

Abstract
The heat shock transcription factors (Hsfs) activate the stress-inducible expression of heat shock proteins (Hsps) and other molecular chaperones in response to stress and, therefore, play an essential role in protein disaggregation and protein folding. In humans, missense mutation in the hsf4 gene causes cataract, and mice bearing a targeted disruption of the hsf4 gene exhibit defects in lens fiber cell differentiation and early cataract formation. Here, we show that Hsf4b is a direct target of the mitogen-activated protein (MAP) kinase extracellular signal-related kinase (ERK) and that phosphorylation of Hsf4b by ERK leads to increased ability of Hsf4b to bind DNA. Surprisingly, Hsf4b also interacts with an ERK-specific dual-specificity tyrosine phosphatase named DUSP26 identified from a yeast two-hybrid screen. While activated ERK phosphorylates Hsf4b, DUSP26 controls the activity of ERK, leading to phosphorylation/dephosphorylation of Hsf4b, altering its ability to bind DNA. Therefore, DUSP26 interaction with Hsf4b places this transcription factor within a regulatory circuit in the MAP kinase signaling pathway.
Four genes encoding heat shock transcription factors (Hsfs; Hsf1, Hsf2, Hsf4, and Hsfy) have been identified in mammalian cells, and the tissue distribution, regulation, and function of each Hsf gene and gene product are distinct. Hsf1 is ubiquitously expressed and is activated in response to a variety of stresses, having cytoprotective and antiapoptotic functions (27, 32, 34, 49, 52). Hsf2 is expressed in developing neuroectoderm and developing testis and functions in proper development of the central nervous system and during spermatogenesis (23, 31, 36, 37, 44, 45). Hsf2 is activated following the inhibition of the ubiquitin proteosome pathway (30). Hsfy is a heat shock factor-like protein expressed in the testis with unknown regulation and function (40). The signaling pathways regulating heat shock factors are complex and only partially understood. Hsf1 is phosphorylated by multiple protein kinases (extracellular signal-related kinase [ERK], c-Jun NH2-terminal kinase [JNK], p38, calcium calmodulin protein kinase, and perhaps others) (11-13, 19, 20, 26, 27, 46, 48) and interacts with many partner proteins (Hsp90, Hsp70, 14-3-3, RalBP1, and HSBP1) (1, 5, 22, 38). Hsf2 is not phosphorylated and interacts with different sets of proteins such as protein phosphatase 2A (21).
The heat shock factor Hsf4 possesses alternative splice variants leading to generation of two isoforms: Hsf4a that has transcriptional repressor activity and Hsf4b that has transcriptional activator properties (35, 41). Human cell lines expressing exogenous Hsf4a exhibit lower levels of basal and inducible expression of Hsp-90, -70, and -25 (35, 41, 51). In addition, in an in vitro reconstitution system, the mechanism underlying Hsf4a transcriptional repression appears to be through its interaction with the basal transcription factor TFIIF, leading to inhibition of proper assembly of basal transcription machinery (17). In contrast, the Hsf4b isoform is a relatively weak transcriptional activator compared to Hsf1, although Hsf4b can complement survival defects of Saccharomyces cerevisiae cells that lack Hsf (41). Interestingly, missense mutations in the DNA binding domain of hsf4 gene have been detected in humans with Marner and lamellar cataracts (7). Similarly, targeted disruption of the hsf4 gene in mice results in lens degeneration and cataract development early during postnatal development (18, 33). Hsf4 binding to the heat shock element (HSE) can be detected in the lens between postnatal day 1 to 5, and it remains active into adulthood. The results suggest that Hsf4b plays a critical role in the lens fiber cell maturation. While the developmental role of Hsf4 is becoming clear, the mode of Hsf4 regulation of transcription is not yet understood.
To examine the molecular mechanisms controlling the transcriptional activity of Hsf4b, we used Hsf4b as a bait in a yeast two-hybrid screening system and have identified a novel dual-specificity tyrosine phosphatase (genomic designation DUSP26) that interacts with Hsf4b. Dual-specificity tyrosine phosphatases (DSPs) are similar to tyrosine phosphatases by possessing the tyrosine phosphatase signature motif (I/V)HCXAGXGR(S/T) involved in their catalytic activity (4, 25). Thus far, many dual-specificity tyrosine phosphatases have been identified, and they recognize T-X-Y motifs that are phosphorylated by MEK1/2, MKK3/6, or MKK7 located upstream of mitogen-activated protein (MAP) kinases, ERK1/2, JNK, and the p38 protein kinases (α, β, γ, δ) (8, 25). The human genome encodes 38 dual-specificity tyrosine phosphatases. Among them there are 11 MAP kinase phosphatases, 17 atypical dual-specificity phosphatases, 4 PRL phosphatases, 3 Cdc14 phosphatases, and 3 Cdc25 phosphatases (16). A partial list of the dual-specificity tyrosine phosphatases identified to date includes MKP-1 (DUSP1), PAC-1 (DUSP2), hVH2/MKP2 (DUSP4), MKP-X (DUSP7), MKP-4 (DUSP9), MKP-5 (DUSP10), hVH5/M3/6 (DUSP8), MKP-6 (DUSP14), MKP-7 (DUSP16), and MKP-8 (DUSP26), all of which can dephosphorylate serine/threonine and tyrosine residues on different MAP kinase family members, leading to their inactivation (8, 25, 43). These phosphatases share a common structure consisting of a catalytic domain with high levels of sequence similarity to vaccinia virus dual-specificity tyrosine phosphatase (VH-1). These enzymes also contain a more divergent amino-terminal noncatalytic domain containing two short segments of sequence similarity with the Cdc25 phosphatase catalytic domain. As dual-specificity phosphatases are activated by stimuli that trigger MAP kinase pathways (such as heat shock, hypoxia, and mitogens), it has been proposed that DSPs negatively regulate MAP kinase signaling (8, 25, 29). DSPs are able to dephosphorylate one or more MAP kinases. For example, MKP-3 dephosphorylates ERK, M3/6 dephosphorylates JNK, MKP-1 dephosphorylates JNK, ERK, and p38 (16), and MKP-8 (DUSP26) was recently shown to dephosphorylate p38 (43). Numbers of DSPs (MKP-5/DUSP10, MKP-X/DUSP7, and MKP-2/DUSP4) are located in the human genome that are often found lost in tumors, suggesting that some of these phosphatases may possess tumor suppressor properties (16).
Many MAP kinases are known to bind scaffold proteins (such as JIP1-3, β-arrestin-2, MP1, and KSR) (24, 28, 39). These scaffold proteins act to control the activity of MAP kinases and thus ultimately regulate phosphorylation of downstream MAP kinase targets. For example, β-arrestin-2 acts as a scaffold protein which controls the activity of JNK3 and thereby desensitization of the heterotrimeric guanine nucleotide binding protein-coupled receptors (10).
Here, we show that Hsf4b binds to a dual-specificity tyrosine phosphatase similar to a group of DSPs that lack the amino-terminal CH2 domain. We also show that Hsf4b is phosphorylated by ERK1/2. Coexpression of Hsf4b with DUSP26 leads to dephosphorylation of Hsf4b as well as a reduction in its ability to bind HSEs. Hsf4b is not directly dephosphorylated by DUSP26, but Hsf4b interaction with DUSP26 brings this phosphatase into close proximity of an ERK-binding region on Hsf4b, leading to control of the activities of both ERK and Hsf4b. Therefore, DUSP26 interaction with Hsf4b places this transcription factor in a regulatory circuit within the MAP kinase signaling pathway.
METHODS AND MATERIALS
Plasmids and constructs.
The plasmid pSos-Hsf4b was constructed by PCR amplification of Hsf4b cDNA using human Hsf4b as a template, followed by insertion of the fragment into pSos bait vector using MluI and NcoI restriction enzymes. The plasmids pGEX-Hsf4b, pGEX-Hsf4b (1-230), pGEX-Hsf4b (1-323), and pGEX-Hsf4b (196-493) were generated by using PCR amplification of human Hsf4b cDNA as a template. PCR products were inserted into the pGEX-2T vector using BamHI and EcoRI restriction enzymes. The constitutively active form of Xenopus MEK in the vector pcDL-SRσ 296 was the gift of M. Iwashima (Medical College of Georgia). The plasmid pcDNA3-Flag (NH2-terminal)-DUSP26 was constructed using DUSP26 cDNA from the plasmid pMyr-DUSP26 that was isolated from the yeast two-hybrid screening using the restriction enzymes KpnI and XhoI. The expression vector DUSP26-EGFP was constructed by insertion of PCR-amplified DUSP26 cDNA into plasmid pEGFP-N1 (Clontech) at BamHI and EcoRI restriction enzyme sites. The plasmid pcDNA3-Flag-Hsf4b was generated by inserting a Flag tag sequence at the NH2 terminus of Hsf4b cDNA followed by its insertion into pcDNA3 plasmid. The plasmids pEBG-Hsf4b (1-230), pEBG-Hsf4b (1-323), pEBG-Hsf4b (196-493), pEBG-Hsf4b (246-323), and pEBG-Hsf4b were constructed by inserting the corresponding Hsf4b cDNA fragments into the vector pEBG (gift of R. Feig, Toft University School of Medicine) using BamHI and ClaI restriction sites. The plasmid pEBG-Hsf4b allows expression of the glutathione S-transferase (GST)-Hsf4b fusion protein in mammalian cells.
Yeast two-hybrid screening.
For yeast two-hybrid screening, we used a CytoTrap two-hybrid system (Stratagene) as described previously (3, 22). The full-length human Hsf4b was subcloned into the pSos vector and was used to screen a human heart tissue cDNA library that had been subcloned into the pMyr vector. Ten micrograms of the bait construct and 10 μg of the cDNA library were cotransfected into the competent yeast Cdc25H strain. The transformed yeast were plated into SD-(uracil-leucine-deficient) glucose plates and allowed to grow at 25°C for 48 to 72 h. Yeast colonies that appeared on the plates were replica plated on SD-(uracil-leucine-deficient) galactose and SD-(uracil-leucine-deficient) glucose plates and incubated at 37°C for 6 days. Colonies positive for growth on galactose but not glucose were grown in 5 ml of SD-glucose broth at 25°C. Plasmid DNA was extracted, and transformed Escherichia coli DH5α and selected on chloramphenicol-containing agar plates. Plasmid DNA was isolated and sequenced. All control plasmids were supplied by the manufacturer and used as positive controls.
Cell culture and heat treatment.
The H1299 human lung carcinoma cell line was maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Cultured cells were heated at 80% confluence in a circulating water bath, as appropriate.
Transient-transfection assays.
Transient transfections were performed using Trans IT-Lt1 (Mirus Corporation). Transfected DNA mixes included 4 to 8 μg of expression plasmid DNA and, when required, empty plasmid DNA added to a total of 8 μg. The DNA mix was added to 5 ×105 to 6 ×105 cells. The transfection efficiency varied between 60 to 70% in all experiments, as determined by immunofluorescence analysis (22).
Immunoprecipitation, immunoblotting, immunocomplex kinase assays, pull-down assays.
Cells (6 × 105) were cotransfected with appropriate plasmids, allowed to recover for 48 h, rinsed with phosphate-buffered saline (PBS), and appropriately treated and harvested. Cells were lysed with lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris, pH 7.5, containing 1× cocktail of protease inhibitors [Sigma]). The protein concentration of the supernatant was estimated using a bicinchoninic acid protein assay kit (Bio-Rad). One milligram of each of the cell lysates was mixed with 40 μl of a 50% solution of protein A-agarose and incubated at 4°C for 1 h. The protein A-agarose was then centrifuged, and the precleared supernatant was incubated with 2.5 μg of primary antibody and incubated at 4°C for 2 h or overnight. Forty microliters of 50% solution of protein A-agarose was then added at 4°C, for 2 h. The protein A complexes were centrifuged at 10,000 × g for 1 min, and the pellet was washed with lysis buffer three times. One hundred microliters of 2× sodium dodecyl sulfate (SDS) sample buffer was added, and samples were heated at 100°C for 5 min. Samples were fractionated on SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by immunoblotting using appropriate antibodies. The membrane was immunoblotted using one of the following primary antibodies as described in the text. The corresponding horseradish peroxidase-conjugated secondary antibodies were used, and signals were developed using the enhanced chemiluminescence method (ECL kit; Amersham Pharmacia) (22). For immunocomplex kinase assays, H1299 cells were treated as described in the text. Cells were then lysed in lysis buffer (25 mM Tris-HCl, pH 7.4, 15 mM NaCl, 1.5 mM MgCl2, 2 mM EGTA, 125 mM sodium phosphate, 1% Triton X-100, 10 mM NaF, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM sodium vanadate, and 20 μg/ml aprotinin). The protein kinases were immunoprecipitated by incubation with 2 μg of appropriate antibodies (polyclonal rabbit antibody to ERK1/2, JNK1, or p38) (Santa Cruz) as described above. Immunoprecipitated samples were rinsed twice with buffer containing 20 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 1 mM PMSF, 2 mM dithiothreitol (DTT), and 1 mM sodium orthovanadate and then rinsed once with Tris buffer (Tris-HCl, pH 7.4). The beads were then mixed with 5 μg of the substrate in kinase reaction buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 100 μM ATP, 2 μCi of [γ-32P]ATP) at 37°C for 15 min. The reaction was stopped by the addition of SDS sample buffer. Substrate phosphorylation was detected by autoradiography after SDS-PAGE (19).
For in vitro pull-down assays, 800 μg of cell lysate prepared in buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 10% glycerol) following transient transfection with expression plasmids encoding Flag-Hsf4b were incubated with 20 μl of 50% slurry of glutathione-conjugated Sepharose 4b beads, and precleared supernatant was then incubated with 20 μg of purified GST-DUSP26 expressed in E. coli BL21 and incubated at 4°C for 6 h. The GST-DUSP26 proteins were precipitated with the 50 μl of a 50% slurry of glutathione-conjugated Sepharose 4b beads. After rinsing three times with lysis buffer and once in buffer containing 50 mM Tris-HCl, pH 7.4, 200 mM NaCl, beads were boiled in the SDS sample buffer. Proteins were separated via SDS-PAGE, transferred to the membrane, and immunoblotted. For in vivo pull-down assays, H1299 cells were cotransfected with expression construct pEBG-Hsf4b (1-230), pEBG-Hsf4b (1-323), pEBG-Hsf4b (196-493), or pEBG-Hsf4b (full-length) together with plasmids containing DUSP26-enhanced green fluorescent protein (EGFP) fusion protein or pcDNA3-Flag-DUSP26. Cells were lysed after 48 h in buffer (as above), and 800 μg of cell lysate was incubated with 30 μl of a 50% slurry of glutathione-conjugated Sepharose 4B beads. Beads were washed three times, and bound proteins were separated on 12% SDS-PAGE gels as described above and immunoblotted. Antibodies to Flag, hemagglutinin (HA), phospho-JNK, and GST were purchased from Sigma.
Phosphatase assay.
Purified protein prepared from the construct encoding GST-DUSP26 was incubated at indicated concentrations for increasing times at 37°C in a reaction buffer containing 50 mM imidazole (pH 7.5), 20 mM p-nitrophenol phosphate (pNPP), and 5 mM dithiothreitol. The reaction was stopped by the addition of 0.1 N NaOH, and pNPP hydrolysis was measured by absorbance at 405 nm (15).
Gel mobility shift assays.
Electrophoretic mobility shift analysis (EMSA) using whole-cell extracts has been described previously (19). Briefly, after each treatment, cells were rinsed with PBS and lysed in 100 μl of extraction buffer (10 mM HEPES, pH 7.9, 0.4 mM NaCl, 0.1 mM EDTA, 0.5 mM DTT, 5% glycerol, 0.5 mM PMSF). The protein concentration of samples was estimated by the bicinchoninic acid method (Pierce). Equal amounts of protein (10 μg) in extraction buffer (volume not exceeding 15 μl) were added to the reaction mixture, which contained 4 μl of binding buffer (37.5 mM NaCl, 15 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 0.5 mM DTT, 5% glycerol), 10 μg of yeast tRNA, 1 μg of sheared E. coli DNA, 10 μg of poly(dI-dC), and 1 ng of 32P-labeled HSE oligonucleotide. The mixture was incubated for 15 min at 25°C and resolved on a 4.5% nondenaturing polyacrylamide gel. After electrophoresis, gels were fixed in 7% (vol/vol) acetic acid for 5 min, rinsed once in distilled water, dried under a vacuum, and exposed to X-ray film. The nucleotide sequence used for HSE was as follows: 5′-GTCGACGGATCCGAGCGCCTCGAATGTTCTAGAAAAGG-3′. The double-stranded oligonucleotide was labeled using Klenow fragment of DNA polymerase I, deoxynucleotide triphosphates, and [α-32P]dCTP.
Two-dimensional single amino acid analysis.
H1299 cells were cotransfected with expression plasmids pcDNA3-Flag-Hsf4b and pcDNA3-HA-DUSP26. At 36 h after transfection, cells were washed twice with serum-free and phosphate-free Dulbecco's modified Eagle's medium. Cells were incubated in phosphate-free medium for 1 h prior to labeling with 200 μCi/ml of 32P for 4 h at 37°C. Cells were washed twice with PBS and lysed in NP-40 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 10 mM NaF, 1 mM PMSF, 2 mM sodium vanadate, and 20 μg/ml aprotinin). The 32P-labeled Flag-Hsf4b was immunoprecipitated using antibody to Flag. The immunoprecipitated complex was then separated by SDS-PAGE, transferred to a membrane, and exposed to X-ray film to quantitate the labeled Hsf4b. The area of the membrane containing the 32P-labeled Flag-Hsf4b was excised, rinsed with methanol for 30 s and then water for 30 s, treated with 6 N HCl, and boiled at 100°C for 60 min. Samples were dried, and Cerenkov counts were determined. Samples were resuspended in 5 to 10 μl of buffer (pH 1.9) loaded onto thin-layer chromatography plates and resolved by chromatography using 1.5-kV constant voltage for 20 min in buffer containing formic acid (pH 1.9), glacial acetic acid, and deionized water (1:3:36). The plates were rinsed and resolved in buffer containing glacial acetic acid, pyridine, and deionized water (pH 3.5) (1:10:189) for 16 min. Unlabeled standard amino acids were visualized 30 min after spraying the plates with 0.25% ninhydrin in acetone at 65°C. The thin-layer chromatography plates were exposed to X-ray film and quantitated using PhosphorImager analyses (6).
RESULTS
Hsf4b interacts with a dual-specificity tyrosine phosphatase.
To examine the molecular mechanisms controlling the transcriptional activity of Hsf4b, we used Hsf4b as a bait in a yeast two-hybrid screen and identified a dual-specificity tyrosine phosphatase named DUSP26 (genomic designation, also known as MKP-8, MGC1136, MGC2627, NATA1, and SKRP3) interacting with Hsf4b (Fig. 1A to C). DUSP26 contains 211 amino acids with a predicted molecular mass of 22 kDa (43). DUSP26 is located in human chromosome 8p12 and contains a carboxyl-terminal signature motif for the DSPs, but lacks the amino-terminal Rhodanase fold or the Cdc25 homology domain (CH2 domain) (Fig. 1B and C) (8, 25). DUSP26 is therefore in the same class as JSP-T, hSKRP1, hVHR (DUSP3), and MKP-6 (DUSP14) dual-specificity phosphatases that also lack the Rhodanase fold required for direct docking to MAP kinase family members (Fig. 1C) (2, 8, 25, 50).
FIG. 1.

Identification of a dual-specificity tyrosine phosphatase interacting with Hsf4b. (A) Schematic diagram of full-length Hsf4b cDNA fused to pSos-Hsf4b bait plasmids used in two-hybrid screening to isolate Hsf4b interacting protein. DBD is the DNA binding domain; HRA/B are the amino-terminal hydrophobic heptad repeats. DHR is downstream of C-terminal hydrophobic heptad repeats (HR/C) present in other Hsfs but is missing in Hsf4 (34, 47). Lower panel: yeast two-hybrid screen showing Hsf4b interaction with DUSP26. First row: yeast containing pSos-Hsf4b and pMyr-DUSP26. Second row: yeast containing pSos-MafB and pMyr-MafB (positive control). Third row: yeast containing pSos and pMyr-cDNA library (negative control). Galactose induces the expression of the proteins encoded by pMyr plasmids. The yeast strain used here contains a temperature-sensitive mutation in the yeast homolog of Sos. Introduction of a Sos fusion protein with membrane localization myristylation protein rescues cell growth at 37°C. (B) The signature motif conserved in the DUSP26 and other DSPs is shaded/underlined. (C) DUSP26 structure, showing that it contains the carboxyl-terminal catalytic domain and lacks the CH2 domain (43). Structures of the few DSPs so far identified have also been presented for comparison. (D) In vivo-expressed GST-Hsf4b interacts with Flag-DUSP26. Lysates from H1299 cells expressing Flag-DUSP26 were incubated with cell lysate expressing full-length GST-Hsf4b or deletion mutants. GST pull-down products were resolved on SDS-PAGE and immunoblotted using antibody to Flag to detect DUSP26 (upper panel). Lower panel shows immunoblotting of lysates expressing constructs indicated 1 through 4 that were used for the pull-down experiment. The expression of β-actin is presented as a control. (E) GST-DUSP26 interacts with Hsf4b using in vitro pull-down assays. Purified GST-DUSP26 protein (lane 1) or GST (lane 2) was used to pull-down Hsf4b from cell lysates of H1299 cells expressing Hsf4b. Upper panel shows immunoblotting experiments to detect Hsf4b pulled down with GST-DUSP26. Middle panels show immunoblotting (IB) of cell lysates expressing Hsf4b used in pull-down assay and β-actin as loading control. Lower panel shows Coomassie blue staining of purified GST-DUSP26 and GST proteins. (F) Hsf4b and DUSP26 interact in vivo. H1299 cells were transiently cotransfected with EGFP, DUSP26-EGFP fusion protein, or Flag-Hsf4b. Coimmunoprecipitation experiments were performed with cell lysates from control cells expressing plasmids encoding EGFP and Flag-Hsf4b (lane 1) or cells expressing plasmids encoding DUSP26-EGFP and Flag-Hsf4b (lane 2) using an antibody to Flag. DUSP26-EGFP was detected using an antibody to EGFP. Lanes 3 and 4 show immunoblotting using antibodies to EGFP from cell lysates expressing DUSP26-EGFP or EGFP, respectively. Lower panel shows the expression of Flag-Hsf4b in cell lysates.
To confirm that Hsf4b and DUSP26 can interact in vitro or in vivo when expressed in mammalian cells, we performed in vivo (Fig. 1D) and in vitro (Fig. 1E) GST pull-down assays and coimmunoprecipitation (Fig. 1F) experiments. The result presented in Fig. 1D indicates that only full-length Hsf4b and a mutant form of Hsf4b encoding amino acid residues 196 to 493 interact with DUSP26 (Fig. 1D, lanes 3 and 4). An Hsf4b mutant spanning amino acid residues 1 to 230 failed to interact with DUSP26 (Fig. 1D, lane 2). The results presented in Fig. 1E indicate that purified GST-DUSP26 protein can pull down Hsf4b from cell lysates in vitro, while no interaction was observed using purified GST protein alone. The interaction of Hsf4b and DUSP26 in mammalian cells was also confirmed using coimmunoprecipitation experiments. The results showed that Hsf4b and DUSP26 fused to EGFP interact in vivo, while Hsf4b did not interact with plasmids encoding EGFP alone (Fig. 1F). Similarly, coimmunoprecipitation experiments using H1299 cells coexpressing Hsf4b and HA-DUSP26 showed that Hsf4b interacts with HA-DUSP26 in vivo (data not shown).
The finding that Hsf4b binds a predicted DSP raises the possibility that Hsf4b activity may be regulated by phosphorylation. Thus, it was necessary to establish that DUSP26 possesses protein phosphatase activity and that Hsf4b exists as a phosphoprotein within cells. Hence, DUSP26 phosphatase activity was determined in vitro. The results show that GST-DUSP26 and not the mutant form of GST-DUSP26, with cysteine-to-alanine substitution at a critical residue conserved within this family of phosphatases lacked any phosphatase activity (Fig. 2A and B). The quantities of the purified GST, wild-type GST-DUSP26, and mutant GST-DUSP26 (C152A) proteins are presented in Fig. 2C. A comparative analysis in terms of DUSP26 activity per microgram of protein toward pNPP used as a substrate indicate that DUSP26 is at least as active as other DSPs such as SKRP1 (50).
FIG. 2.
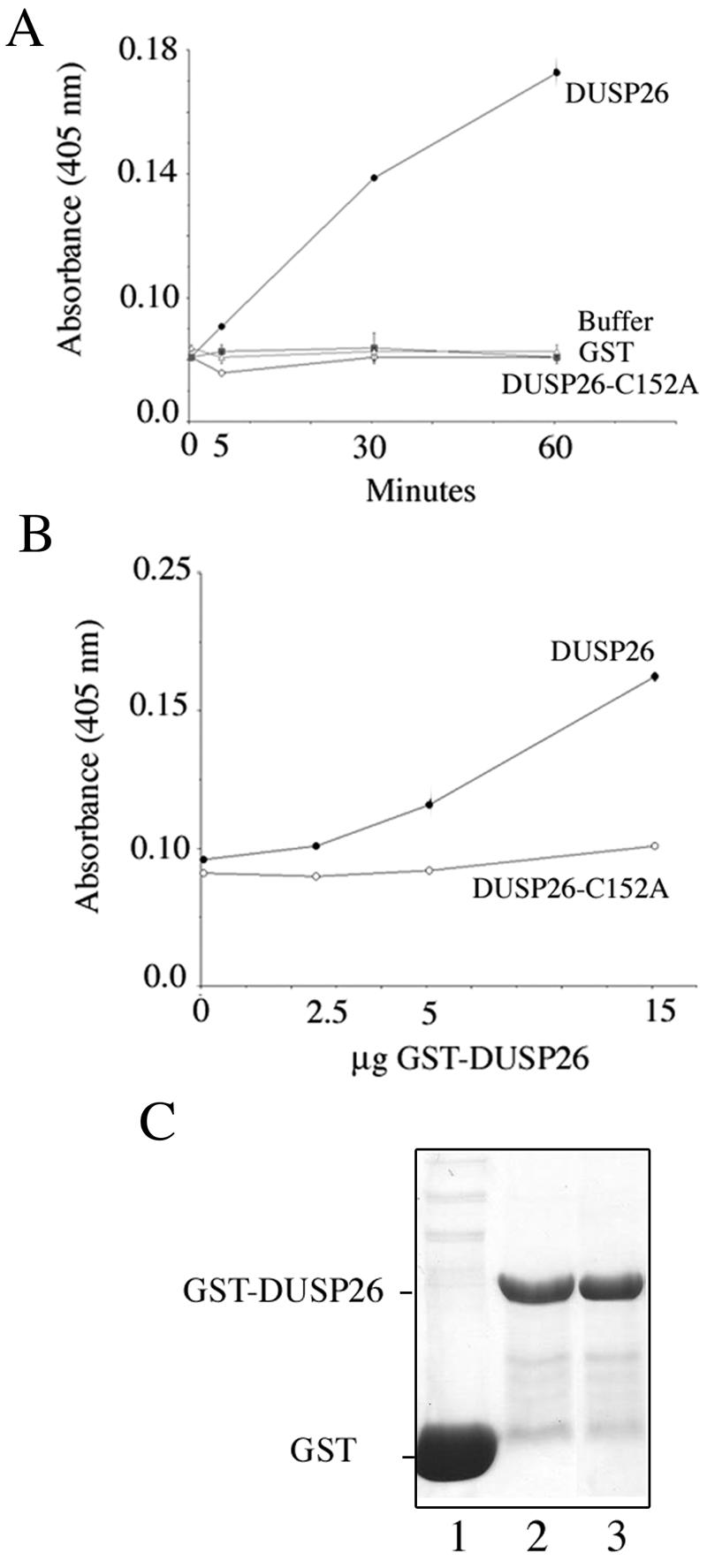
DUSP26 possesses phosphatase activity. (A) Fifteen micrograms of bacterially expressed and purified GST-DUSP26 or GST-DUSP26 (C152A) mutant was used to determine phosphatase activity towards pNPP following incubation at 37°C for 0 to 60 min (15). The color reaction was quantitated at a 405-nm wavelength. Control reactions were also performed using buffer or GST alone. (B) Reaction conditions were the same as for panel A, but 0, 2.5, 5, or 15 μg of GST-DUSP26 or GST-DUSP26-C152A fusion protein was added to each reaction mixture and incubated at 37°C for 30 min. (C) Coomassie blue staining of purified GST or GST fusion proteins. Lane 1 is purified GST alone. Lane 2 is purified wild-type GST-DUSP26. Lane 3 is purified GST-DUSP26 with C152A mutation.
Hsf4b is a phosphoprotein, and phosphorylation alters its ability to bind heat shock element.
To determine the phosphorylation status of Hsf4b in the presence of DUSP26, H1299 cells were cotransfected with Hsf4b and increasing amounts of plasmids encoding DUSP26. As expected, Hsf4b coexpressed with higher concentrations of DUSP26 resolved in two separate bands on SDS-PAGE (Fig. 3A). These results suggest that Hsf4b is phosphorylated but becomes dephosphorylated in the presence of DUSP26. To examine the phosphorylation state of Hsf4b, we performed immunoblotting experiments using antibody to phosphothreonine, phosphoserine (antibody not sufficient quality) (data not shown), and phosphotyrosine as well as phosphoamino acid analyses (Fig. 3B and 3C). To detect possible phosphorylation of Hsf4b by immunoblotting, cells were transiently transfected with Hsf4b and treated with increasing concentrations of sodium vanadate, a general protein tyrosine phosphatase inhibitor. Hsf4b was immunoprecipitated, and immunoblotting experiments were performed using antibody to phosphorylated amino acids. The results indicate that Hsf4b is phosphorylated on threonine residues but not on tyrosine residues (Fig. 3B). More direct evidence for Hsf4b phosphorylation status was obtained by phosphoamino acid analysis experiments. The results show that 32P-Hsf4b was phosphorylated on serine and threonine residues. Additionally, cotransfection of DUSP26 together with Hsf4b resulted in marked reduction of 32P-Hsf4b. No tyrosine phosphorylation was detected on Hsf4b (Fig. 3C). The amount of Hsf4b expression in cells in the absence of DUSP26 (Fig. 3C, lower panel, lane 1) or presence of DUSP26 (Fig. 3C, lower panel, lane 2) is shown following immunoblotting. Note that Hsf4b levels in cells are reduced when it is coexpressed with DUSP26 (compare Fig. 3C, lower panel, lanes 2 and 1).
FIG. 3.

Hsf4b is phosphorylated on serine and threonine residues. (A) H1299 cell lysates expressing empty vector or Flag-Hsf4b and different amounts of plasmids encoding HA-DUSP26 were analyzed by immunoblotting using antibody to Flag-Hsf4b, HA-DUSP26, or β-actin. (B) Hsf4b is a threonine-phosphorylated protein. H1299 cells were transiently transfected with Flag-Hsf4b before treatment with 0 to 100 μM of sodium vanadate for 2 h. Flag-Hsf4b was immunoprecipitated (500 μg of lysate) using anti-Flag antibody and visualized with antibodies to Flag-Hsf4b, phosphothreonine, or phosphotyrosine as indicated. β-Actin is shown to indicate that an equal amount of cell lysate was used for immunoprecipitation. (C) H1299 cells were transiently transfected with plasmids containing Flag-Hsf4b, with or without cotransfection with plasmids containing HA-DUSP26. Following 3 h of metabolic labeling with 32P at 37°C, 32P-Flag-Hsf4b was immunoprecipitated from equal amounts of cell lysate using antibody to Flag, and immunoprecipitated material was prepared for phosphoamino acid analysis in either the absence (left panel) or presence (right panel) of coexpressed HA-DUSP26. The positions of the phosphorylated threonine, serine, or tyrosine are indicated by arrowheads. Lower panel: immunoblotting experiments showing expression of Flag-Hsf4b (lanes 1 and 2) and HA-DUSP26 in the cell lysate. Hsf4b usually appears to be lower in quantity (dephosphorylated) when coexpressed with DUSP26 (lane 2). β-Actin is shown to indicate an equal amount of cell lysate in each lane. (D) DUSP26 affects Hsf4b binding to HSE. H1299 cells were cotransfected with Flag-Hsf4b and increasing amounts of HA-DUSP26 (same samples as in panel A, lanes 1 to 4). After 48 h, 10 μg of cell lysate was analyzed by EMSA. Lane 5 is the same cell lysate as in lane 1 plus antibody to Flag to show supershift of Hsf4b. Expression of proteins expressed in the cell lysates are as in panel A.
Phosphorylation or dephosphorylation of transcription factors has been postulated to regulate nuclear translocation, DNA binding, and transcriptional activity. Since we show evidence that Hsf4b is a phosphorylated protein and can be dephosphorylated in the presence of DUSP26, we asked whether dephosphorylation of Hsf4b affects its DNA binding ability. Consistent with previous observations, these results indicate that Hsf4b binds HSE constitutively when expressed in cells (Fig. 3D, lane 1). Interestingly, Hsf4b's DNA binding ability was reduced threefold when cells were cotransfected with increasing concentrations of expression constructs harboring DUSP26 (Fig. 3D, lane 4). These data indicate that dephosphorylation of Hsf4b in the presence of DUSP26 reduces its ability to bind HSE, a regulatory mechanism for Hsf4b transcriptional ability.
We also examined whether DUSP26 has any effect on the endogenous or overexpressed Hsf4b intracellular localization by immunostaining. Endogenous or overexpressed Hsf4b expression was detected mainly in the nuclei in the absence of DUSP26-EGFP or HA-DUSP26. However, some colocalization of Hsf4b and DUSP26 was observed both in the nucleus and in the perinuclear region in 95% of the cells (data not shown).
Since Hsf4b is a transcription factor and the targets for Hsf4b exist in the nucleus, we propose that DUSP26 could potentially act to inhibit Hsf4b transcriptional ability by promoting Hsf4b cytosolic localization.
Hsf4b is phosphorylated by MAP kinases, and its phosphorylation is indirectly regulated by DUSP26.
Among others, data presented in Fig. 3 suggest either that Hsf4b can be directly dephosphorylated by DUSP26 or that DUSP26 may inactivate a kinase, such as one of the MAP kinase family members which are known substrates of DSPs, and thereby reduce phosphorylation of Hsf4b by such kinases. To address these possibilities, we first tested whether in vivo-labeled 32P-Hsf4b can be dephosphorylated by immunoprecipitated DUSP26 in vitro. The quantitation of the data shown in Fig. 4 indicate a twofold reduction in the 32P-Hsf4b in cells expressing DUSP26, but the amount of Flag-Hsf4b that was immunoprecipitated was also twofold lower, indicating that DUSP26 does not directly dephosphorylate Flag-Hsf4b.
FIG. 4.
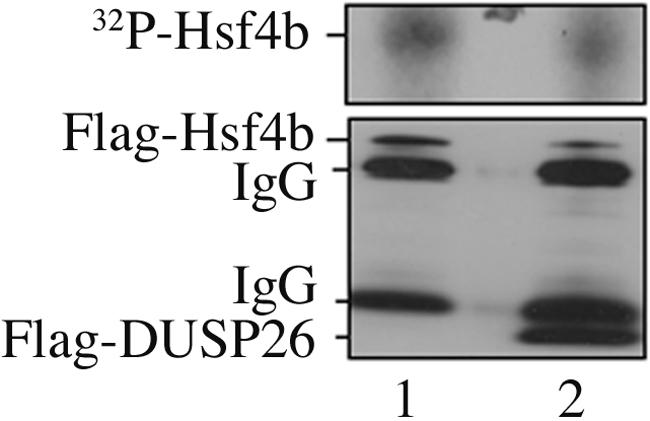
Hsf4b is not a direct target of DUSP26 for dephosphorylation. H1299 cells were transiently transfected with plasmids encoding Flag-Hsf4b, and cells were metabolically labeled with 32P for 4 h at 37°C. 32P-Hsf4b was immunoprecipitated using antibody to Flag, with one group used as a control (lane 1), and the second incubated with immunoprecipitated Flag-DUSP26 in vitro for 30 min at 25°C (lane 2). The mixture was analyzed by SDS-PAGE, and the gel was exposed to X-ray film to detect 32P-Hsf4b (upper panel). Separate fractions were analyzed by immunoblotting to Flag to detect immunoprecipitated Hsf4b or DUSP26. Note that the reduction in 32P counts in the Hsf4b plus DUSP26 sample in the upper panel was twofold reduced but was proportional to the twofold reduction (1150 cpm versus 490 cpm) in the immunoprecipitated Hsf4b detected in the immunoblotting shown in the lower panel.
Since the DSPs so far identified target one or more of the MAP kinase family members, we performed immunocomplex kinase assays using GST-Hsf4b or GST-Jun as a target to determine the substrate specificity of DUSP26 among the MAP kinases. We show that GST-Hsf4b is not efficiently phosphorylated by ERK1/2 under physiological conditions but can be phosphorylated by ERK when ERK1/2 is immunoprecipitated from cells transiently expressing a constitutively active form of Mek (Fig. 5A and B). Furthermore, the ability of ERK to phosphorylate Hsf4b is reduced when ERK1/2 is immunoprecipitated from cells expressing both constitutively active Mek and DUSP26 (Fig. 5A and B). Similarly, both GST-Jun and GST-Hsf4b can be phosphorylated by JNK1 immunoprecipitated from cells following heat shock, which is known to activate JNK (Fig. 5C) (13). Furthermore, JNK1 immunoprecipitated from heated cells expressing DUSP26 showed a seven- and fourfold reduction in JNK1 activity, as detected by its ability to phosphorylate GST-Jun or GST-Hsf4b, respectively, in vitro. These results indicate that Hsf4b is the substrate of both MAP kinases ERK1/2 and JNK1. Additionally, our results suggest that DUSP26 may effectively regulate JNK1 phosphorylation of c-Jun or Hsf4b. Cell lysates used for immunoprecipitation experiments were also used to detect the phosphorylated forms of JNK1 (Fig. 5D), showing an increase in 46- and 52-kDa phospho-JNK1 following exposure of the cells to heat shock, and a reduction in phospho-JNK1 when DUSP26 is coexpressed in cells (Fig. 5D, compare lanes 3 and 4). These results suggest that DUSP26 is a phosphatase that appears to be specific to both ERK1/2 and JNK1 MAP kinases. We also determined whether Hsf4b is a substrate for p38 MAP kinase in vitro, and whether DUSP26 can affect p38 activity in vivo. Our results show that although Hsf4b can be phosphorylated by p38 in vitro, DUSP26 does not affect the activity of p38 toward Hsf4b when DUSP26 was expressed in cells pretreated with sorbitol to activate the p38 signaling pathway (data not shown). The fact that DUSP26 regulates the activity of both ERK1/2 and JNK1 but not of p38 is not surprising, since all of the known dual-specificity tyrosine phosphatases target one or more MAP kinase family members (25).
FIG. 5.

Hsf4b is phosphorylated by MAP kinase family members in vitro. (A and B) Immune complex kinase assays. H1299 cells were transiently transfected with (+) or without (−) plasmids containing Flag-DUSP26 or constitutive active Mek to activate ERK1/2. At 48 h after transfection, ERK1 was immunoprecipitated and used in an immune complex kinase assay using Hsf4b (residues 196 to 493) as a substrate. (B) Immunoblotting showing the expression of endogenous (upper band) and expressed (lower band) Mek, ERK1/2, and Flag-DUSP26 in the cell lysates used. Lanes 1 to 3 correspond to the same samples in panels A and B. (C and D) Immune complex kinase assays. H1299 cells were transiently transfected with or without plasmids containing Flag-DUSP26 as indicated. At 48 h after transfection, cells were left at 37°C or exposed to 45°C for 30 min to activate JNK1. JNK1 was immunoprecipitated and used in an immune complex kinase assay using purified GST-Jun or GST-Hsf4b as a substrate. Cell lysates from panel C were immunoblotted with antibody to phosphorylated JNK1, total JNK1, or Flag to detect DUSP26 (D). Lanes 1 to 4 corresponds to the same samples in panels C and D.
As MAP kinase signaling pathways are involved in regulation of cell growth, differentiation, and survival (14), we wished to determine whether activators of MAP kinases, ERK, JNK, and p38 have an effect on Hsf4b protein. Thus, we treated cells transiently expressing Hsf4b with serum following serum deprivation or with anisomycin or sorbitol. Addition of serum to cells leads to stimulation of growth factor receptors and activation of ERK1/2, while treatment of cells with anisomycin or sorbitol activates JNK1/2, and p38, respectively. Cell lysates were immunoblotted to detect Hsf4b. The results indicate that there are over twofold increases in the level of Hsf4b protein following treatment of cells with serum or sorbitol (Fig. 6A). However, following activation of the JNK signaling pathway, Hsf4b protein levels remained relatively unchanged. These data suggest that phosphorylation of Hsf4b by ERK1/2 or p38 can stabilize Hsf4b protein (Fig. 6A). However, the activation of JNK signaling pathway does not have the same effect on Hsf4b as do the ERK1/2 and p38. These results indicate that although Hsf4b may be phosphorylated by multiple MAP kinase family members, Hsf4b regulation by individual MAP kinases differ. Interestingly, following activation of the ERK and p38 signaling pathways where the amount of Hsf4b is elevated, a higher-molecular-mass species is detected in the Hsf4b immunoblots (Fig. 6A), suggesting that Hsf4b is modified further following activation of MAP kinase ERK and p38 signaling pathways.
FIG. 6.

Hsf4b's ability to bind HSE is altered following activation of the MAP kinase signaling pathway. (A) H1299 cells were transiently transfected with plasmids containing Flag-Hsf4b. Forty-eight hours later, cells pretreated with medium supplemented with 0.5% serum for 20 h were then treated with medium plus 10% serum for 20 min to activate ERK1/2, or cells were treated with anisomycin (20 μg/ml) or 7 mM sorbitol for 20 min to activate JNK or p38, respectively. Immunoblotting was performed to detect Hsf4b using antibody to Flag. β-Actin was used as a control for equal loading. (B) H1299 cells were transiently transfected with plasmids containing Flag-Hsf4b and cotransfected (+) with plasmids containing HA-DUSP26 or Mek to activate ERK1/2. Cells lysates were used in EMSA to detect Hsf4b's binding ability to HSE (upper panel) or in immunoblotting assays to detect Hsf4b using antibody to Flag (lower panel). Lanes 1 to 4 in the upper panel correspond to lanes 1 to 4 in the lower panel. Lanes 5, 6, and 7 in the upper panel are the same as lane 1, but cell lysates were incubated for 20 min in the presence of antibody to Hsf1, Flag-Hsf4, or Hsf2, respectively, to confirm specificity of Hsf4b for HSE.
Since, among the MAP kinases, we found that only ERK1/2 can bind Hsf4b and phosphorylate it, leading to increased levels of Hsf4b protein, and that ERK1/2 also can be regulated by DUSP26, we focused our studies to determine the role of ERK1/2 and DUSP26 in Hsf4b regulation. Thus, to determine whether activators of ERK1/2 signaling pathways affect Hsf4b DNA binding activity, H1299 cells were transiently cotransfected with expression constructs harboring Hsf4b, a constitutively active Mek, or DUSP26. The ability of Hsf4b to bind HSE was determined by EMSA, and expression of Hsf4b was detected by immunoblotting. The data indicate that while Hsf4b DNA binding activity increased by 30% in cells expressing both Hsf4b and constitutively active Mek (Fig. 6B, compare lanes 2 and 1), Hsf4b activity was reduced in cells expressing DUSP26 and Hsf4b by 50% (Fig. 6B; compare lanes 4 and 1) or in cells coexpressing DUSP26, Hsf4b, and constitutively active Mek by 30% (Fig. 6B, compare lanes 3 and 1). These data indicate that activated ERK1/2 and DUSP26 regulate Hsf4b DNA binding activity when coexpressed in H1299 cells.
ERK MAP kinase and DUSP26 bind Hsf4b.
DUSP26 lacks the CH2 domain, and as predicted, it does not bind ERK1/2 directly, and this was confirmed by coimmunoprecipitation experiments (see Fig. 8, below). However, since DUSP26 appears to affect ERK1/2 activity toward Hsf4b, we hypothesized that DUSP26, while bound to Hsf4b, may regulate the activity of ERK1/2. Thus, coimmunoprecipitation experiments were performed to detect the interaction between Hsf4b and ERK1/2. Results show that Hsf4b interacts with ERK1/2 in vivo (Fig. 7A). To determine the region of Hsf4b interacting with ERK1/2, in vivo-expressed full-length GST-Hsf4b or deletion mutants were used to pull down ERK1/2, which was detected by immunoblotting. The results presented in Fig. 7B indicate that full-length Hsf4b and Hsf4b residues 196 to 493 or 1 to 323 were sufficient to pull down ERK1/2. Since Hsf4b residues 1 to 230 were not required for ERK1/2 binding, we concluded that residues 196 to 493 are sufficient for ERK1/2 binding to Hsf4b. The full-length Hsf4b sequence contains three potential MAP kinase binding motifs: the first is located between amino acid residues 207KRKLSLML214 (critical amino acids are underlined); the second is between residues 285RREKGLAL292; the third is located between amino acid residues 361DRGPLGLES369. In vivo GST pull-down experiments using a mutant Hsf4b where the first MAP kinase binding motif had been disrupted by 207GRGLSLML214 indicate that interaction of ERK1/2 with Hsf4b was not affected, suggesting that this domain is not required for ERK1/2 binding to Hsf4b (data not shown). We then performed in vivo GST pull-down experiments where we used wild-type or mutated Hsf4b containing various fragments encoding MAP kinase binding region two or three, mentioned above. Our results show that ERK1 binds to Hsf4b containing amino acid residues 196 to 493 (Fig. 7C, lane 2), 323 to 371 (Fig. 7C, lane 4), and 246 to 323 (Fig. 7C, lane 6), encoding both MAP kinase binding regions. In addition, mutations of the critical amino acid residues in regions two (285GGEKGLAL292) (Fig. 7C, lane 5) and three (361DRGPAGAES369) (Fig. 7C, lane 3) lead to abolishment of binding of ERK1 to Hsf4b. These data suggest that ERK1 can bind Hsf4b in two different regions.
FIG. 8.

Hsf4b interacts with both ERK1 and DUSP26. H1299 cells were transiently transfected with plasmids containing GST alone or GST-Hsf4b (196 to 493) and cotransfected with plasmids encoding HA-ERK1 and Flag-DUSP26 as indicated. At 48 h after transfection, cell lysates were subjected to immunoprecipitation experiments using antibody to Flag-DUSP26 (left panels). The immunoprecipitated proteins were subjected to immunoblotting using antibodies to HA to detect ERK1 or to GST to detect GST alone or GST-Hsf4b. Right panels presents immunoblotting of cell lysates to detect the expression of HA-ERK1, Flag-DUSP26, and GST or GST-Hsf4b using antibodies to HA, Flag, or GST, respectively. Lanes 1, 2, and 3 indicate the same groups in the left and right panels.
FIG. 7.

ERK1/2 and DUSP26 interact with Hsf4b. (A) Hsf4b interacts with endogenous ERK1/2 in vivo. H1299 cells were transiently transfected with plasmids containing HA-Hsf4b and immunoprecipitated using no primary antibody (lane 1) or antibody to HA (lane 2), and the immunoprecipitated materials were fractionated on SDS-PAGE and immunoblotted using antibody to ERK1/2 (top panel). Total ERK1/2 and expressed HA-Hsf4b are also presented as controls (lower two panels). (B) In vivo pull-down of GST-Hsf4b and ERK1/2. H1299 cells were transiently transfected with plasmids containing GST-Hsf4b deletion mutants or GST alone as indicated (1 to 5). At 48 h after transfection, cell lysates were subjected to GST pull down, and the pull-down fractions were immunoblotted using antibody to ERK1/2 (upper panel). Cell lysates used for GST pull-down were immunoblotted using antibody to ERK1/2 (lower panel). Lanes 1 to 5 show the Hsf4b deletion constructs and GST alone that was used for GST pull down. (C) In vivo pull down of wild-type and mutant of GST-Hsf4b fragments containing regions two and three that bind HA-ERK1. H1299 cells were transiently transfected with plasmids containing GST-Hsf4b wild-type or mutated fragments and cotransfected with HA-ERK1 as indicated. At 48 h after transfection, cell lysates were subjected to GST pull down, and the pull-down fractions were immunoblotted using antibody to HA to detect ERK1 (upper panel). Middle and lower panels present immunoblotting of cell lysates to show expression of HA-ERK1, GST alone, or GST fusion proteins using antibody to HA or GST, respectively. Lanes 1 to 6 correspond to all panels and show plasmids expressed in cells (+) and proteins present in the pull down. (D) In vivo pull-down of GST-Hsf4b, HA-ERK1, and Flag-DUSP26. H1299 cells were transiently transfected with plasmids containing GST alone or GST-Hsf4b fragments of the wild type or mutants and cotransfected with HA-ERK1 and Flag-DUSP26 as indicated. At 48 h after transfection, cell lysates were subjected to GST pull down, and the pull-down fractions were immunoblotted using antibodies to HA to detect ERK1 (upper panel), to Flag to detect DUSP26 (middle panel), or to GST to detect GST alone or GST fusion proteins (lower panel). Lanes 1 to 5 present the GST alone and Hsf4b deletion constructs that were used in GST pull-down experiments. Immunoblotting of cell lysates to show expressions of HA-ERK1 and Flag-DUSP26 in different groups are presented as controls (lower panel).
We previously (Fig. 1D) showed that DUSP26 binds Hsf4b between amino acid residues 196 to 493 (Fig. 7D, lane 3). To determine whether both ERK and DUSP26 can be immunoprecipitated with different fragments of Hsf4b, we used in vivo GST pull-down experiments. The data are presented in Fig. 7D and indicate that, while GST-Hsf4b fragment encoding residues 322 to 493 can only pull down ERK1 (Fig. 7D, lane 4), the fragment encoding amino acid residues 246 to 323 can pull down both ERK1 and DUSP26 (Fig. 7D, lane 2). The fragment encoding amino acid residues 246 to 323 encode ERK binding region two defined above, and mutation of this region abolishes binding of both ERK and DUSP26 to Hsf4b (Fig. 7D, lane 5). These data indicate that the Hsf4b ERK binding region two can bind to both ERK1 and DUSP26, while ERK binding region three binds ERK1 only, which may indicate that, in the presence of both ERK and DUSP26, Hsf4b is able to bind to both ERK1 and DUSP26.
Thus far, we have presented evidence that ERK1 interacts with Hsf4b in two separate regions (regions two and three) and DUSP26 interacts with Hsf4b in region two (Fig. 7), and as we mentioned earlier, consistent with other DSPs that lack the CH2 domain, DUSP26 does not bind ERK1/2 directly. The question remaining was whether both ERK1 and DUSP26 interact with Hsf4b simultaneously. Thus, we performed the following experiment: cells were transiently transfected with GST-Hsf4b (amino acid residues 196 to 493) or GST alone and cotransfected with HA-ERK1 and Flag-DUSP26. Immunoprecipitation experiments were performed using antibody to Flag-DUSP26 and immunoblotted with antibody to HA-ERK1 (Fig. 8, upper left panel) and GST (Fig. 8, lower left panel). The results show that Flag-DUSP26 is in the same complex as HA-ERK1 and GST-Hsf4b (Fig. 8, upper left panel, lane 2) and not in cells expressing GST alone (Fig. 8, upper left panel, lane 1), indicating that both ERK1 and DUSP26 interact with Hsf4b simultaneously. Under the experimental conditions utilized, the interaction of GST-Hsf4b, Flag-DUSP26, and HA-ERK1 is the only likely possibility, since DUSP26 and ERK1 do not interact with each other, as shown in the immunoprecipitation group containing GST alone or the group containing only HA-ERK1 and Flag-DUSP26 (Fig. 8, upper left panel, lane 1 or 3, respectively). The right panel in Fig. 8 shows an immunoblot of the same cell lysates using antibody to HA-ERK1, Flag-DUSP26, and GST.
DUSP26 and Hsf4b express in neurons.
To determine which cell types may express both Hsf4b and DUSP26, we performed PCR analyses using cDNA synthesized from several tissues obtained from adult mice. The results indicate that while Hsf4b expression was detected in the cerebellum, brain, liver, lung, and eye, DUSP26 expression could only be detected in skeletal muscle, cerebellum, and brain (Fig. 9A). Low levels of expression of DUSP26 and Hsf4b were also detected in the spleen. These results indicate that high levels of Hsf4b and DUSP26 express in the mouse brain. These results were also confirmed using immunoblotting of whole brain using antibody specific to mouse Hsf4b or DUSP26 (Fig. 9B, upper panel). The primary cultures of murine astrocytes show no DUSP26 expression but they do express Hsf4b (Fig. 9B). To determine whether endogenous Hsf4b and DUSP26 interact in vivo, we performed coimmunoprecipitation experiments using antibody to Hsf4b to pull down DUSP26 using the same cell population obtained from brain or primary astrocyte cultures (shown in Fig. 9B). The results indicate that Hsf4b and DUSP26 interact in brain cell lysates, while no interaction could be observed in the cell lysates obtained from astrocytes (Fig. 9C).
FIG.9.


Hsf4b and DUSP26 coexpress in the mouse brain. (A) cDNAs were prepared from the indicated tissues and PCR amplified (20 cycles) using primers specific to Hsf4b, DUSP26, or HPRT. (B) Immunoblotting experiments were performed using 30 μg of cell lysate prepared from whole murine brain or primary astrocyte cultures. The membranes were blotted using antibody to DUSP26, Hsf4b, or β-actin. (C) The same cell lysates from panel B were used to detect interactions between endogenous DUSP26 and Hsf4b. Cell lysates were subjected to immunoprecipitation using antibody to Hsf4b and blotted using antibody to DUSP26. These experiments were performed three times, and results were consistent. IgG, immunoglobulin G. (D) Immunohistochemical staining of paraffin-embedded sections of the cerebellum showing the endogenous expression of DUSP26, Hsf4-EGFP (targeted knock-in mice) (33), glial fibrillary acidic protein (GFAP) (astrocyte specific), and NeuN (neuron specific) using appropriate antibodies. Arrows indicate the areas where Hsf4b, DUSP26, and NeuN potentially coexpress. (E) Rat primary cortical neuronal cultures from postnatal day 0 were grown for 3 days; cells were then fixed with 4% paraformaldehyde and were immunostained using antibody to NeuN and Hsf4b or NeuN and DUSP26. Nuclei were stained using 4′,6′-diamidino-2-phenylindole (DAPI). Coimmunostaining of both Hsf4b and DUSP26 was not performed because antibodies to both Hsf4b and DUSP26 were rabbit polyclonal. However, almost all of the NeuN-positive cells were immunostained with both Hsf4b and DUSP26 (right panels).
Since both Hsf4b and DUSP26 express in the mouse brain and cerebellum (Fig. 9A), we performed immunohistochemistry to locate Hsf4b-EGFP (33) or DUSP26 in the cerebellum of hsf4 heterozygous mice. The results indicate that both proteins could be detected in the cerebellum in the same regions. For comparative purposes, consecutive sections of the cerebellum was also stained with glial fibrillary acidic protein- and NeuN-specific markers for astrocytes and neurons, respectively. The results indicate that both Hsf4b and DUSP26 may specifically coexpress in the neurons (Fig. 9D). To further determine whether Hsf4b and DUSP26 express in cultured neurons, established primary cortical neuronal cultures prepared from postnatal day 0 were immunostained using antibodies to Hsf4b, DUSP26, or NeuN. The results are presented in Fig. 9E and indicate that more than 90% of the primary neurons are positive for the expression of NeuN, Hsf4b, and DUSP26. These results indicate that Hsf4b regulation by DUSP26 may occur in a tissue-specific manner.
DISCUSSION
The transcriptional regulation and phosphorylation status of Hsf4 is not understood. Here, we present evidence that Hsf4b is phosphorylated on both threonine and serine residues under physiological growth conditions, and this phosphorylation at least in part is mediated in vitro by MAP kinases ERK1/2, JNK1, and p38. We show that Hsf4b contains three MAP kinase binding motifs, only two of which are critical for ERK1/2 binding (Fig. 7). However, we were able only to demonstrate Hsf4b interaction with ERK1/2 and p38 but not with JNK1 in vivo (Fig. 7 and data not shown). Hsf4b contains one canonical MAP kinase phosphorylation site located at amino acid residue 299 (PNSP) in close proximity to the MAP kinase docking sites located at amino acid residues 285 (RREKGLAL) and 361 (DRGPLGLES). As we show in Fig. 7, mutation of the residues in these regions lead to reduction in binding of ERK1 to Hsf4b. Hsf4b also contains a number of potential MAP kinase phosphorylation sites (SP or TP) scattered throughout the protein, and authentication of such potential phosphorylation sites is currently under way. In these studies, we also present evidence that Hsf4b is a downstream target of MAP kinase signaling and that activation of ERK1/2 and p38 leads to Hsf4b accumulation in cells (Fig. 6A), resulting in its increased DNA binding activity. Although Hsf4b is phosphorylated by JNK1 in vitro, activation of the JNK signaling pathway following treatment of cells with anisomycin does not cause similar Hsf4b accumulation, leading us to hypothesize that phosphorylation of Hsf4b by JNK1 results in divergent downstream effects from Hsf4b phosphorylation by ERK1/2.
To identify Hsf4b-interacting partner proteins, we screened a human heart cDNA library using a yeast two-hybrid system and identified a dual-specificity tyrosine phosphatase (DUSP26) binding to Hsf4b. DUSP26 belongs to the low-molecular-weight (LMW) type of dual-specificity tyrosine phosphatases, since it lacks the Cdc25 homology domain thought to be required for binding to MAP kinases (25, 43). Among the DSPs identified so far, only JKAP, VHR, SKRP1, MKP6/DUSP14, and JSP1 belong to the LMW type, and their effects on MAP kinases remain controversial (25). The most direct evidence for an effect of LMW DSP on MAP kinase signaling comes from the targeted disruption of JKAP, which illustrates its role both as a positive regulator for the JNK signaling pathway and in JNK pathway activation by cytokine stimulation (9). JKAP specifically activates JNK but not p38 or ERK2. In contrast, vaccinia virus VH1-related VHR phosphatase down regulates the JNK signaling by binding and dephosphorylating JNK and also has some specificity toward ERK1 and 2 (42). Another DSP that is VH1/VHR related is the VHX phosphatase, which mainly dephosphorylates ERK2 (2). The stress-activated protein kinase pathway-regulated phosphatase 1 (SKRP1) inactivates JNK signaling and has been shown to interact with MKK7 upstream of JNK (50). DUSP26 has been recently shown to inactivate p38 protein kinase (43). However, in our study, DUSP26 appears to be specific to ERK1/2 and JNK1, since expression of DUSP26 in cells leads to inactivation of only ERK1/2 and JNK1 (Fig. 5 and data not shown). Due to the structure of DUSP26 that lacks the CH2 domain, perhaps not surprisingly, we did not observe DUSP26 binding to ERK1/2, JNK1, p38, or the ERK1/2 upstream activator Mek in vivo (Fig. 8 and data not shown). In addition, using coimmunoprecipitation studies, we find that DUSP26 specifically binds to Hsf4b and not to Hsf1 or Elk1 transcription factors (data not shown).
Interestingly, although Hsf4b expression is detected in many tissues, among the tissues tested, DUSP26 expression is limited to skeletal muscle and neurons. Immunoblotting, immunoprecipitation, immunohistochemical, and immunofluorescence analyses indicate that DUSP26 may regulate Hsf4b activity in neurons. The targeted disruption of the hsf4 gene indicates that there are abnormalities in the brain of hsf4-deficient mice, and this is under investigation in our laboratory. Additional studies are required to determine the specific signaling and regulation of Hsf4b by DUSP26 and the downstream targets of Hsf4b in neurons.
In conclusion, our data describe a novel pathway for transcription factor regulation by MAP kinase signaling and suggest that transcription factors (in this case, Hsf4b) may act as a scaffold for MAP kinases as well as their phosphatases. Thus, we hypothesize a scenario where ERK1/2 and DUSP26 bind Hsf4b in close proximity and while ERK1/2 phosphorylates Hsf4b and leads to its increased DNA binding activity, DUSP26 inactivates ERK1/2 leading to dephosphorylation of Hsf4b and a reduction in its DNA binding activity. Consistent with this hypothesis, we have found that phosphorylation of Hsf4b in cells leads to an increase in the amount of Hsf4b protein, which in turn manifests in an elevated level of Hsf4b DNA binding activity. Since previous studies indicated that accumulation of Hsf4 protein in cells is sufficient to affect transcription of downstream target genes (35, 41, 51), we suggest that the effect of ERK and DUSP26 on Hsf4b DNA binding activity is sufficient to also affect transcription. Surprisingly, stimulation of cells by sorbitol, which activates p38 (as well as JNK) MAP kinases, also leads to accumulation of Hsf4b. In contrast, treatment of cells with anisomycin, which preferentially activates JNK, did not result in an increased Hsf4b protein level. Therefore, although we could not detect an effect on p38 MAP kinase activity by DUSP26, it is conceivable that Hsf4b is phosphorylated by all three MAP kinase family members but that their effect on Hsf4b transcriptional activity could be very different and tissue specific. As far as the reason why ERK1/2 can bind in two different regions on Hsf4b and DUSP26 can only bind to one of the classical ERK1/2 binding regions, this may indicate that, in the absence of DUSP26 expression (e.g., specific tissues), ERK1/2 may bind to both regions on Hsf4b depending on its binding affinity; however, in the presence of DUSP26, only one region on Hsf4b may be available for ERK1/2 binding. More detailed analyses are required to determine the function of Hsf4b in neurons (that express both Hsf4b and DUSP26) (Fig. 9) that may require control of Hsf4b activity by MAP kinases and their phosphatases.
Acknowledgments
We thank the following investigators for providing valuable materials: M. Iwashima for constitutive active Mek and R. Feig for expression vector pEBG.
This work was supported by National Institute of Health grants GM0707451 and CA62130.
REFERENCES
- 1.Abravaya, K., M. P. Meyers, and R. I. Morimoto. 1992. The human heat shock protein HSP-70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 6:1153-1164. [DOI] [PubMed] [Google Scholar]
- 2.Alonso, A., J. J. Merlo, S. Na, N. Kholod, L. Jaroszewski, A. Kharitonenkov, S. Williams, A. Godzik, J. D. Posada, and T. Mustelin. 2002. Inhibition of T cell antigen receptor signaling by VHR-related MKPX (VHX), a new dual specificity phosphatase related to VH1 related (VHR). J. Biol. Chem. 277:5524-5528. [DOI] [PubMed] [Google Scholar]
- 3.Aronheim, A., E. Zandi, H. Hennenmann, S. J. Elledge, and M. Karin. 1997. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interaction. Mol. Cell. Biol. 17:3094-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barford, D., A. K. Das, and M.-P. Egloff. 1998. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27:133-164. [DOI] [PubMed] [Google Scholar]
- 5.Bharadwaj, S., A. Ali, and N. Ovsenek. 1999. Multiple components of the HSP90 chaperone complex function in regulation of HSF1 in vivo. Mol. Cell. Biol. 19:8033-8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle, W. J., P. van der Geer, and T. Hunter. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110-149. [DOI] [PubMed] [Google Scholar]
- 7.Bu, L., Y. Jin, Y. Shi, R. Chu, A. Ban, H. Eiberg, L. Andres, H. Jiang, G. Zheng, M. Qian, B. Cui, Y. Xia, J. Liu, L. Hu, G. Zhao, M. R. Hayden, and X. Kong. 2002. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat. Genet. 31:276-278. [DOI] [PubMed] [Google Scholar]
- 8.Camps, M., A. Nichols, and S. Arkinstall. 2000. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB 14:6-16. [PubMed] [Google Scholar]
- 9.Chen, A. J., G. Zhou, T. Juan, S. M. Colicos, J. P. Cannon, et. al. 2002. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 277:36592-36601. [DOI] [PubMed] [Google Scholar]
- 10.Chen, W., K. C. Kirkbride, T. How, C. D. Nelson, J. Mo, J. P. Frederick, X. F. Wang, R. J. Lefkowitz, and G. C. Blobe. 2000. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling. Science 290:1574-1577. [DOI] [PubMed] [Google Scholar]
- 11.Chu, B., F. Soncin, B. D. Price, M. A. Stevenson, and S. K. Calderwood. 1996. Sequential phosphorlyation by mitogen activated protein kinase and glycogen synthase kinase-3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 271:30847-30857. [DOI] [PubMed] [Google Scholar]
- 12.Chu, B., R. Zhong, F. Soncin, M. A. Stevenson, and S. K. Calderwood. 1998. Transcriptional activity of heat shock factor 1 at 37°C is repressed through phosphorylation on two distinct serine residues by GSK-3z and PKCα and z. J. Biol. Chem. 273:18640-18646. [DOI] [PubMed] [Google Scholar]
- 13.Dai, R., B. He, W. Freitag, Y. Zhang, and N. F. Mivechi. 2000. JNK targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J. Biol. Chem. 275:18210-18218. [DOI] [PubMed] [Google Scholar]
- 14.Davis, R. J. 1993. The mitogen activated protein kinase signal transduction pathway. J. Biol. Chem. 268:14553-14556. [PubMed] [Google Scholar]
- 15.Dickinson, R. J., D. J. Williams, D. N. Slack, J. Williamson, O. M. Seternes, and S. M. Keyse. 2002. Characterization of a murine gene encoding a developmentally regulated cytoplasmic dual specificity mitogen-activated protein kinase phosphatase. Biochemical J. 364:145-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducruet, A. P., A. Vogt, P. Wipf, and J. S. Lazo. 2005. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer's disease. Annu. Rev. Med. 45:725-750. [DOI] [PubMed] [Google Scholar]
- 17.Frejtag, W., Y. Zhang, R. Dai, M. G. Anderson, and N. F. Mivechi. 2001. Heat shock factor-4 (HSF-4a) represses basal transcription through interaction with TFIIF. J. Biol. Chem. 276:14685-14694. [DOI] [PubMed] [Google Scholar]
- 18.Fujimoto, M., H. Izu, K. Seki, K. Fukuda, T. Nishida, S. Yamada, K. Kato, S. Yonemura, S. Inouye, and A. Nakai. 2004. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 23:4297-4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He, B., Y.-H. Meng, and N. F. Mivechi. 1998. GSK-3 and ERK MAPK inactivate HSF-1 by facilitating the disappearance of transcriptionally active granules after heat shock. Mol. Cell. Biol. 18:6624-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmberg, C. I., V. Hietakangas, A. Mikhailov, J. O. Rantanen, M. Kallio, A. Meinander, J. Hellman, N. Morrice, C. Mackintosh, R. I. Morimoto, J. E. Eriksson, and L. Sistonen. 2001. Phosphorylation of serine 230 promotes inducible transcriptional activity of HSF1. EMBO J. 20:3800-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong, Y., and K. D. Sarge. 1999. Regulation of protein phosphatase 2A activity by heat shock transcription factor 2. J. Biol. Chem. 274:12967-12970. [DOI] [PubMed] [Google Scholar]
- 22.Hu, Y., and N. F. Mivechi. 2003. HSF-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J. Biol. Chem. 278:17299-17306. [DOI] [PubMed] [Google Scholar]
- 23.Kallio, M., Y. Chang, M. Manuel, T. P. Alstalo, M. Rallu, Y. Gitton, L. Pirkkala, M. Y. Loones, L. Paslaru, S. Larney, S. Hirad, M. Morange, L. Sistonen, and V. Mezger. 2002. Brain abnormalities, defective meiotic chromosome synapsis and female subfertility in Hsf2 null mice. EMBO J. 21:2591-2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelkar, N., S. Gupta, M. Dickens, and R. J. Davis. 2000. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 20:1030-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyse, S. M. 2000. Protein phosphatases and the regulation of mitogen-activated protein kinase signaling. Curr. Opin. Cell Biol. 12:186-192. [DOI] [PubMed] [Google Scholar]
- 26.Kim, J., A. Nueda, Y-H. Meng, W. S. Dynan, and N. F. Mivechi. 1997. Analysis of the phosphorylation of human heat shock transcription factor-1 by MAP kinase family members. J. Cell. Biochem. 67:43-54. [DOI] [PubMed] [Google Scholar]
- 27.Kline, M. P., and R. I. Morimoto. 1997. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol. Cell. Biol. 17:2107-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kortum, R. L., and R. E. Lewis. 2004. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol. Cell. Biol. 24:4407-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandl, M., D. N. Slack, and S. M. Keyse. 2005. Specific interaction and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual specificity protein phosphatases DUSP5. Mol. Cell. Biol. 25:1830-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathew, A., S. K. Mathur, and R. I. Morimoto. 1998. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteosome pathway. Mol. Cell. Biol. 18:5091-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMillan, D. R., E. Christians, M. Forster, X.-Z. Xiao, P. Connell, J.-C. Plumier, X. Zuo, J. Richardson, S. Morgan, and I. J. Benjamin. 2002. Heat shock transcription factor 2 is not essential for embryonic development, fertility, or adult cognitive and psychomotor function in mice. Mol. Cell. Biol. 22:8005-8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMillan, D. R., X. Xiao, L. Shao, K. Graves, and I. J. Benjamin. 1998. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 273:7523-7528. [DOI] [PubMed] [Google Scholar]
- 33.Min, J.-N., Y. Zhang, D. Moskophidis, and N. F. Mivechi. 2004. Unique contribution of Hsf4 in ocular lens development and fiber cell differentiation. Genesis 40:205-217, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Morimoto, R. I. 1998. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12:3788-3796. [DOI] [PubMed] [Google Scholar]
- 35.Nakai, A., M. Tanabe, Y. Kawazoe, J. Inazawa, R. I. Morimoto, and K. Nagata. 1997. HSF-4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell. Biol. 17:469-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rallu, M., M. Loones, Y. Lallemand, R. Morimoto, M. Morange, and V. Mezger. 1997. Function and regulation of HSF2 during mouse embryogenesis. Proc. Natl. Acad. Sci. USA 94:2392-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarge, K. D., K. Park-Sarge, J. D. Kirby, K. E. Mayo, and R. I. Morimoto. 1994. Expression of heat shock factor 2 in mouse testis: potential role as a regulator of heat shock protein gene expression during spermatogenesis. Biol. Reprod. 50:1392-1407. [DOI] [PubMed] [Google Scholar]
- 38.Satyal, S. H., D. Chen, S. G. Fox, J. M. Kramer, and R. I. Morimoto. 1998. Negative regulation of the heat shock transcriptional response by HSBP1. Genes Dev. 12:1962-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaeffer, H. J., A. D. Catling, S. T. Eblen, L. S. Collier, A. Krauss, and M. J. Weber. 1998. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281:1668-1671. [DOI] [PubMed] [Google Scholar]
- 40.Shinka, T., Y. Sato, G. Chen, T. Naroda, K. Kinoshita, Y. Unemi, K. Tsuji, T. Iwamoto, and Y. Nakahori. 2004. Molecular characterization of heat shock-like factor encoded on the human chromosome, and implications for male infertility. Biol. Reprod. 71:297-306. [DOI] [PubMed] [Google Scholar]
- 41.Tanabe, M., N. Sasai, K. Nagata, X.-D. Liu, P. C. C. Liu, D. J. Thiele, and A. Nakai. 1999. The mammalian HSF-4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J. Biol. Chem. 274:27845-27856. [DOI] [PubMed] [Google Scholar]
- 42.Todd, J. L., J. D. Rigas, L. A. Rafty, and J. M. Denu. 2002. Dual-specificity protein tyrosine phosphatase VHR down-regulates c-Jun N-terminal kinase (JNK). Oncogene 21:2573-2583. [DOI] [PubMed] [Google Scholar]
- 43.Vasudevan, S. A., J. Skoko, K. Wang, S. M. Burlingame, P. N. Patel, J. S. Lazo, J. G. Nuchtern, and J. Yang. 2005. MKP-8, a novel MAPK phosphatase that inhibits p38 kinase. Biochem. Biophys. Res. Commun. 330:511-518. [DOI] [PubMed] [Google Scholar]
- 44.Wang, G., Z. Ying, X. Jin, N. Tu, Y. Zhang, M. Phillips, D. Moskophidis, and N. F. Mivechi. 2004. Essential requirement for both hsf1 and hsf2 transcriptional activity in spermatogenesis and male fertility. Genesis 38:66-80. [DOI] [PubMed] [Google Scholar]
- 45.Wang, G., J. Zhang, D. Moskophidis, and N. F. Mivechi. 2003. Targeted disruption of the Hsf2 gene results in increased embryonic lethality, neuronal defects, and reduced spermatogenesis. Genesis 36:48-61. [DOI] [PubMed] [Google Scholar]
- 46.Wang, X., N. Grammatikakis, A. Siganou, and S. K. Calderwood. 2003. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3e binding, and cytoplasmic sequestration of heat shock factor 1. Mol. Cell. Biol. 23:6013-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, C. 1995. Heat shock transcription factors: structure and regulation. Annu. Rev. Cell Dev. Biol. 11:441-469. [DOI] [PubMed] [Google Scholar]
- 48.Xia, W., Y. Guo, N. Vilaboa, J. Zuo, and R. Voellmy. 1998. Transcriptional activation of heat shock factor HSF-1 probed by phosphopeptide analysis of factor 32P-labeled in vivo. J. Biol. Chem. 273:8749-8755. [DOI] [PubMed] [Google Scholar]
- 49.Xiao, X., X. Zuo, A. A. Davis, D. R. McMillan, B. B. Curry, J. A. Richardson, and I. J. Benjamin. 1999. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 18:5943-5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zama, T., R. Aoki, T. Kamimoto, K. Inoue, Y. Ikeda, and M. Hagiwara. 2002. A novel dual specificity phosphatase SKRP1 interacts with the MAPK kinase MKK7 and inactivates the JNK MAPK pathway. Implication for the precise regulation of the particular MAPK pathway. J. Biol. Chem. 277:23909-23918. [DOI] [PubMed] [Google Scholar]
- 51.Zhang, Y., W. Frejtag, R. Dai, and N. F. Mivechi. 2001. Heat shock factor-4 (HSF-4a) is a repressor of HSF-1 mediated transcription. J. Cell. Biochem. 82:692-703. [DOI] [PubMed] [Google Scholar]
- 52.Zhang, Y., L. Huang, J. Zhang, D. Moskophidis, and N. F. Mivechi. 2002. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem. 86:376-393. [DOI] [PubMed] [Google Scholar]