ with GLUT4
In L6myc cells, stimulation with insulin caused translocation of both GLUT4 and PKCζ from the LDM to PM (Figure 2). In L6myc PKCζ cells, overexpression of PKCζ caused insulinlike effects on actin remodeling (Figure 5) and glucose transport (Figures 6 and 7). We next examined the association of PKCζ with GLUT4. Before insulin treatment, L6myc cells showed perinuclear staining of GLUT4 and PKCζ with limited colocalization (Figure 8, a-c). After insulin treatment, both GLUT4 and PKCζ clustered into new actinlike structures beneath the dorsal surface of L6myc cells (Figure 8, d and e). Moreover, insulin stimulated the colocalization of PKCζ and GLUT4 (Figure 8f). Both WM and PS completely inhibited the insulin-induced relocation and colocalization of PKCζ and GLUT4 in L6myc cells (our unpublished data).
Figure 8.

Association of protein kinase Cζ with GLUT4 and the inhibition effect of WM and PKCζ PS by laser confocal microscopy. L6myc cells (a-f) and L6myc PKCζ cells (g-r) were serum starved 3 h and were left untreated (a-c, g-i), or treated with 100 nmol/l insulin (d-f, p-r) for 5 min at 37°C, 100 nmol/l WM (i and j) for 25 min, or 5 μmol/l PS (m-o) for 55 min at 37°C. Afterward, the cells were fixed, permeabilized, and double stained for PKCζ (anti-PKCζ antibody followed by Alexa 488 green-conjugated second antibody) and GLUT4myc (anti-myc antibody followed by Alexa 546 red-conjugated second antibody) as described in Materials and Methods. Cells were scanned along the z-axis. Bar, 10 μm. The images are representative of five experiments.
In L6myc PKCζ cells, overexpression of PKCζ caused colocalization of relocated PKCζ and GLUT4 in a pattern of actin mesh before insulin treatment (Figure 8, g-i), and insulin treatment enhanced this tendency (Figure 8, p-r). PS (Figure 8, j-l), but not WM (Figure 8, m-o), abolished both the relocation and the colocalization of GLUT4 and PKCζ enriched in the new actin structures and after insulin stimulation (our unpublished data). These data suggest that insulin effect on GLUT4 translocation and actin remodeling is mediated by PKCζ at the downstream of PI3-K in insulin signaling pathway.
Association of PKCζ with Rac-1
Evidence suggests an involvement of Rac-1 in insulin-induced actin remodeling in muscle cells (JeBailey et al., 2004). Because PKCζ activity is regulated by Rac-1(Qiu et al., 2000), Rac-1 may participate in insulin-induced actin remodeling through PKCζ in muscle cells. Before insulin treatment, L6myc cells showed perinuclear immunoreactivities of endogenous PKCζ and Rac-1 (Figure 9, a and b). Colocalization PKCζ and Rac-1 were not observed (Figure 9c). After insulin treatment, both of endogenous PKCζ and Rac-1 reorganized into actin mesh-like structures (Figure 9, d and e). Moreover, insulin stimulated colocalization of PKCζ with Rac-1 (Figure 9f). There data suggest an insulin-mediated spatial association between PKCζ and Rac-1 in L6myc cells.
Figure 9.

Interaction of PKCζ with Rac-1 on actin remodeling shown by laser confocal microscopy. L6myc myoblasts were prepared in medium containing 2% (vol/vol) FBS on six-well plates with coverslips. Plasmids contained HA-tagged Rac-1 (CA or DN) gene were introduced to the cells with Lipofectmine 2000 at the start of day 4 after seeding, and cells were maintained for another 72 h. Then, cells were serum-starved 3 h and stimulated with or without 100 nmol/l insulin for 5 min at 37°C. Afterward, the cells were fixed, permeabilized, and double stained for endogenous PKCζ (anti-PKCζ antibody followed by Alexa 488 green-conjugated second antibody; a, d, g, and j), endogenous Rac-1 (anti-Rac-1 antibody followed by Alexa 546 red-conjugated second antibody; b and e), or HA-tag for transfected Rac-1 CA (h) and Rac-1 DN (k) (anti-HA antibody followed by Alexa 546 red-conjugated second antibody) as described in Materials and Methods. Serial optical sections of 0.5 μm thickness along the z-axis were generated as described in Materials and Methods. Bar, 10 μm. The images are representative of three experiments.
To further characterize the association of PKCζ with Rac-1, differentiated L6myc cells were transiently transfected with constitutive active Rac-1 (Rac-1 CA) or dominant negative Rac-1 (Rac-1 DN). Confocal laser microscopy revealed relocation and colocalization of transfected Rac-1 and endogenous PKCζ in cells transfected with Rac-1 CA (Figure 9, g-i). The overexpression of Rac-1 CA recruited PKCζ to membrane scaffold where GLUT4 was compartmentalized by filamentous actin. The colocalization of Rac-1 CA and PKCζ was blocked by PS, but not WM, in both basal and insulin-treated conditions (our unpublished data). In cells transfected with Rac-1 DN, insulin failed to induce the formation of new actin structures and staining of both endogenous PKCζ and Rac-1 DN was diffuse (Figure 9, j-l). These data suggest that actin remodeling induced by either insulin or Rac-1 is mediated by PKCζ at the downstream of PI3-K in insulin signaling pathway. At the same time, either Rac-1 CA or DN was transfected into L6myc cells, and their stable expression was confirmed by immunofluorescence microscopy after clone selection. The transfection of Rac-1 CA could bring 20% increase on glucose uptake in basal state and the introduction of Rac-1 DN could eliminate insulin-induced glucose uptake ∼67%.
DISCUSSION
The participation of PKCζ in insulin-stimulated glucose transport has been documented in skeletal muscle (Bandyopadhyay et al., 1997a; Braiman et al., 2001; Beeson et al., 2003) and adipocytes (Standaert et al., 1997; Bandyopadhyay et al., 1999, 2002; Tsuru et al., 2002). However, the underlying mechanism between activation of PKCζ, translocation of GLUT4 and hence glucose transport in skeletal muscle has not been clarified. We have previously shown that insulin promotes formation of new actin-rich structures where redistribution of insulin signaling molecule PI3-K and GLUT4 vesicles as well as t- and v-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins takes place (Khayat et al., 2000; Tong et al., 2001). Here, we show that aPKCζ induces actin remodeling, resulting in colocalization of PKCζ and GLUT4 in the new actin structures. Using the constitutively active and dominant inhibitory mutants, we further demonstrate that the small GTPase Rac-1 may control the remodeling of actin through PKCζ. To our knowledge, this is the first study to illustrate the relationship of insulin-induced PKCζ activation and actin remodeling.
In L6myc cells, PKCζ was detected in the LDM where most of the GLUT4 located. Insulin stimulation caused a translocation of PKCζ from the LDM to the PM, accompanied by an increase in the enzyme activity. Using a cell surface staining approach, we demonstrate that the redistribution and activation of PKCζ correlated with translocation of GLUT4 from intracellular compartment to the cell surface of the muscle cells. A 1.3-fold increase of GLUT4 protein in PM was associated with twofold increase of surface GLUT4 translocation as determined by the densitometric assay. The inhibitory effects of WM on GLUT4 translocation and glucose uptake in L6myc cells not only confirm the key role of PI3-K in insulin signaling pathway but also demonstrate that PI3-K is required for insulin-mediated PKCζ activation (Standaert et al., 1997; Standaert et al., 1999). PKB, also as known as Akt, has been reported to regulate GLUT4 translocation and glucose uptake in L6 myoblasts (Tanti et al., 1997; Wang et al., 1999). The elimination of insulin-mediated GLUT4 translocation and glucose uptake by PS, which has no effect on PKB activity, suggests that PKCζ is the key downstream molecule from PI3-K in the propagation of insulin signals in L6myc cells. In this study, overexpression of PKCζ had no influence on PKB/Akt phosphorylation, although this would not preclude the possibility that PKB/Akt may activate PKCζ. It is also probable that PKCζ and PKB/Akt are parallel signaling molecules that branch off from PI3-K and thereafter function without dependence upon each other during the insulin action. Together, our results confirm previous findings that PKCζ is essential for glucose transport effects of insulin (Bandyopadhyay et al., 1997a,b, 1999; Standaert et al., 1997).
F-actin has been reported to be required for insulin stimulated GLUT4 translocation and glucose uptake (Khayat et al., 2000; Tong et al., 2001). It is well recognized that insulin causes a rapid and marked actin remodeling beneath the plasma membrane, promoting membrane ruffling in muscle cells (Tsakiridis et al., 1994). The new actin structures have been shown to contain PI3-K (Khayat et al., 2000), Akt/PKB (Peyrollier et al., 2000), and GLUT4 (Tong et al., 2001). Here, we provide structural evidence that PKCζ participates in the regulation of actin remodeling by insulin. Using confocal microscopy, a portion of the intracellular PKCζ became concentrated in the newly formed actin-rich structures at the dorsal surface of the muscle cells after insulin stimulation. The colocalization of PKCζ and GLUT4 in the new actin structures confirms the association of PKCζ with GLUT4-containing compartment in the cell fractionation experiments. The involvement of PKCζ in insulin action is further supported by the inhibitory effect of PS on actin remodeling, GLUT4 translocation and hence glucose transport after insulin stimulation. Both PKCζ and PKB/Akt have been implicated in insulin-stimulated GLUT4 translocation. However, the dominant negative forms of PKB/Akt could not prevent insulin-induced actin remodeling (Wang et al., 1999). Therefore, PKCζ and PKB/Akt may contribute to insulin-stimulated GLUT4 translocation through different mechanisms.
Confocal microscopy revealed colocalization between endogenous PKCζ and Rac-1 in L6myc cells after insulin treatment with the staining pattern similar to that of new actin structures induced by insulin. The close association between PKCζ and Rac-1 is further supported by 1) changes in the PK Cζ staining pattern resembling new actin structures after transfection of Rac-1 CA into L6myc cells without insulin treatment; and 2) the prevention of actin reorganization and hence relocalization of PKCζ after transient transfection of the Rac-1 DN, despite insulin stimulation. Rac-1 may regulate the activity of PKCζ through interaction with the adapter protein Par6 (Qiu et al., 2000; Noda et al., 2001). These results support the notion that aPKCζ induces actin reorganization through a Rho-dependent pathway, and PKCζ functions downstream of Rac-1 (Uberall et al., 1999; Brandt et al., 2002). In addition to Rac-1, the GTP-binding protein Cdc42 might also be associated with actin remodeling through PKCζ (Joberty et al., 2000). Cdc42 stimulates the formation of actin-rich filopodia to maintain cell polarity (Nobes and Hall, 1995), whereas Rac-1 induces actin filaments to form lamellipodia. In muscle cells, Rac-1 has been implicated in the insulin-stimulated actin remodeling characterized by membrane ruffles/lamellipodia (Ridley and Hall, 1992; Ridley et al., 1992). Previously, we have shown that Rac-1 is required for actin remodeling mediated by insulin and Rac-1 DN blocked insulin-induced actin remodeling and GLUT4 translocation to the cell surface (Khayat et al., 2000). Here, the confocal results combined with glucose uptake after Rac-1 transfection reveal that actin remodeling caused by Rac-1 is necessary but insufficient in insulin signaling pathway. Maybe other mechanism such as GLUT4 activity is needed (Konrad et al., 2002). Even Rac-1 is essential to the insulin-induced actin remodeling in L6myc cells, and the morphology of actin remodeling induced by Rac-1 and Cdc42 may be different, thus we cannot exclude the involvement of Cdc42 in this process. It has been reported that insulin-induced glucose uptake depends on the activation of TC10 and Cdc42 in adipocytes, (Chiang et al., 2001; Usui et al., 2003); whether cdc42 participates insulin-induced actin remodeling and GLUT4 translocation in L6myc cells needs further research.
Given the close association between PKCζ activation, actin remodeling, and GLUT4 translocation, it is conceivable that PKCζ may participate in insulin-mediated glucose transport through actin remodeling. We propose that a portion of intracellular PKCζ molecules may tether to the GLUT4 vesicles in the LDM fraction in basal state. On binding to its receptor, insulin activates PI3-K, acting through Rac-1, leads to actin remodeling through PKCζ. The reorganized actin structures provide a scaffold for interactions between insulin-sensitive GLUT4-containing vesicles and the target SNARE proteins. Intriguingly, PKCζ has been shown to induce serine phosphorylation of VAMP2 and promote glucose uptake in rat skeletal muscle (Braiman et al., 2001). PKCζ may also interacts with 80K-H to release the clamp action of Munc18c on syntaxin-4, allowing VAMP2 to bind t-SNAREs (Hodgkinson et al., 2005). Hence, PKCζ may facilitate translocation of GLUT4 in skeletal muscle after insulin stimulation by reorganizing actin filaments into new structures where phosphorylation of proteins such as VAMP2 and 80K-H occurs. The inhibition of WM, PS, and Rac-1 DN on redistribution of PKCζ, actin remodeling, GLUT4 translocation, and glucose transport after insulin stimulation suggests that the PI3-K activation of PKCζ plays a key role in insulin-mediated glucose metabolism in rat skeletal muscle cells.
Acknowledgments
We are grateful to Dr. Amira Kip for kindly supplying the L6myc cells and to Dr. Robert Farese for supplying PKCζ gene. We thank Laureen Kwok for the technical assistance in confocal laser microscopy. The study was supported by the grants from the Research Grant Committee, Hong Kong, and by the Major State Basic Research Development Program of China (973 Program 2004CB518602, 720004, and 2004CCA01400) and by the National Natural Science Foundation of China (30471930 and 30370668).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-10-0969) on March 8, 2006.
References
- Bandyopadhyay, G., Sajan, M. P., Kanoh, Y., Standaert, M. L., Quon, M. J., Lea-Currie, R., Sen, A., and Farese, R. V. (2002). Protein kinase C-zeta mediates insulin effects on glucose transport in cultured preadipocyte-derived human adipocytes. J. Clin. Endocrinol. Metab. 87, 716-723. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay, G., Standaert, M. L., Galloway, L., Moscat, J., and Farese, R. V. (1997a). Evidence for involvement of protein kinase C-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology 138, 4721-4731. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay, G., Standaert, M. L., Kikkawa, U., Ono, Y., Moscat, J., and Farese, R. V. (1999). Effects of transiently expressed atypical (zeta, lambda), conventional (alpha, beta) and novel (delta, epsilon) protein kinase C isoforms on insulin-stimulated translocation of epitope-tagged GLUT4 glucose transporters in rat adipocytes: specific interchangeable effects of protein kinases C-zeta and C-lambda. Biochem. J. 337, 461-470. [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay, G., Standaert, M. L., Zhao, L., Yu, B., Avignon, A., Galloway, L., Karnam, P., Moscat, J., and Farese, R. V. (1997b). Activation of protein kinase C (alpha, beta, and zeta) by insulin in 3T3/L1 cells. Transfection studies suggest a role for protein kinase C-zeta in glucose transport. J. Biol. Chem. 272, 2551-2558. [DOI] [PubMed] [Google Scholar]
- Beeson, M., et al. (2003). Activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes 52, 1926-1934. [DOI] [PubMed] [Google Scholar]
- Braiman, L., Alt, A., Kuroki, T., Ohba, M., Bak, A., Tennenbaum, T., and Sampson, S. R. (2001). Activation of protein kinase C zeta induces serine phosphorylation of VAMP2 in the GLUT4 compartment and increases glucose transport in skeletal muscle. Mol. Cell. Biol. 21, 7852-7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt, D., Gimona, M., Hillmann, M., Haller, H., and Mischak, H. (2002). Protein kinase C induces actin reorganization via a Src- and Rho-dependent pathway. J. Biol. Chem. 277, 20903-20910. [DOI] [PubMed] [Google Scholar]
- Bryant, N. J., Govers, R., and James, D. E. (2002). Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell. Biol. 3, 267-277. [DOI] [PubMed] [Google Scholar]
- Chiang, S. H., Baumann, C. A., Kanzaki, M., Thurmond, D. C., Watson, R. T., Neudauer, C. L., Macara, I. G., Pessin, J. E., and Saltiel, A. R. (2001). Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410, 944-948. [DOI] [PubMed] [Google Scholar]
- Hill, M. M., Clark, S. F., Tucker, D. F., Birnbaum, M. J., James, D. E., and Macaulay, S. L. (1999). A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol. 19, 7771-7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson, C. P., Mander, A., and Sale, G. J. (2005). Identification of 80K-H as a protein involved in GLUT4 vesicle trafficking. Biochem. J. 388, 785-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JeBailey, L., Rudich, A., Huang, X., Di Ciano-Oliveira, C., Kapus, A., and Klip, A. (2004). Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol. Endocrinol. 18, 359-372. [DOI] [PubMed] [Google Scholar]
- Joberty, G., Petersen, C., Gao, L., and Macara, I. G. (2000). The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2, 531-539. [DOI] [PubMed] [Google Scholar]
- Kanai, F., Nishioka, Y., Hayashi, H., Kamohara, S., Todaka, M., and Ebina, Y. (1993). Direct demonstration of insulin-induced GLUT4 translocation to the surface of intact cells by insertion of a c-myc epitope into an exofacial GLUT4 domain. J. Biol. Chem. 268, 14523-14526. [PubMed] [Google Scholar]
- Khayat, Z. A., Tong, P., Yaworsky, K., Bloch, R. J., and Klip, A. (2000). Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113, 279-290. [DOI] [PubMed] [Google Scholar]
- Kim, Y.-B., Kotani, K., Ciaraldi, T. P., Henry, R. R., and Kahn, B. B. (2003). Insulin-stimulated protein kinase C λ/ζ activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversal with weight reduction. Diabetes, 1935-1942. [DOI] [PubMed]
- Kim, Y. B., Nikoulina, S. E., Ciaraldi, T. P., Henry, R. R., and Kahn, B. B. (1999). Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Investig. 104, 733-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klip, A., Logan, W. J., and Li, G. (1982). Hexose transport in L6 muscle cells. Kinetic properties and the number of [3H]cytochalasin B binding sites. Biochim. Biophys. Acta 687, 265-280. [DOI] [PubMed] [Google Scholar]
- Klip, A., Ramlal, T., Bilan, P. J., Marette, A., Liu, Z., and Mitsumoto, Y. (1993). What signals are involved in the stimulation of glucose transport by insulin in muscle cells? Cell Signal 5, 519-529. [DOI] [PubMed] [Google Scholar]
- Kohn, A. D., Summers, S. A., Birnbaum, M. J., and Roth, R. A. (1996). Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271, 31372-31378. [DOI] [PubMed] [Google Scholar]
- Konrad, D., Bilan, P. J., Nawaz, Z., Sweeney, G., Niu, W., Liu, Z., Antonescu, C. N., Rudich, A., and Klip, A. (2002). Need for GLUT4 activation to reach maximum effect of insulin-mediated glucose uptake in brown adipocytes isolated from GLUT4myc-expressing mice. Diabetes 51, 2719-2726. [DOI] [PubMed] [Google Scholar]
- Kotani, K., et al. (1998). Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol. Cell. Biol. 18, 6971-6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D., Edwards, A. S., Fawcett, J. P., Mbamalu, G., Scott, J. D., and Pawson, T. (2000). A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat. Cell Biol. 2, 540-547. [DOI] [PubMed] [Google Scholar]
- Mitsumoto, Y., and Klip, A. (1992). Development regulation of the subcellular distribution and glycosylation of GLUT1 and GLUT4 glucose transporters during myogenesis of L6 muscle cells. J. Biol. Chem. 267, 4957-4962. [PubMed] [Google Scholar]
- Nobes, C. D., and Hall, A. (1995). Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53-62. [DOI] [PubMed] [Google Scholar]
- Noda, Y., Takeya, R., Ohno, S., Naito, S., Ito, T., and Sumimoto, H. (2001). Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells 6, 107-119. [DOI] [PubMed] [Google Scholar]
- Peyrollier, K., Hajduch, E., Gray, A., Litherland, G. J., Prescott, A. R., Leslie, N. R., and Hundal, H. S. (2000). A role for the actin cytoskeleton in the hormonal and growth-factor-mediated activation of protein kinase B. Biochem. J. 352, 617-622. [PMC free article] [PubMed] [Google Scholar]
- Qiu, R. G., Abo, A., and Steven Martin, G. (2000). A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signaling and cell transformation. Curr. Biol. 10, 697-707. [DOI] [PubMed] [Google Scholar]
- Ridley, A. J., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389-399. [DOI] [PubMed] [Google Scholar]
- Ridley, A. J., Paterson, H. F., Johnston, C. L., Diekmann, D., and Hall, A. (1992). The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 70, 401-410. [DOI] [PubMed] [Google Scholar]
- Sajan, M. P., Standaert, M. L., Miura, A., Bandyopadhyay, G., Vollenweider, P., Franklin, D. M., Lea-Currie, R., and Farese, R. V. (2004). Impaired activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(PO4)3 in cultured preadipocyte-derived adipocytes and myotubes of obese subjects. J. Clin. Endocrinol. Metab. 89, 3994-3998. [DOI] [PubMed] [Google Scholar]
- Saltiel, A. R., and Pessin, J. E. (2002). Insulin signaling pathways in time and space. Trends Cell Biol. 12, 65-71. [DOI] [PubMed] [Google Scholar]
- Standaert, M. L., et al. (1999). Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J. Biol. Chem. 274, 25308-25316. [DOI] [PubMed] [Google Scholar]
- Standaert, M. L., Galloway, L., Karnam, P., Bandyopadhyay, G., Moscat, J., and Farese, R. V. (1997). Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J. Biol. Chem. 272, 30075-30082. [DOI] [PubMed] [Google Scholar]
- Standaert, M. L., Ortmeyer, H. K., Sajan, M. P., Kanoh, Y., Bandyopadhyay, G., Hansen, B. C., and Farese, R. V. (2002). Skeletal muscle insulin resistance in obesity-associated type 2 diabetes in monkeys is linked to a defect in insulin activation of protein kinase C-zeta/lambda/iota. Diabetes 51, 2936-2943. [DOI] [PubMed] [Google Scholar]
- Sumitani, S., Ramlal, T., Somwar, R., Keller, S. R., and Klip, A. (1997). Insulin regulation and selective segregation with glucose transporter-4 of the membrane aminopeptidase vp165 in rat skeletal muscle cells. Endocrinology 138, 1029-1034. [DOI] [PubMed] [Google Scholar]
- Tanti, J. F., Grillo, S., Gremeaux, T., Coffer, P. J., Van Obberghen, E., and Le Marchand-Brustel, Y. (1997). Potential role of protein kinase B in glucose transporter 4 translocation in adipocytes. Endocrinology 138, 2005-2010. [DOI] [PubMed] [Google Scholar]
- Tong, P., Khayat, Z. A., Huang, C., Patel, N., Ueyama, A., and Klip, A. (2001). Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J. Clin. Investig. 108, 371-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridis, T., Vranic, M., and Klip, A. (1994). Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J. Biol. Chem. 269, 29934-29942. [PubMed] [Google Scholar]
- Tsuru, M., Katagiri, H., Asano, T., Yamada, T., Ohno, S., Ogihara, T., and Oka, Y. (2002). Role of protein kinase C isoforms in glucose transport in 3T3-L1 adipocytes: insignificance of atypical protein kinase C. Am. J. Physiol. 283, E338-E345. [DOI] [PubMed] [Google Scholar]
- Uberall, F., Hellbert, K., Kampfer, S., Maly, K., Villunger, A., Spitaler, M., Mwanjewe, J., Baier-Bitterlich, G., Baier, G., and Grunicke, H. H. (1999). Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J. Cell Biol. 144, 413-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama, A., Yaworsky, K. L., Wang, Q., Ebina, Y., and Klip, A. (1999). GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am. J. Physiol. 277, E572-E578. [DOI] [PubMed] [Google Scholar]
- Usui, I., Imamura, T., Huang, J., Satoh, H., and Olefsky, J. M. (2003). Cdc42 is a Rho GTPase family member that can mediate insulin signaling to glucose transport in 3T3-L1 adipocytes. J. Biol. Chem. 278, 13765-13774. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Khayat, Z., Kishi, K., Ebina, Y., and Klip, A. (1998). GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett. 427, 193-197. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Somwar, R., Bilan, P. J., Liu, Z., Jin, J., Woodgett, J. R., and Klip, A. (1999). Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol. Cell. Biol. 19, 4008-4018. [DOI] [PMC free article] [PubMed] [Google Scholar]