

Abstract
We found that mutating the RNP1 motif in the predicted RRM domain in yeast eukaryotic initiation factor 3 (eIF3) subunit b/PRT1 (prt1-rnp1) impairs its direct interactions in vitro with both eIF3a/TIF32 and eIF3j/HCR1. The rnp1 mutation in PRT1 confers temperature-sensitive translation initiation in vivo and reduces 40S-binding of eIF3 to native preinitiation complexes. Several findings indicate that the rnp1 lesion decreases recruitment of eIF3 to the 40S subunit by HCR1: (i) rnp1 strongly impairs the association of HCR1 with PRT1 without substantially disrupting the eIF3 complex; (ii) rnp1 impairs the 40S binding of eIF3 more so than the 40S binding of HCR1; (iii) overexpressing HCR1-R215I decreases the Ts− phenotype and increases 40S-bound eIF3 in rnp1 cells; (iv) the rnp1 Ts− phenotype is exacerbated by tif32-Δ6, which eliminates a binding determinant for HCR1 in TIF32; and (v) hcr1Δ impairs 40S binding of eIF3 in otherwise wild-type cells. Interestingly, rnp1 also reduces the levels of 40S-bound eIF5 and eIF1 and increases leaky scanning at the GCN4 uORF1. Thus, the PRT1 RNP1 motif coordinates the functions of HCR1 and TIF32 in 40S binding of eIF3 and is needed for optimal preinitiation complex assembly and AUG recognition in vivo.
Polypeptide synthesis in eukaryotes commences when the methionyl initiator tRNA (Met-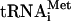 ) is placed in the P site of the 80S ribosome and base pairs with the AUG codon in mRNA. The steps required for this assembly comprise the translation initiation pathway and involve numerous initiation factors that stimulate one or more reactions in the process (16). First, Met-
) is placed in the P site of the 80S ribosome and base pairs with the AUG codon in mRNA. The steps required for this assembly comprise the translation initiation pathway and involve numerous initiation factors that stimulate one or more reactions in the process (16). First, Met-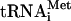 in a complex with eukaryotic initiation factor 2 (eIF2) and GTP, the ternary complex (TC), is delivered to the 40S ribosome, creating the 43S preinitiation complex (PIC) in a reaction stimulated by eIF1, eIF1A, eIF3, and eIF5 (2, 6, 17, 19, 21, 30). The mRNA, prebound to the cap-binding complex eIF4F and the poly(A) binding protein, is then recruited to the 43S PIC to form the 48S PIC. The 48S PIC scans the mRNA and when the AUG codon is recognized, the GTP in TC is hydrolyzed in a reaction stimulated by eIF5. The eIF2-GDP is released from the 40S ribosome, leaving Met-
in a complex with eukaryotic initiation factor 2 (eIF2) and GTP, the ternary complex (TC), is delivered to the 40S ribosome, creating the 43S preinitiation complex (PIC) in a reaction stimulated by eIF1, eIF1A, eIF3, and eIF5 (2, 6, 17, 19, 21, 30). The mRNA, prebound to the cap-binding complex eIF4F and the poly(A) binding protein, is then recruited to the 43S PIC to form the 48S PIC. The 48S PIC scans the mRNA and when the AUG codon is recognized, the GTP in TC is hydrolyzed in a reaction stimulated by eIF5. The eIF2-GDP is released from the 40S ribosome, leaving Met-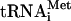 in the P site (16). Finally, joining of the 60S ribosomal subunit occurs in a reaction stimulated by eIF5B (29), and the remaining initiation factors dissociate from the resulting 80S initiation complex (38).
in the P site (16). Finally, joining of the 60S ribosomal subunit occurs in a reaction stimulated by eIF5B (29), and the remaining initiation factors dissociate from the resulting 80S initiation complex (38).
The mechanism of scanning is poorly understood; however, eIF1, eIF1A, and eIF4F all play important roles (11, 24, 27, 28), and eIF3 may also contribute (23) in the process. The stringent recognition of AUG has been shown to involve eIF2, eIF5, eIF3, and also eIF4G, in addition to eIF1 in vivo (10, 15, 43). In vitro, eIF1 prevents 48S PIC formation at near-cognate AUG triplets or AUG triplets in a poor sequence context (27, 28), and it may hinder the ability of eIF5 to hydrolyze GTP in the TC (20, 38, 43), and the subsequent release of Pi from eIF2-GDP (1), at non-AUG triplets.
eIF3 is the most complex initiation factor, consisting of 13 different subunits in mammals. The Saccharomyces cerevisiae eIF3, by contrast, contains only five essential subunits—eIF3a/TIF32, eIF3b/PRT1, eIF3c/NIP1, eIF3g/TIF35, and eIF3i/TIF34—and the nonessential subunit eIF3j/HCR1. Although eIF3 is much less complex in yeast, it has nevertheless been implicated in 43S and 48S PIC formation, which initially was demonstrated for mammalian eIF3 (17, 30, 31). Yeast eIF3 has also been implicated in scanning and AUG recognition (23, 43), making yeast an excellent model system with which to study the functions of eIF3.
In S. cerevisiae, a multifactor complex (MFC) consisting of eIF3, TC, eIF5, and eIF1 has been shown to be important for optimal translation initiation in vivo (3, 31). The eIF3 interacts directly with eIF5, eIF2, and eIF1 in the MFC, and these interactions may contribute to the stimulatory effect of eIF3 in binding of eIF1 and TC to the 40S subunit that was observed in vitro (19-21). Two interactions between yeast eIF3 and eIF2 have been described thus far: a direct interaction between eIF2β and TIF32/eIF3a and an indirect interaction between eIF2β and NIP1/eIF3c that is bridged by eIF5. In addition to binding eIF5, NIP1 also interacts with eIF1, which additionally interacts with eIF5, TIF32, and eIF2β (3, 4, 36, 42) (see Fig. 2D).
FIG. 2.

prt1-rnp weakens interactions of eIF3 with HCR1, eIF2, and eIF1 in the MFC. (A) Plasmids containing PRT1-His (p4468) or prt1-rnp1-His (p4473) were introduced into prt1Δ::hisG strain H3677, and the resident PRT1-URA3 plasmid was evicted by growth on medium containing 5-FOA. The resulting strains, and H3677, were grown in YPD at 25°C and incubated at 37°C for 1 h. WCEs were incubated with nickel-nitrilotriacetic acid silica at 4°C for 2 h and eluted with imidazole, and the eluates were subjected to Western analysis of the proteins indicated on the left. For each strain, 3% of the input (In), 15 and 30% of the eluates, and 3% of the flowthrough (FT) were resolved in successive lanes. (B) Western signals in the eluate fractions for experiments of the type shown in panels A and C were quantified, and values for each factor were normalized to the corresponding eluate signal for either PRT1-His or prt1-rnp1-His, as appropriate. The resulting values for prt1-rnp1-His were normalized to the corresponding values for PRT1-His, and the final values calculated from three to five independent experiments were averaged to obtain the means and standard errors plotted in the histogram. A value of unity indicates no difference in association of that factor with prt1-rnp1 versus PRT1. (C) Same as panel A except that no heat treatment was performed. (D) Schematic model showing the position of the rnp1 mutation in PRT1 in the context of a previously published three-dimensional hypothetical model of the MFC. Subunits of eIF3 are labeled with their yeast (e.g., TIF32) and universal (e.g., 3a) designations. The subunits of eIF2 are labeled α, β, and γ, with GTP and Met-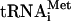 bound to eIF2γ, to comprise the TC. The protein subunits and Met-
bound to eIF2γ, to comprise the TC. The protein subunits and Met-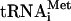 are shown roughly in proportion to their molecular weights. ntd, N-terminal domain; ctd, C-terminal domain; hld, HCR1-like domain. (E) 35S-labeled peptides comprising the N-terminal 136 residues of PRT1 and prt1-rnp1 were synthesized in vitro by using plasmids p3711 and p4470, respectively. Full-length GST-TIF32 (encoded by p2947), GST-HCR1 (p3763), and GST alone (pGEX-5X-3) were expressed in E. coli, immobilized on glutathione-Sepharose beads, and incubated with 10 μl of the 35S-labeled PRT1-NTD peptides at 4°C for 2 h. The beads were washed three times with phosphate-buffered saline, and the bound proteins were eluted and resolved by SDS-polyacrylamide gel electrophoresis. The gel was stained by GelCode Blue stain Reagent (Pierce) (upper panel), dried, and subjected to autoradiography (lower panel). Aliquots containing 20% of the input are also shown. Similar results were obtained in three independent experiments. (F) PRT1 strain H2879 and prt1-rnp1 strain H3674 were transformed with hc vector YEplac195 or its derivatives containing both TIF32 and NIP1 (p3131), NIP1 (p3130) or TIF32 (p3132). Serial 10-fold dilutions of the indicated strains were spotted onto SC-Ura and incubated at 25°C or 34°C for 2 and 6 days, respectively.
are shown roughly in proportion to their molecular weights. ntd, N-terminal domain; ctd, C-terminal domain; hld, HCR1-like domain. (E) 35S-labeled peptides comprising the N-terminal 136 residues of PRT1 and prt1-rnp1 were synthesized in vitro by using plasmids p3711 and p4470, respectively. Full-length GST-TIF32 (encoded by p2947), GST-HCR1 (p3763), and GST alone (pGEX-5X-3) were expressed in E. coli, immobilized on glutathione-Sepharose beads, and incubated with 10 μl of the 35S-labeled PRT1-NTD peptides at 4°C for 2 h. The beads were washed three times with phosphate-buffered saline, and the bound proteins were eluted and resolved by SDS-polyacrylamide gel electrophoresis. The gel was stained by GelCode Blue stain Reagent (Pierce) (upper panel), dried, and subjected to autoradiography (lower panel). Aliquots containing 20% of the input are also shown. Similar results were obtained in three independent experiments. (F) PRT1 strain H2879 and prt1-rnp1 strain H3674 were transformed with hc vector YEplac195 or its derivatives containing both TIF32 and NIP1 (p3131), NIP1 (p3130) or TIF32 (p3132). Serial 10-fold dilutions of the indicated strains were spotted onto SC-Ura and incubated at 25°C or 34°C for 2 and 6 days, respectively.
The importance of these interactions for 43S assembly in yeast is evident from experiments in which mutations in eIF5, TIF32, NIP1, or eIF1 led to reduced 40S binding in vivo of not only the mutated factor but also other MFC constituents (6, 23, 36, 41). Moreover, certain mutations in NIP1 (43), eIF5 (35), and eIF1 (36) that destabilize the MFC disrupt translational control of GCN4 in a manner indicating reduced TC binding to the 40S subunit. Other NIP1 mutations have phenotypes indicating the importance of its interactions with eIF1 or eIF5 in stringent selection of AUG as the start codon. These include Sui− mutations that suppress initiation codon mutations by allowing non-AUG triplets to be used as start codons at higher frequencies than normal and Ssu− (suppressor of Sui−) mutations that suppress a Sui− mutation in eIF2β or eIF5 (43).
Deletion of HCR1 (encoding eIF3j in yeast) produces a slow-growth phenotype that is attributed to its dual function, one in translation initiation for which it was isolated (40) and another in maturation (39) and nuclear export of ribosomal subunits (46). In vitro experiments have demonstrated a requirement of mammalian eIF3j for stable 40S association of the purified eIF3 complex and shown that eIF3j can bind on its own to the 40S subunit (12, 19, 38). This requirement for eIF3j in 40S binding of mammalian eIF3 was suppressed in the presence of TC, eIF1, and eIF1A during formation of 43S complexes, making it unclear whether eIF3j would be an important factor for 40S binding of eIF3 and 43S PIC assembly in vivo. Indeed, yeast eIF3j was found to stabilize the MFC but did not appear to be required for wild-type (WT) 40S binding of eIF3 in yeast cells (44).
We have previously reported that eIF3 could be involved in scanning (23) and AUG recognition (43). Binding to mRNA might therefore be an intrinsic property of yeast eIF3. In support of this, mammalian PRT1/eIF3b and eIF3a/TIF32 can be UV cross-linked to β-globin mRNA in 48S PICs formed in vitro (19, 45). The N-terminal domain (NTD) of PRT1 contains an RNA recognition motif (RRM), which is an obvious region to mutate in the hope of generating mutants that would implicate eIF3 in scanning by disrupting a putative PRT1-mRNA interaction. However, 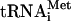 or rRNA could also be targets of the PRT1 RRM. Furthermore, the RRM is located within the binding domain for HCR1 and TIF32 in PRT1 (44), and these protein-protein interactions could be disrupted by mutation of the RRM.
or rRNA could also be targets of the PRT1 RRM. Furthermore, the RRM is located within the binding domain for HCR1 and TIF32 in PRT1 (44), and these protein-protein interactions could be disrupted by mutation of the RRM.
To address the functions of the putative RRM in PRT1, we generated a multiple-alanine substitution of the RNP1 motif of this domain, the rnp1 mutation, which leads to temperature-sensitive (Ts−) translation initiation and impairs recruitment of eIF3 to the 40S subunit in vivo. (We will use the terms recruitment and binding to indicate steady-state interaction of factors with the ribosome, reflecting the equilibrium between association and dissociation reactions.) The rnp1 mutation disrupts binding of the eIF3 core complex to HCR1 but has a much smaller effect on 40S binding of HCR1 versus eIF3 core subunits, suggesting that HCR1 is important for recruiting eIF3 to 40S subunits in yeast cells. In agreement with this, overexpression of HCR1 suppressed the Ts− phenotype and conferred greater 40S binding of eIF3 in the extracts of rnp1 cells, and the deletion of HCR1 reduced 40S binding of eIF3 in otherwise WT extracts. These findings are significant in identifying a molecular contact in yeast eIF3 important for the evolutionarily conserved function of HCR1/eIF3j in the recruitment of eIF3 to 40S subunits. Interestingly, rnp1 mutant cells also show an increase in leaky scanning of an upstream AUG codon in GCN4 mRNA, suggesting that the PRT1 RRM contributes to the efficiency of AUG recognition in addition to its role in ribosome binding by eIF3.
MATERIALS AND METHODS
Plasmid constructions.
Plasmids used in the present study are listed in Table 1. To construct pRS315-PRT1-His (p4468), a ClaI-BglII fragment of 2682 bp, that contains the 5′ untranslated region (UTR) and the open reading frame (ORF) of PRT1, sequences encoding a His8 tag fused to the 3′end of the ORF, but no 3′ UTR, was isolated from p2846 (30) and ligated with an ∼6-kb fragment of pRS315 digested with BamHI and partially digested with ClaI. The resulting plasmid, p4468, complements a prt1Δ mutant to the same degree as does plasmid p2625, which contains untagged PRT1 with both 5′ and 3′ UTRs intact. To construct pRS315-prt1-rnp1 (p4473). the prt1-rnp1 mutations were generated by using the QuikChange II kit (Stratagene) according to the manufacturer's protocol. p4468 was used as the template with the primers 323.1 (5′-GATGAAGCCACTGGTAAGACGGCAGCTGCTGCCGCCGCGGCATGTGGCTCAATGAACGATGC) and 323.3 (5′-GCATCGTTCATTGAGCCACATGCCGCGGCGGCAGCAGCTGCCGTCTTACCAGTGGCTTCATC). Since the strategy involves PCR amplification of the entire plasmid, an ∼740-bp BmgBI-SnaBI fragment containing the rnp1 mutations was isolated from the mutagenized plasmid and used to replace the corresponding fragment in p4468 to produce p4473. The entire BmgBI and SnaBI fragment of p4473 was sequenced to confirm the presence of only the desired mutations. To construct pRS306-prt1-rnp1 (p4469), a 2,729-bp ClaI-SacI fragment from p4473, containing the wild-type 5′ UTR and prt1-rnp1 ORF, was ligated with pRS306 digested with ClaI and SacI to generate an integrating plasmid for prt1-rnp1 with a unique BamHI site for chromosomal integration. To construct YEp24-TC-hcr1-R215I (p4472), a PvuII fragment from p3776 (44) containing HCR1-R215I was ligated to plasmid YEp24-TC (p3000) (4) digested with NruI (both blunt ends). To construct YEp24-hcr1-R215I (p4471), a PvuII fragment from p3776 (44) containing hcr-R215I was ligated to vector YEp24 digested with SmaI (both blunt ends). Plasmid pT-T7-prt1-rnp1-DA (p4470) was constructed as described previously for p3711 (44), except that PCR was performed by using p4473 (prt1-rnp1) as a template. The insert was sequenced to verify the presence of the rnp1 mutation.
TABLE 1.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| YEplac195 | hc URA3 vector | 13 |
| YEplac181 | hc LEU2 vector | 13 |
| YEp24 | hc URA3 vector | 26 |
| pRS316 | lc URA3 vector | 34 |
| p3000 (hc TC) | hc URA3 vector containing SUI2, SUI3, GCD11, and IMT4 | 4 |
| pM199 | lc URA3 vector containing GCN4-lacZ with uORF1 alone | 14 |
| pM226 | lc URA3 vector with containing GCN4-lacZ with elongated uORF1 | 14 |
| p3927 (YEplac-TIF32Δ6-His) | hc URA3 vector containing tif32Δ6-His | 42 |
| p2625 (pRS316-PRT1) | lc URA3 plasmid containing PRT1 | 31 |
| p4468 (pRS315-PRT1-His) | lc LEU2 vector containing PRT1-His | This study |
| p4473 (pRS315-prt1-rnp1-His) | lc LEU2 vector containing prt1-rnp1-His | This study |
| p3131 (YEplac195-TIF32-NIP1) | hc URA3 vector containing TIF32 and NIP1 | 30 |
| p3130 (YEplac195-NIP1) | hc URA3 vector containing NIP1 | 30 |
| p3132 (YEplac195-TIF32) | hc URA3 vector containing TIF32 | 30 |
| p3778 (YEplac181-HCR1) | hc LEU2 vector containing HCR1 | 40 |
| p3780 (YEplac181-hcr1-R215I) | hc LEU2 vector containing hcr1-R215I | 44 |
| p4471 (YEp24-hcr1-R215I) | hc URA3 vector containing hcr1-R215I | This study |
| p4472 (YEp24-TC-hcr1-R215I) | hc URA3 vector with SUI2, SUI3, GCD11, IMT4, hcr1-R215I | This study |
| p2947 (pGEX-TIF32) | GST-TIF32 fusion under tac promoter | 5 |
| p3763 (pGEX-HCR1) | GST-HCR1 fusion under tac promoter | 44 |
| pGEX-5X-3 | E. coli vector for expressing GST fusions under tac promoter | 37 |
| p4470 (pT-T7-prt1-rnp1-ΔA) | prt1-rnp1[codons 1 to 136] under T7 promoter | This study |
| p3711 (pT-T7-PRT1-ΔA) | PRT1[codons 1 to 136] under T7 promoter | 44 |
Yeast strain constructions.
Yeast strains used in the present study are listed in Table 2. Preparation of media was done essentially as described previously (32). To construct H3674 (MATa prt1-rnp1 leu2-3,112 ura3-52), pRS306-prt1-rnp1 (p4469) was digested with BamHI, and the purified DNA fragment was used to transform strain H2879 to Ura+. Ura− segregants were obtained by selecting for growth on medium containing 5-fluoorotic acid (5-FOA), and the resulting strains were tested for the presence of PRT1 or prt1-rnp1 by testing for Ts− growth. To construct H3675 (MATa PRT1 leu2-3,112 ura3-52 hcr1Δ), two primers, 5′-TGATATTATGGTCGTCCCTGTA-3′ and 5′-AAAGCCAGATAACGGTGCAAAA-3′, were used to PCR amplify a DNA fragment containing the hcr1Δ::kanMX allele from strain 6704 obtained from Research Genetics. The purified DNA fragment was used to transform strain H2879 to G418 resistance. The deletion of HCR1 in the resulting strain, H3675, was verified by Western analysis of a WCE with HCR1 antibodies and by showing that its Slg− phenotype was complemented by the single-copy plasmid p3774 containing HCR1. To construct H3676 (MATa prt1-rnp1 leu2-3,112 ura3-52 hcr1Δ), the procedure described above for constructing H3675 was followed to delete HCR1 in H3674. To construct H3677 (MATa trp1Δ leu2-3,112 ura3-52 prt1Δ::hisG p2625 [PRT1 URA3]), gcn2Δ strain YKHN60 (23) was transformed to Ura+ with the integrative GCN2 plasmid pHQ835 (kindly provided by Hongfang Qiu) digested with SnaBI. Ura− segregants were obtained by selecting for growth on medium containing 5-FOA, and the resulting strains were tested for the presence of GCN2 or gcn2Δ by testing growth on medium containing 3-aminotriazole (3-AT). A GCN2 (3-AT-resistant) strain was chosen and transformed with p2625 to Ura+. A Leu− Ura+ derivative lacking pKHN7 was obtained by growth on medium containing leucine.
TABLE 2.
S. cerevisiae strains used in this study
| Straina | Genotype | Source or reference |
|---|---|---|
| H1676* | MATaprt1-1 leu2-3,112 ura3-52 | 31 |
| H2879* | MATaPRT1 leu2-3,112 ura3-52 | 23 |
| H3342* | MATatrp1Δ leu2-3,112 ura3-52 prt1Δ::hisG gcn2Δ::hisG pKHN7 [prt1-1 LEU2] | 23 |
| H3677* | MATatrp1Δ leu2-3,112 ura3-52 prt1Δ::hisG p2625 [PRT1 URA3] | This study |
| H3674* | MATaprt1-rnp1 leu2-3,112 ura3-52 | This study |
| H3675* | MATaPRT1 leu2-3,112 ura3-52 hcr1Δ | This study |
| H3676* | MATaprt1-rnp1 leu2-3,112 ura3-52 hcr1Δ | This study |
| 6704 | MATahcr1Δ::KanMX4 his3-Δ1 leu2-Δ0 ura3-Δ0 met15-Δ0 | Research Genetics |
*, Isogenic strains.
Probes used in Northern analysis.
The probe for IMT4-encoded tRNAiMet was generated by end labeling the oligonucleotide 5′-GGTAGCGCCGCTCGGTTTCGATCCGAGGTC-3′ using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Redivue 6000, Ci/mmol; Amersham) according to the vendor's protocol. The unincorporated radiolabeled nucleotides were removed by using MicroSpin G-25 spin columns (Amersham) according to the vendor's protocol. The probe against RPL41A mRNA was generated by using the Rediprime II random prime labeling system (Amersham) and [α-32P]dCTP (Redivue 6000, Ci/mmol; Amersham) according to the vendor's protocol. The DNA fragment used for random labeling was amplified from genomic DNA by using the primers 5′-TAAAAGAACCAGACCACATCGATTC-3′ and 5′-GACTTGCCAAGCACAATTACAATG-3′ and genomic DNA isolated from strain H1676. The unincorporated nucleotides were removed by using MicroSpin G-25 spin columns as described above.
Antibodies.
Antibodies against PRT1, NIP1, TIF34, eIF1, eIF5 (31), TIF32, TIF35 (30), and TIF11 (25) have been described.
HCHO cross-linking, extract preparation, and fractionation of extracts for analysis of preinitiation complexes.
Yeast cells were cross-linked with HCHO, whole-cell extracts (WCEs) were prepared (23), and WCEs were separated by sedimentation through sucrose gradients (3) as described previously, collecting 0.7-ml fractions while scanning continuously at A254. For polysome profiles a 4.5 to 45% gradient was used; to analyze the factors associated with the 40S ribosome, a 7.5 to 30% gradient was used. A 6× sodium dodecyl sulfate (SDS) loading buffer was added to 0.2 ml of each fraction and boiled for 10 min prior to SDS-polyacrylamide gel electrophoresis and Western analysis, which was sufficient to reverse the cross-linking induced by HCHO. RNA was extracted from fractions essentially as described previously (8), except that two extractions with hot (70°C) phenol for 15 min were conducted, which was sufficient to reverse the cross-linking.
Other biochemical methods.
β-Galactosidase assays were conducted as described previously (22). Western analysis was carried out by using 4 to 20% polyacrylamide gels from Bio-Rad Laboratories and developed using an enhanced chemiluminescence kit (Amersham) and the antibodies described above. Northern analysis was conducted essentially as described previously (23) with the radioactive probes described above. Glutathione S-transferase (GST) pull-down experiments and preparation of recombinant proteins were done essentially as described in reference 5.
RESULTS
The RNP1 motif in PRT1/eIF3b is required for efficient translation initiation at elevated growth temperatures.
We constructed four different mutations in the predicted RNP1 motif of PRT1/eIF3b, targeting a seven-residue stretch (124KGFLFVE130) of sequence similarity with the RNP1 motif of the second RRM domain in poly(A) binding protein (PABP) (9) (Fig. 1A). These included single substitutions of K124 or E130 with oppositely charged residues Asp and Lys, respectively, a double Ala substitution of both F126 and F128, and simultaneous Ala substitutions in all seven residues of the 124KGFLFVE130 sequence. Residues 124, 130, 126, and 128 correspond to amino acids that in human PABP make direct interactions with RNA through their side chains (9), suggesting that mutations at these positions in PRT1 could disrupt a putative interaction with RNA.
FIG. 1.

Effect of prt1-rnp1 on cell growth and translation initiation in vivo. (A) Schematic representation of PRT1, showing the location of prt1-1, the RRM domain, and the RNP1 and RNP2 motifs. Below is a sequence alignment of this region in yeast (S.c., Saccharomyces cerevisiae) PRT1 and residues in RRM2 of human (H.s., Homo sapiens) PABP, together with the seven residues that are changed to alanines in prt1-rnp1. Letters in boldface indicate amino acids in PABP RRM2 that make direct contact with RNA (9). (B, left) Growth curves of isogenic PRT1 (H2879), prt1-rnp1 (H3674), and prt1-1 (H1676) cells in liquid yeast extract-peptone-dextrose (YPD) medium at 25°C. At the time indicated by the arrow, half of each culture was harvested and resuspended in YPD at 37°C and cultured for the indicated time (B, right). (C) prt1-rnp1 strain H3674 was transformed with low-copy (lc) PRT1 plasmid p2625 or vector pRS316, and WT PRT1 strain H2879 was transformed with pRS316. Tenfold serial dilutions of the cells were spotted on solid SC lacking uracil (SC-Ura) and incubated at 25 or 34°C. (D) Western analysis of WCEs from PRT1 (H2879) and prt1-rnp1 (H3674) cells grown in YPD at 25°C and treated for 1 h at 37°C with antibodies to PRT1 and TIF11/eIF1A. Signals were quantified by using NIH image software, PRT1/eIF1A ratios were calculated, and both ratios were normalized to that calculated for the PRT1 strain, with the final value shown below the PRT1 blots. (E) PRT1 (H2879) and prt1-rnp1 (H3674) cells were grown in YPD at 25°C, treated for 1 h at 37°C, and cross-linked with 1% HCHO prior to harvesting. Fifteen optical density units at 260 nm of WCE were separated by velocity sedimentation through a 4.5 to 45% sucrose gradient, and the fractions were collected starting from the top of the gradient while scanning at A254 to visualize ribosomal species and polysomes, as indicated. (F) Same as in panel E except that cells were not heat treated. Doubling times of the strains (Td) at 25°C are indicated.
Only the 7-Ala substitution had an observable effect on cell growth when contained in a plasmid-borne copy of PRT1 as the only source of PRT1 in the cell, conferring a temperature-sensitive phenotype. When this mutation, designated prt1-rnp1 (abbreviated rnp1 below), was introduced into the chromosomal PRT1 allele, the resulting strain showed slow growth (Slg−) at 34°C and growth arrest at 37°C but no growth defect at 25°C (Fig. 1B and C). The Slg− phenotype of rnp1 cells was complemented by plasmid-borne PRT1 (Fig. 1C). Incubation of rnp1 cells at 37°C for only 30 min or 1 h led to severe polysome runoff and accumulation of 80S ribosomes (Fig. 1E and data not shown), indicating a strong decrease in translation initiation, whereas the mutant displayed WT polysome content at 25°C (Fig. 1F). The growth defect and polysome runoff in the rnp1 mutant at 37°C are comparable to that observed previously in prt1-1 cells (Fig. 1B) (23). Western analysis of WCEs showed that the rnp1 protein is present at nearly WT levels after 1 h of incubation at 37°C (Fig. 1D), implying that rnp1 impairs the function of PRT1 in translation initiation. The fact that none of the single or double residue substitutions we made in the RNP1 motif predicted to disrupt interactions with RNA produced any phenotype suggests that the growth defect conferred by the more extensive rnp1 mutation probably does not result from impairing interaction of PRT1 with RNA.
prt1-rnp1 destabilizes interaction of eIF3 with eIF2 and eIF1 in the MFC.
To determine whether the defective translation initiation observed in the prt1-rnp mutant reflects the disruption of eIF3 or destabilization of the MFC, we purified the MFC by Ni affinity chromatography directed against His8-tagged versions of WT and rnp1 mutant PRT1 proteins expressed from plasmid-borne alleles in a prt1Δ strain. Relative to the amount of His8-prt1-rnp1 recovered, we observed relatively small (∼30% or less) reductions in amounts of copurifying eIF3 subunits and other MFC components compared to that seen for His8-PRT1 when cells were grown at 25°C (Fig. 2C; see quantification in Fig. 2B). Note that eIF1A and the 40S protein RPS22 were recovered at low levels relative to MFC components (Fig. 2C), indicating that primarily the MFC free of ribosomes was purified, as previously shown (31). With cells grown at 37°C, we recovered lower amounts of all MFC components from the prt1-rnp1-His8 cells, including the mutant protein itself. However, after normalizing for the amounts of His8-PRT1 or His8-prt1-rnp purified from the two strains, it was evident that recovery of the nonessential HCR1/j subunit of eIF3, eIF2γ, and eIF1 was more strongly reduced (by >70%) than was eIF5 or the other core eIF3 subunits by the rnp1 mutation (Fig. 2A and B). Thus, while the core eIF3 complex appears to be largely intact, interactions of eIF3 with HCR1, eIF2, and eIF1 in the MFC are strongly impaired by rnp1 at 37°C.
prt1-rnp1 impairs direct binding of the PRT1 NTD to TIF32 and HCR1.
The defect in HCR1 association with eIF3 in prt1-rnp1 extracts (Fig. 2A and B) is consistent with our previous finding that the RRM domain in PRT1 mediates a direct interaction with HCR1. The RRM domain in PRT1 also interacts with TIF32/eIF3a (44) (Fig. 2D). Hence, we sought to determine whether rnp1 disrupts the binary interactions of PRT1-NTD with HCR1 or TIF32. 35S-labeled polypeptides containing residues 1 to 136 from WT PRT1 (PRT1-NTD) or the rnp1 mutant (prt1-rnp1-NTD) were synthesized in vitro and tested for binding to GST fusions made to full-length TIF32 or HCR1 expressed in Escherichia coli. We found that PRT1-NTD bound specifically to both GST-TIF32 and GST-HCR1 in a manner impaired by rnp1 (Fig. 2E). Although the yield of full-length GST-TIF32 is quite low compared to GST-HCR1 and GST alone, it shows relatively greater binding of the PRT1-NTD polypeptide, a finding consistent with previous results (44). Thus, rnp1 impairs direct binding of PRT1 to both TIF32 and HCR1.
Genetic evidence that PRT1-TIF32 interaction is weakened by prt1-rnp1 in vivo.
Although we found that rnp1 reduces binding of the PRT1-NTD to TIF32 and HCR1 in vitro (Fig. 2E) and there is previous evidence that HCR1 promotes interaction between TIF32 and the RRM domain in PRT1 (44), we did not observe a large reduction in the amount of TIF32 copurifying with His8-prt1-rnp (Fig. 2A and B). Hence, we took a genetic approach to demonstrate that rnp1 weakens the PRT1-TIF32 interaction in vivo. We showed previously that the TIF32-NIP1 binary subcomplex of eIF3 subunits (a and c) forms in vivo when both proteins are overexpressed (30); hence, we reasoned that overexpressing NIP1 might titrate TIF32 from the mutant eIF3 complex containing prt1-rnp1 (but not from WT eIF3) and thereby exacerbate the growth defect in prt1-rnp1 cells. If so, then simultaneously overexpressing TIF32 should suppress the inhibitory effect of NIP1 overexpression. Supporting these predictions, a high-copy (hc) plasmid bearing NIP1 (hc NIP1) exacerbated the Slg− phenotype of rnp1 cells at 34°C, but an hc plasmid containing both NIP1 and TIF32 did not (Fig. 2F, rows 1, 2, and 4). Overexpressing TIF32 alone did not suppress the Slg− phenotype of the rnp1 mutant (Fig. 2F, row 3). Thus, it appears that rnp1 weakens association of TIF32 with other eIF3 subunits, but this defect is not severe enough to cause dissociation of TIF32 from eIF3 in otherwise WT cells. Disrupting the PRT1-TIF32 interaction could still impair eIF3 function in translation initiation, as discussed below.
The prt1-rnp1 mutation decreases 40S binding of eIF3, eIF1, and eIF5.
To determine whether the disruption of eIF3 interactions with HCR1 and other MFC components by the rnp1 mutation impairs assembly of 43S PICs in vivo, we measured binding of eIF3 subunits and other MFC components to 40S subunits in WCEs of mutant cells treated with formaldehyde. This treatment cross-links eIFs to 40S ribosomes in vivo, minimizing dissociation of PICs during sedimentation through sucrose gradients without the addition of heparin as a stabilizing agent. The cross-links are reversed by heat treatment prior to subjecting fractions to Western analysis of eIFs and Northern analysis of mRNA (23). As shown in the supplemental material, extracts of non-cross-linked WT cells showed few or no eIFs in the 40S region of the gradient, whereas treatment with HCHO led to significant amounts of eIFs in the 40S fractions (see Fig. S1A in the supplemental material). Using this technique, we observed a large decrease in 40S binding of eIF3 subunits in rnp1 cells compared to WT after a 30-min incubation at 37°C (Fig. 3A). In contrast, rnp1 cells grown at 25°C displayed a much smaller reduction in 40S-bound eIF3 (Fig. 3C). From quantification of Western data from five replicate experiments (Fig. 3B and data not shown), we determined that rnp1 reduces 40S-bound eIF3 subunits by ∼90% after 30 min or 1 h at 37°C in cell extracts. Smaller reductions of 40 to 60% in 40S binding of eIF1 and eIF5 were also observed; however, 40S binding of eIF2, 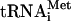 , eIF1A, and RPL41A mRNA occurred at essentially WT levels in extracts of rnp1 cells incubated at 37°C (Fig. 3B). It is noteworthy that 40S binding of HCR1, while reduced, was considerably higher than that of the five essential eIF3 subunits for rnp1 cells incubated at 37°C (Fig. 3A and B). This last result is in keeping with our previous findings that heat inactivation of eIF3 in cell extracts of the prt1-1 mutant led to much greater reductions in 40S binding of eIF3 core subunits than of HCR1 (30). Thus, rnp1 impairs 40S binding of only a subset of MFC constituents. The fact that rnp1 reduces the level of 40S-bound eIF3 without decreasing the level of TC bound to 40S subunits can be explained by proposing that rnp1 has a stronger effect on the conversion of 48S PICs to 80S initiation complexes than on TC recruitment (see Discussion).
, eIF1A, and RPL41A mRNA occurred at essentially WT levels in extracts of rnp1 cells incubated at 37°C (Fig. 3B). It is noteworthy that 40S binding of HCR1, while reduced, was considerably higher than that of the five essential eIF3 subunits for rnp1 cells incubated at 37°C (Fig. 3A and B). This last result is in keeping with our previous findings that heat inactivation of eIF3 in cell extracts of the prt1-1 mutant led to much greater reductions in 40S binding of eIF3 core subunits than of HCR1 (30). Thus, rnp1 impairs 40S binding of only a subset of MFC constituents. The fact that rnp1 reduces the level of 40S-bound eIF3 without decreasing the level of TC bound to 40S subunits can be explained by proposing that rnp1 has a stronger effect on the conversion of 48S PICs to 80S initiation complexes than on TC recruitment (see Discussion).
FIG. 3.

40S binding of eIF3 core subunits is reduced in prt1-rnp1 cells at the nonpermissive temperature. (A) PRT1 (H2879) or prt1-rnp1 (H3674) cells were grown in YPD at 25°C, heated for 30 min at 37°C, and cross-linked with 2% HCHO prior to harvesting. Twenty optical density units at 260 nm of WCE were separated by velocity sedimentation on a 7.5 to 30% sucrose gradient, and 0.7-ml fractions, numbered from the top of the gradient (7.5%), were divided into 0.2- and 0.5-ml aliquots and analyzed by Western and Northern analysis, respectively, to detect eIFs, 40S subunit protein RPS22, RPL41A mRNA, and 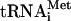 . The two to three fractions containing 40S subunits and 43S/48S preinitiation complexes are labeled above the Western blot as “40S.” Aliquots of the starting WCEs were analyzed in parallel (Input). (B) Amounts of each factor and RNA in the 40S fractions were quantified by phosphorimaging or fluorescence imaging analyses from five replicate experiments, and results for the rnp1 mutant were normalized to the corresponding PRT1 values and averaged. The data for the five eIF3 core subunits were combined and averaged (eIF3). The means and standard errors were plotted in the histogram. (C) Same as in panel A except for no heat treatment.
. The two to three fractions containing 40S subunits and 43S/48S preinitiation complexes are labeled above the Western blot as “40S.” Aliquots of the starting WCEs were analyzed in parallel (Input). (B) Amounts of each factor and RNA in the 40S fractions were quantified by phosphorimaging or fluorescence imaging analyses from five replicate experiments, and results for the rnp1 mutant were normalized to the corresponding PRT1 values and averaged. The data for the five eIF3 core subunits were combined and averaged (eIF3). The means and standard errors were plotted in the histogram. (C) Same as in panel A except for no heat treatment.
Overexpressing HCR1 alleviates the initiation defect in prt1-rnp1 cells.
Consistent with our finding that rnp1 leads to substantial dissociation of HCR1 from eIF3 in vivo, we found that overexpressing HCR1 partially suppressed the Slg− phenotype of rnp1 cells at 33°C. Even stronger suppression was achieved by overexpressing a mutant version of HCR1 (HCR1-R215I) shown previously to suppress a different prt1 mutation more effectively than did hc WT HCR1 (44) (Fig. 4A). The hc HCR1-R215I plasmid also increased the amounts of eIF3 subunits, and of HCR1 itself, in the 40S fractions of the rnp1 mutant incubated at 37°C for 1 h (Fig. 4B). Quantification of multiple cross-linking experiments indicated a 2.5-fold increase in 40S-bound eIF3 produced by hc HCR1-R215I in the rnp1 mutant extract (Fig. 4D), whereas hc HCR1-R215I had no effect on 40S binding of eIF3 for the PRT1 strain (Fig. 4C and data not shown). Although we observed small increases in 40S binding of eIF5 and eIF1, the levels of these factors, and of eIF3 as well, bound to 40S subunits remained below the WT levels in extracts of rnp1 cells overexpressing HCR1-R215I, in accordance with the residual Slg− phenotype of rnp1 cells containing hc HCR1-R215I. These results suggest that the Slg− phenotype of the rnp1 mutant derives at least partly from reduced 40S-binding of eIF3 resulting from the defective interaction of HCR1 with the core eIF3 complex.
FIG.4.

Overexpression of HCR1-R215I reduces the Ts− phenotype and defective 40S binding of eIF3 in prt1-rnp1 cells. (A) Tenfold serial dilutions of PRT1 (H2879) and prt1-rnp1 (H3674) cells transformed with YEplac181, YEplac181-HCR1 (p3778), or YEplac181-HCR1-R215I (p3780) were spotted on SC-Leu medium and incubated at 25 or 33°C for 2 days. (B) Analysis of 40S-bound eIFs in HCHO cross-linked cells of prt1-rnp1 strain H3674 containing vector YEplac181 or YEplac181-HCR1-R215I (p3780) was conducted as described in Fig. 3A except that cells were grown in SC-Leu and treated at 37°C for 1 h. (C) Same as panel B but using PRT1 cells of strain H2879. (D) The experiment in panel B was repeated three to five times, and the results were quantified as described in Fig. 3B.
We showed previously that overexpressing a truncated form of TIF32 (tif32-Δ6-His) lacking a C-terminal segment that interacts with HCR1, PRT1, eIF2β and 18S rRNA produces a dominant Slg− phenotype. Replacement of WT TIF32 with the tif32-Δ6-His protein in the eIF3 complex leads to formation of a defective MFC lacking HCR1 and eIF2, and loss of one of the binding sites for the 40S subunit in eIF3 (41, 42). We found here that introducing a hc tif32-Δ6-His plasmid intensifies the Slg− phenotype in rnp1 cells (Fig. 5A). It also exacerbates the defects in 40S binding of eIF3 and possibly of eIF5 and eIF1 as well (Fig. 5B and C) compared to that shown above for the rnp1 mutation alone (Fig. 3A and B). These findings indicate that the rnp1 and tif32-Δ6-His mutations have additive effects in disrupting 40S binding by eIF3.
FIG. 5.

Overexpression of dominant-negative tif32-Δ6-His exacerbates the Ts− phenotype and defective 40S binding of eIF3 in prt1-rnp1 cells. (A) Serial dilutions of PRT1 (H2879) cells harboring vector YEp24 or prt1-rnp1 (H3674) cells harboring vector YEp24 or hc tif32-Δ6-His (p3927) were spotted on SC-Ura and incubated at 25 or 34°C for 3 and 6 days, respectively. (B and C) Analysis of 40S-bound eIFs in HCHO cross-linked cells of prt1-rnp1 strain H3674 containing empty vector or hc tif32-Δ6-His (p3927) was conducted and quantified essentially as described in Fig. 3A and B, based on three independent experiments.
Deletion of HCR1 reduces 40S binding by eIF3 and exacerbates the translation initiation defect in prt1-rnp1 cells.
Our findings that overexpressing HCR1-R215I partially suppressed the Slg− phenotype and defective 40S binding of eIF3 in extracts of rnp1 cells (Fig. 4A and B) suggested that HCR1 promotes 40S binding of eIF3 in vivo. Furthermore, the fact that greater 40S binding of HCR1 versus other eIF3 subunits was retained in rnp1 extracts (Fig. 3A and B) suggested that HCR1 makes a direct contact with the 40S subunit that facilitates recruitment of the core eIF3 complex. If so, deletion of HCR1 should reduce 40S binding by eIF3 in otherwise WT cells. In agreement with this prediction, hcr1Δ produced a marked decrease in 40S binding by the core eIF3 subunits and moderate reductions in eIF5 and eIF1 binding to 40S subunits (Fig. 6B and C).
FIG. 6.

Deletion of HCR1 exacerbates the Ts− phenotype in prt1-rnp1 cells and reduces 40S binding of eIF3 in PRT1 cells. (A) Serial dilutions of PRT1 (H2879), prt1-rnp1 (H3674), hcr1Δ (H3675), and prt1-rnp1 hcr1Δ (H3676) cells were spotted on YPD and incubated at 25 or 33°C for 2 and 2.5 days, respectively. (B and C) Analysis of 40S-bound eIFs in HCHO cross-linked cells of HCR1 (H2879) and hcr1Δ (H3675) strains was conducted and quantified essentially as described in Fig. 3A and B, based on four independent experiments. (D) The prt1-rnp1 mutation leads to leaky scanning that is not suppressed by hc HCR1-R215I. PRT1 strain H2879, prt1-rnp1 strain H3674, or transformants of H3674 harboring vector YEplac181, hc-HCR1 (p3778), or hc-HCR1-R215I (p3780) were transformed with pM199 containing a GCN4-lacZ construct with uORF1 alone or pM226 containing an elongated uORF1 that overlaps GCN4-lacZ in a different reading frame (shown schematically as constructs 1 and 2, respectively). Cells were grown in SC-Ura at 33°C, and β-galactosidase activities were measured in WCEs and are expressed in units of nanomoles of o-nitrophenyl-β-d-galactopyranoside hydrolyzed per minute per milligram of protein. Τhe activity measured for pM226 was normalized to that for pM199, and the normalized values from at least three independent transformants were averaged to obtain the means and standard errors plotted in the histogram.
Interestingly, we found that hcr1Δ intensifies the growth defect of rnp1 cells. As shown in Fig. 6A, combining the hcr1Δ and rnp1 mutations is nearly lethal at 33°C and produces a strong synthetic growth defect even at 25°C, a defect much greater than that conferred by hcr1Δ alone (Fig. 6A). These genetic data may indicate that HCR1 and the RNP1 motif in PRT1 make independent, additive contributions to 40S binding by eIF3 in vivo (see Discussion).
prt1-rnp1 increases leaky scanning in vivo.
Finally, we sought to determine whether the disruption of contacts between eIF3, eIF2, and eIF1 in the MFC produced by the rnp1 mutation (Fig. 2A to C) has an impact on scanning by the 48S complex apart from its deleterious effect on eIF3 binding to the 40S subunit. Hence, we investigated whether rnp1 elicits leaky scanning of an upstream AUG codon in GCN4 mRNA. Four short upstream ORFs (uORF1 to -4) mediate increased GCN4 translation in response to eIF2α phosphorylation in amino acid-starved cells. The 5′-proximal uORF1 is recognized by essentially all 43S complexes scanning from the cap and, after uORF1 translation, ∼50% of the 40S subunits can remain bound to the mRNA and resume scanning. Virtually all of these rescanning 40S subunits reinitiate at GCN4 when uORF2 to -4 are deleted from the leader, allowing a high constitutive level of GCN4 translation from constructs bearing uORF1 alone. In contrast, very few of the rescanning 40S complexes can reinitiate at GCN4 if uORF1 is elongated to overlap the beginning of the GCN4 ORF, resulting in low constitutive GCN4 translation (see Fig. 6D for schematics of these two constructs containing uORF1 alone upstream from GCN4.) Mutations that cause leaky scanning of the uORF1 AUG codon allow a fraction of 43S complexes scanning from the cap to bypass the elongated uORF1 and initiate at the GCN4 start codon instead. This phenotype was described previously for deletion of the FUN12 gene encoding eIF5B (7, 33). Because such a large fraction of ribosomes resume scanning after translating the WT version of uORF1 and subsequently reinitiate at GCN4, an increase in leaky scanning of the uORF1 AUG codon has little impact on the overall level of GCN4 translation for the construct containing WT uORF1. Hence, we calculated the ratio of GCN4 translation for the elongated-uORF1 versus WT-uORF1 constructs (elong-uORF1/WT-uORF1) in the prt1-rnp1 mutant and compared it to the corresponding ratio measured in PRT1 cells to normalize for the general reduction in translation efficiency conferred by rnp1.
The elong-uORF1/WT-uORF1 ratio is very low (0.02) in WT cells grown at 33°C, as expected from very low levels of leaky scanning of elongated uORF1. The elong-uORF1/WT-uORF1 ratio is ∼4-fold higher in the rnp1 mutant under the same conditions, suggesting a 4-fold increase in leaky scanning (Fig. 6D). This defect in uORF1 recognition was not suppressed by introducing the hc HCR1-R215I (or hc HCR1) plasmid (Fig. 6D) shown above to reduce the growth defect and deficient eIF3 binding to 40S subunits conferred by rnp1 at 33°C (Fig. 4A and B). These findings suggest that rnp1 impairs AUG recognition by a mechanism distinct from the defective binding of eIF3 subunits that occurs in this mutant.
DISCUSSION
To address the possible role of the PRT1 RRM in eIF3 function, we examined the in vivo consequences of mutating conserved residues in the RNP1 motif of the RRM. At the nonpermissive temperature, the rnp1 mutation confers a strong defect in cell growth and translation initiation (Fig. 1) that is associated with a marked decrease in the 40S binding of all essential core eIF3 subunits and moderate reductions in the 40S binding of MFC components eIF1 and eIF5 in extracts of cross-linked cells (Fig. 3). Affinity purification of the MFC showed that the essential core of eIF3 was nearly intact, but its association with eIF1, eIF2, and HCR1 was markedly reduced by rnp1 at 37°C (Fig. 2A to C). The reduced association of HCR1 with the eIF3 core is consistent with our finding that rnp1 impairs direct interaction of PRT1-NTD with both HCR1 and TIF32 in vitro (Fig. 2E). We also presented genetic evidence that the TIF32 interaction with PRT1-NTD is impaired by rnp1 in vivo (Fig. 2F) even though TIF32 is largely retained in the mutant eIF3 complex (Fig. 2A), presumably by its interactions with more C-terminal regions of PRT1 and with NIP1 (Fig. 2D) (42, 44). The reduced interaction of eIF1 and eIF2 with the rnp1 mutant eIF3 complex (Fig. 2A to C) could arise indirectly from an altered conformation of the TIF32-CTD, since this segment of TIF32 interacts with both eIF2β and eIF1 (42). The dissociation of HCR1 from the eIF3 core produced by rnp1 might play an additional role in weakening eIF2 and eIF1 interactions with eIF3 since deletion of HCR1 was shown previously to destabilize the MFC, especially impairing eIF1-eIF3 interaction (44).
It has been shown that mammalian HCR1/eIF3j is required for the stable 40S binding of purified eIF3 to 40S subunits in vitro (12, 19). Several findings indicate that the impaired interaction of HCR1 with prt1-rnp1 is partly responsible for the defective 40S binding by eIF3 core subunits observed here in rnp1 yeast cells. First, HCR1 binding to the 40S subunit was less impaired by rnp1 than was the 40S binding of eIF3 core subunits (Fig. 3A and B), suggesting that HCR1 can bind to 40S subunits independently of eIF3 in vivo. Second, overexpression of HCR1-R215I reduced the growth defect and increased the 40S binding of eIF3 core subunits in rnp1 cells (Fig. 4). Given that hc HCR1-R215I increased the amount of 40S-bound HCR1, it appears that reinstating the 40S-HCR1 interaction by mass action restores the ability of HCR1 to recruit eIF3 to 40S subunits in rnp1 cells. Third, we found that deletion of HCR1 reduced the 40S association of eIF3 core subunits (Fig. 6B and C), providing direct evidence that HCR1 contributes to 40S binding by yeast eIF3 in vivo.
We reported previously that 40S binding of eIF3 was not reduced in hcr1Δ cells when 43S PICs were stabilized with heparin (44) rather than fixed in vivo by HCHO cross-linking as done here (Fig. 6B and C). We have verified our previous finding that heparin treatment of extracts from non-cross-linked hcr1Δ cells reveals no defect in the 40S binding of eIF3 subunits, whereas extracts of HCHO cross-linked hcr1Δ cells examined in parallel display a clear reduction in 40S-bound eIF3 (Fig. S2A and B in the supplemental material). In view of the fact that mammalian eIF3j promotes 40S association of eIF3 in vitro (12, 19) and the genetic and biochemical data presented here indicating a role for HCR1/j in eIF3 binding to 40S subunits, we believe that the HCHO cross-linking data in Fig. 6B provide a reliable indication of the deleterious effect of hcr1Δ on the 40S binding of eIF3 in vivo. Considering that the requirement for mammalian eIF3j in 40S binding of eIF3 could be suppressed by the presence of single-stranded oligonucleotides that can bind to the 40S subunit (19), perhaps heparin serves the same role described for oligonucleotides and bypasses the requirement for HCR1/j for efficient 40S binding by yeast eIF3 in cell extracts.
It was demonstrated that TC, eIF1, and eIF1A can bypass the requirement for mammalian eIF3j in 40S binding of eIF3 in vitro (38). Thus, it is unclear whether eIF3j is important for eIF3 recruitment and PIC assembly in mammalian cells. Our findings on HCR1 are significant in showing that the yeast homolog of eIF3j is required for high-level 40S binding of eIF3 and the formation or stability of 43S PICs in the presence of native levels of all other initiation factors in vivo.
Our observation that deletion of HCR1 exacerbates the growth defect of prt1-rnp1 cells (Fig. 6A) suggests that the PRT1 RRM contributes to 40S association of eIF3 by an HCR1-independent mechanism in addition to promoting HCR1 binding to the eIF3 core complex. rnp1 reduces association of eIF2 with eIF3 in the MFC (Fig. 2A to C) but does not decrease the level of 40S-bound eIF2 (Fig. 3). We found recently that depletion of eIF2β in vivo leads to an ∼40% reduction in the 40S binding of eIF3 (18). Hence, by weakening eIF2-eIF3 association, rnp1 may decrease the ability of eIF2 to stabilize binding of the eIF3 core complex to 40S subunits in addition to impairing HCR1-eIF3 interaction on the ribosome. Decreasing both interactions simultaneously in the hcr1Δ prt1-rnp1 double mutant at 33°C could produce a nearly lethal reduction in the 40S binding of eIF3. Consistent with this hypothesis, we found that overexpressing the TC partially suppresses the Slg− phenotype and produces a small increase in the 40S binding of eIF3, in rnp1 cells at 33°C (see Fig. S3A, B, and D in the supplemental material). Both effects were intensified by simultaneously introducing hc HCR1-R215I (see Fig. S3C and E in the supplemental material), as expected if eIF2 and HCR1 independently promote eIF3 recruitment to the 40S subunit.
This last conclusion is also consistent with our finding that overexpressing the tif32-Δ6 dominant-negative allele exacerbates both the growth defect and the impairment of 40S binding of eIF3 in extracts of rnp1 cells (Fig. 5). Replacement of TIF32 with tif32-Δ6, which lacks the TIF32 CTD, yields an eIF3 complex that does not stably associate with eIF2 or HCR1 (42) and shows reduced 40S binding in vivo (41). Thus, overexpression of tif32-Δ6 would be expected to exacerbate defects in both HCR1- and eIF2-dependent mechanisms of eIF3 recruitment in rnp1 cells. The tif32-Δ6 mutation may also eliminate a direct contact between eIF3 and the 40S subunit (41).
Our findings that prt1-rnp1 and hcr1Δ led to reduced 40S binding of eIF3 might seem inconsistent with our previous conclusion that the N-terminal half of TIF32, NIP1, and eIF5 comprise a minimal ribosome binding unit that can interact with 40S subunits in vivo (41). However, deletion of the HCR1/eIF2β binding domain in the extreme C terminus of TIF32 in that study (the tif32-Δ6 mutation) produced a lower level of 40S binding by the resulting eIF3/eIF1/eIF5 mutant complex compared to WT MFC, and further deleting the adjacent PRT1 binding domain in TIF32 led to an additional reduction in 40S binding by the resulting TIF32-NTD/NIP1/eIF5 subcomplex (the minimal ribosome binding unit). Thus, these previous findings are fully consistent with the present conclusion that HCR1 and the PRT1 RRM both enhance but are not essential for the 40S binding of eIF3.
It was surprising that the prt1-rnp1 and hcr1Δ mutations did not reduce the 40S binding of the TC in view of other findings from our laboratory indicating interdependence in the 40S binding of eIF3 and TC. Thus, we reported that the 40S binding of TC was reduced between 40 and 80% by mutations in the NIP1/c-NTD (43) and when tif32-Δ6 was overexpressed in cells harboring the tif5-7A mutation in eIF5 that weakens eIF2-eIF3 interaction in the MFC (23). More recently, we found that depletion of the entire eIF3 complex in cells leads to an ∼50% reduction in 40S-bound TC (18). The discrepancy between these results and those presented in Fig. 3 for prt1-rnp1 can be resolved by proposing that the different mutations vary in their relative effects on TC recruitment versus conversion of PICs to 80S initiation complexes. We propose that the rnp1 and hcr1Δ mutations do not prevent eIF3 from associating with 40S subunits but, rather, increase its rate of dissociation as the principle way of reducing the steady-state binding of eIF3 to 40S subunits. This transient association with the 40S allows eIF3 to stimulate TC recruitment and, following dissociation of eIF3, a fraction of TC remains bound to 40S subunits, stabilized by the mRNA, eIF1A, and residual eIF5 and eIF1 present in the complexes. Due to the absence of eIF3, however, these incomplete PICs are converted very slowly to 80S complexes. The accumulation of such defective PICs containing TC would offset the decrease in formation of new PICs containing TC, yielding no net reduction in 40S-bound TC. In cells fully depleted of eIF3, by contrast, its function in stimulating TC recruitment would be completely eliminated, and this defect would outweigh the accumulation of PICs that results from inefficient PIC to 80S conversion in the absence of eIF3, to yield a net loss of 40S-bound TC. The steady-state reductions in 40S-bound TC produced by the NIP1-NTD mutations and by the hc tif32-Δ6 tif5-7A double mutation could be explained similarly by proposing that these lesions impair TC recruitment more than they impede conversion of 43S/48S PICs to 80S complexes.
Apart from the effects of rnp1 on 40S binding of eIF3, eIF1, and eIF5, we also obtained evidence that this mutation impairs AUG recognition during the scanning process, as manifested by an ∼4-fold increase in leaky-scanning of the GCN4 uORF1 start codon (Fig. 6D). This defect could be explained if the RNP1 motif in PRT1 interacts with mRNA and that weakening this interaction impairs recognition of the AUG codon by the scanning 48S complex. This model may seem unlikely considering that the RNP1 motif in PRT1 seems to mediate protein-protein interactions in eIF3. Another possibility is that the impaired 40S binding of eIF5 produced by rnp1 reduces the ability of eIF5 to stimulate GTP hydrolysis in the TC, allowing the resumption of scanning rather than subunit joining at the uORF1 start codon. This would be consistent with the proposal presented above that rnp1 slows conversion of PICs to 80S complexes. The fact that overexpressing HCR1-R215I did not reduce the degree of leaky scanning does not eliminate this second possibility because hc HCR1-R215I did not effectively rescue eIF5 binding to the 40S subunit in rnp1 cells. It remains to be determined whether the leaky scanning phenotype in rnp1 cells arises only from the defective 40S binding of eIF3 and attendant reductions in eIF5 and eIF1 recruitment or instead reflects a more direct function of eIF3 in AUG recognition or subunit joining.
Supplementary Material
Acknowledgments
We thank Ernest Hannig and Jan van't Riet for kindly providing GCD11 antiserum and RPS22 antiserum, respectively; Tom Dever, Mikkel A. Algire, and Jon R. Lorsch for suggestions and critical reading of the manuscript; and members of the Hinnebusch and Dever laboratories for helpful discussions.
This research was supported (in part) by the Intramural Research Program of the NIH, NICHD.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Algire, M. A., D. Maag, and J. R. Lorsch. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20:251-262. [DOI] [PubMed] [Google Scholar]
- 2.Algire, M. A., D. Maag, P. Savio, M. G. Acker, S. Z. Tarun, Jr., A. B. Sachs, K. Asano, K. H. Nielsen, D. S. Olsen, L. Phan, A. G. Hinnebusch, and J. R. Lorsch. 2002. Development and characterization of a reconstituted yeast translation initiation system. RNA 8:382-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano, K., J. Clayton, A. Shalev, and A. G. Hinnebusch. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14:2534-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asano, K., T. Krishnamoorthy, L. Phan, G. D. Pavitt, and A. G. Hinnebusch. 1999. Conserved bipartite motifs in yeast eIF5 and eIF2Bɛ, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 18:1673-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asano, K., L. Phan, J. Anderson, and A. G. Hinnebusch. 1998. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J. Biol. Chem. 273:18573-18585. [DOI] [PubMed] [Google Scholar]
- 6.Asano, K., L. Phan, L. Valasek, L. W. Schoenfeld, A. Shalev, J. Clayton, K. Nielsen, T. F. Donahue, and A. G. Hinnebusch. 2001. A multifactor complex of eIF1, eIF2, eIF3, eIF5, and tRNA(i)Met promotes initiation complex assembly and couples GTP hydrolysis to AUG recognition. Cold Spring Harbor Symp. Quant. Biol. 66:403-415. [DOI] [PubMed] [Google Scholar]
- 7.Choi, S. K., J. H. Lee, W. L. Zoll, W. C. Merrick, and T. E. Dever. 1998. Promotion of Met-tRNAiMet binding to ribosomes by yIF2, a bacterial IF2 homolog in yeast. Science 280:1757-1760. [DOI] [PubMed] [Google Scholar]
- 8.Cigan, A. M., M. Foiani, E. M. Hannig, and A. G. Hinnebusch. 1991. Complex formation by positive and negative translational regulators of GCN4. Mol. Cell. Biol. 11:3217-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deo, R. C., J. B. Bonanno, N. Sonenberg, and S. K. Burley. 1999. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98:835-845. [DOI] [PubMed] [Google Scholar]
- 10.Donahue, T. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae, p. 487-502. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 11.Fekete, C. A., D. J. Applefield, S. A. Blakely, N. Shirokikh, T. Pestova, J. R. Lorsch, and A. G. Hinnebusch. 2005. The eIF1A C-terminal domain promotes initiation complex assembly, scanning, and AUG selection in vivo. EMBO J. 24:3588-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser, C. S., J. Y. Lee, G. L. Mayeur, M. Bushell, J. A. Doudna, and J. W. Hershey. 2004. The j-subunit of human translation initiation factor eIF3 is required for the stable binding of eIF3 and its subcomplexes to 40S ribosomal subunits in vitro. J. Biol. Chem. 279:8946-8956. [DOI] [PubMed] [Google Scholar]
- 13.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 14.Grant, C. M., P. F. Miller, and A. G. Hinnebusch. 1994. Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol. Cell. Biol. 14:2616-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He, H., T. von der Haar, C. R. Singh, M. Ii, B. Li, A. G. Hinnebusch, J. E. McCarthy, and K. Asano. 2003. The yeast eukaryotic initiation factor 4G (eIF4G) HEAT domain interacts with eIF1 and eIF5 and is involved in stringent AUG selection. Mol. Cell. Biol. 23:5431-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hershey, J. W. B., and W. C. Merrick. 2000. Pathway and mechanism of initiation of protein synthesis, p. 33-88. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 17.Hinnebusch, A. G. 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes, p. 185-243. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 18.Jivotovskaya, A. V. 2006. eIF3 and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol. Cell 26:1355-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolupaeva, V. G., A. Unbehaun, I. B. Lomakin, C. U. Hellen, and T. V. Pestova. 2005. Binding of eukaryotic initiation factor 3 to ribosomal 40S subunits and its role in ribosomal dissociation and anti-association. RNA 11:470-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maag, D., C. A. Fekete, Z. Gryczynski, and J. R. Lorsch. 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell 17:265-275. [DOI] [PubMed] [Google Scholar]
- 21.Majumdar, R., A. Bandyopadhyay, and U. Maitra. 2003. Mammalian translation initiation factor eIF1 functions with eIF1A and eIF3 in the formation of a stable 40S preinitiation complex. J. Biol. Chem. 278:6580-6587. [DOI] [PubMed] [Google Scholar]
- 22.Moehle, C. M., and A. G. Hinnebusch. 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 11:2723-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nielsen, K. H., B. Szamecz, L. Valasek, A. Jivotovskaya, B. S. Shin, and A. G. Hinnebusch. 2004. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23:1166-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohlmann, T., D. Prevot, D. Decimo, F. Roux, J. Garin, S. J. Morley, and J. L. Darlix. 2002. In vitro cleavage of eIF4GI but not eIF4GII by HIV-1 protease and its effects on translation in the rabbit reticulocyte lysate system. J. Mol. Biol. 318:9-20. [DOI] [PubMed] [Google Scholar]
- 25.Olsen, D. S., S. E. M., A. Mathew, F. Zhang, T. Krishnamoorthy, L. Phan, and A. G. Hinnebusch. 2003. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parent, S. A., C. M. Fenimore, and K. A. Bostian. 1985. Vector systems for the expression, analysis and cloning of DNA sequences in Saccharomyces cerevisiae. Yeast 1:83-138. [DOI] [PubMed] [Google Scholar]
- 27.Pestova, T. V., S. I. Borukhov, and C. U. T. Hellen. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394:854-859. [DOI] [PubMed] [Google Scholar]
- 28.Pestova, T. V., and V. G. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestova, T. V., I. B. Lomakin, J. H. Lee, S. K. Choi, T. E. Dever, and C. U. T. Hellen. 2000. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403:332-335. [DOI] [PubMed] [Google Scholar]
-
30.Phan, L., L. W. Schoenfeld, L. Valášek, K. H. Nielsen, and A. G. Hinnebusch. 2001. A subcomplex of three eIF3 subunits binds eIF1 and eIF5 and stimulates ribosome binding of mRNA and
 . EMBO J. 20:2954-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
. EMBO J. 20:2954-2965. [DOI] [PMC free article] [PubMed] [Google Scholar] - 31.Phan, L., X. Zhang, K. Asano, J. Anderson, H. P. Vornlocher, J. R. Greenberg, J. Qin, and A. G. Hinnebusch. 1998. Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol. Cell. Biol. 18:4935-4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherman, F., G. R. Fink, and C. W. Lawrence. 1974. Methods of yeast genetics, p. 61-64. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 33.Shin, B. S., D. Maag, A. Roll-Mecak, M. S. Arefin, S. K. Burley, J. R. Lorsch, and T. E. Dever. 2002. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111:1015-1025. [DOI] [PubMed] [Google Scholar]
- 34.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh, C. R., C. Curtis, Y. Yamamoto, N. S. Hall, D. S. Kruse, H. He, E. M. Hannig, and K. Asano. 2005. Eukaryotic translation initiation factor 5 is critical for integrity of the scanning preinitiation complex and accurate control of GCN4 translation. Mol. Cell. Biol. 25:5480-5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh, C. R., H. He, M. Ii, Y. Yamamoto, and K. Asano. 2004. Efficient incorporation of eukaryotic initiation factor 1 into the multifactor complex is critical for formation of functional ribosomal preinitiation complexes in vivo. J. Biol. Chem. 279:31910-31920. [DOI] [PubMed] [Google Scholar]
- 37.Smith, D. B., and K. S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31-40. [DOI] [PubMed] [Google Scholar]
- 38.Unbehaun, A., S. I. Borukhov, C. U. Hellen, and T. V. Pestova. 2004. Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev. 18:3078-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valášek, L., J. Hašek, K. H. Nielsen, and A. G. Hinnebusch. 2001. Dual function of eIF3j/Hcr1p in processing 20 S Pre-rRNA and translation initiation. J. Biol. Chem. 276:43351-43360. [DOI] [PubMed] [Google Scholar]
- 40.Valášek, L., J. Hašek, H. Trachsel, E. M. Imre, and H. Ruis. 1999. The Saccharomyces cerevisiae HCRI gene encoding a homologue of the p35 subunit of human translation eukaryotic initiation factor 3 (eIF3) is a high copy suppressor of a temperature-sensitive mutation in the Rpg1p subunit of yeast eIF3. J. Biol. Chem. 274:27567-27572. [DOI] [PubMed] [Google Scholar]
- 41.Valášek, L., A. Mathew, B. S. Shin, K. H. Nielsen, B. Szamecz, and A. G. Hinnebusch. 2003. The yeast eIF3 subunits TIF32/a and NIP1/c and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev. 17:786-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valášek, L., K. H. Nielsen, and A. G. Hinnebusch. 2002. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21:5886-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valasek, L., K. H. Nielsen, F. Zhang, C. A. Fekete, and A. G. Hinnebusch. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 24:9437-9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valášek, L., L. Phan, L. W. Schoenfeld, V. Valásková, and A. G. Hinnebusch. 2001. Related eIF3 subunits TIF32 and HCR1 interact with an RNA recognition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J. 20:891-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westermann, P., and O. Nygard. 1984. Cross-linking of mRNA to initiation factor eIF-3, 24-kDa cap binding protein and ribosomal proteins S1, S3/3a, S6 and S11 within the 48S pre-initiation complex. Nucleic Acids Res. 12:8887-8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yarunin, A., V. G. Panse, E. Petfalski, C. Dez, D. Tollervey, and E. Hurt. 2005. Functional link between ribosome formation and biogenesis of iron-sulfur proteins. EMBO J. 24:580-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.