

Abstract
The expression of histone deacetylase-related protein (HDRP) is reduced in neurons undergoing apoptosis. Forced reduction of HDRP expression in healthy neurons by treatment with antisense oligonucleotides also induces cell death. Likewise, neurons cultured from mice lacking HDRP are more vulnerable to cell death. Adenovirally mediated expression of HDRP prevents neuronal death, showing that HDRP is a neuroprotective protein. Neuroprotection by forced expression of HDRP is not accompanied by activation of the phosphatidylinositol 3-kinase-Akt or Raf-MEK-ERK signaling pathway, and treatment with pharmacological inhibitors of these pathways fails to inhibit the neuroprotection by HDRP. Stimulation of c-Jun phosphorylation and expression, an essential feature of neuronal death, is prevented by HDRP. We found that HDRP associates with c-Jun N-terminal kinase (JNK) and inhibits its activity, thus explaining the inhibition of c-Jun phosphorylation by HDRP. HDRP also interacts with histone deacetylase 1 (HDAC1) and recruits it to the c-Jun gene promoter, resulting in an inhibition of histone H3 acetylation at the c-Jun promoter. Although HDRP lacks intrinsic deacetylase activity, treatment with pharmacological inhibitors of histone deacetylases induces apoptosis even in the presence of ectopically expressed HDRP, underscoring the importance of c-Jun promoter deacetylation by HDRP-HDAC1 in HDRP-mediated neuroprotection. Our results suggest that neuroprotection by HDRP is mediated by the inhibition of c-Jun through its interaction with JNK and HDAC1.
Apoptosis is an essential aspect of normal nervous system development, but when aberrantly activated, apoptosis leads to undesirable neuronal death, and such inappropriate neuronal loss is the hallmark of a variety of neurodegenerative diseases and neurological conditions, such as stroke or traumatic brain injury. The mechanisms underlying the regulation of apoptosis are beginning to be understood. Among the molecules that have recently been implicated are the histone deacetylases (HDACs). HDACs are the catalytic subunits of multiprotein complexes that deacetylate histones (11, 42). The action of HDACs is opposed by histone acetyltransferases (HATs) such as CREB-binding protein and p300, which catalyze the transfer of an acetyl moiety from acetyl-coenzyme A to specific lysine residues of histones (25). Acetylation of histones relaxes the chromatin structure to a state that is transcriptionally active, while histone deacetylation transforms chromatin to a transcriptionally repressed state (25). Hence, gene expression is regulated, in part, by the balance of HDAC and HAT activities. Although best studied for their effects on histones and transcriptional activity, it is now known that HDACs and HATs regulate the acetylation of a number of other nonhistone proteins, such as p53, p65/RelA, E2F1, GATA1, and MyoD, suggesting complex functions of HDACs in different cellular processes (11, 42). Precisely which cellular functions are involved is currently the subject of intense investigation.
Vertebrates express at least 18 distinct HDACs, which have been grouped into three classes based on their similarities with Saccharomyces cerevisiae HDACs (11, 42). Class I HDACs (HDACs 1, 2, 3, 8, and 11) are homologous to yeast Rpd3. Class II HDACs (HDACs 4, 5, 6, 7, 9, and 10) are homologous to yeast Hda1. The highly conserved C termini of class I and class II HDACs contain a catalytic domain, which associates with transcriptional corepressors such as N-CoR, SMRT, and B-CoR within the nucleus. The third class of deacetylases, called sirtuins (SIRT1-7), have catalytic domains similar to that of the yeast NAD+-dependent deacetylase Sir2 (8). While serving as HDACs in yeast, in mammalian cells SIRTs are involved in the deacetylation of other proteins, rather than histones, and hence are not considered classical HDACs.
Class I HDACs consist of little more than a deacetylase domain and function as transcriptional repressors. They generally are nuclear proteins expressed in most tissue and cell types (11, 42). On the other hand, members of the class II HDAC subfamily display cell type-restricted patterns of expression and contain a large extended N-terminal extension with which a variety of signaling proteins interact, including MEF2, HP1α, Bcl6, CtBP, calmodulin, and 14-3-3 (11, 42). Phosphorylation of conserved serine residues in class II HDACs by calcium/calmodulin-dependent kinase (CaMK) or protein kinase D in response to specific stimuli creates docking sites for the 14-3-3 family of protein chaperones (11, 28, 31, 42). Binding of 14-3-3 results in the export of these HDACs from the nucleus and disrupts their interactions with transcriptional corepressor proteins, resulting in derepression of their target genes.
Several classes of small-molecule HDAC inhibitors have been identified (11, 29). Treatment of cultured cells with such HDAC inhibitors has a variety of effects, including transformation, differentiation, cell survival, and cell death, implicating HDACs in many different biological processes (11, 29). Because of their ability to induce the death of transformed cells, HDAC inhibitors are in clinical trials for the treatment of cancers. It is noteworthy, however, that while there are small differences in the sensitivities of individual class I and class II HDACs to different inhibitors, most of the commonly used inhibitors inhibit all HDACs efficiently. The significance of individual HDACs in any biological effect has thus been difficult to ascertain using inhibitors. Despite this limitation, a number of laboratories have used such pharmacological inhibitors to investigate the involvement of HDACs in the regulation of neuronal survival both in culture and in animal models of neurological disease (24). These studies have provided conflicting results. For example, the administration of HDAC inhibitors reduced neuronal loss in a Drosophila and a mouse model of Huntington's disease (13, 19). Treatment of cultured cortical neurons with HDAC inhibitors has also been reported to have a protective effect (36). While neuroprotection by HDAC inhibitors in experimental systems has prompted their consideration as therapeutic agents in the treatment of neurological diseases, it has been reported that treatment of cultured cerebellar granule neurons (CGNs) with HDAC inhibitors actively promotes cell death (4, 5, 37).
In this study, we have investigated the role of HDACs in the regulation of neuronal survival, using cultured cerebellar granule neurons. We report that an alternatively spliced form of a class II HDAC lacking intrinsic enzymatic activity, histone deacetylase-related protein (HDRP)/MITR, has neuroprotective functions. We provide evidence indicating that HDRP acquires deacetylase activity by the recruitment of HDAC1. Together, HDRP and HDAC1 inhibit low-potassium-medium (LK medium)-induced acetylation of the c-Jun gene promoter, thus inhibiting apoptosis-associated c-Jun expression. Furthermore, HDRP interacts with c-Jun N-terminal kinase (JNK) and inhibits its ability to phosphorylate and activate c-Jun.
MATERIALS AND METHODS
Antibodies and other reagents.
Cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA). All chemicals were purchased from Sigma (St. Louis, MO), unless specified otherwise. Antibodies to the following proteins were used: c-Myc, c-Jun, HDAC9, green fluorescent protein (GFP), and α-tubulin (all from Santa Cruz Biotechnology, Santa Cruz, CA); p-Akt, p-ERK, p-GSK3, HDAC1, p-JNK, and JNK (all from Cell Signaling Technology, Beverly, MA); and acetyl-histone H3 (Upstate Cell Signaling Solutions, Lake Placid, NY). Primary antibodies were used at a 1:1,000 dilution. Secondary antibodies (Santa Cruz Biotechnology) were peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; 1:10,000), donkey anti-goat IgG (1:5,000), and goat anti-mouse IgG (1:10,000). Chromatin immunoprecipitation assay kits were purchased from Upstate Cell Signaling Solutions, and JNK assay kits were obtained from Cell Signaling. A JNK inhibitor peptide was purchased from AXXORA Biochemicals (San Diego, CA).
Plasmids, clones, and adenoviruses.
Glutathione S-transferase (GST)-JNK1, -2, and -3 vectors were a gift from Chia-Yi Kuan, Cincinnati Children's Hospital. Full-length HDRP cDNA and the partial domain cDNAs “N” (corresponding to amino acids 1 to 343) and “N1” (corresponding to amino acids 175 to 343) were obtained by PCR from a human fetal brain library and were subcloned into the pGBKT7 vector (Clontech). A recombinant adenovirus expressing GFP was a kind gift from Kim A. Heidenreich (University of Colorado Health Sciences Center, Denver, CO). The HDRP (c-Myc tag) adenoviral vector was created in the lab of Eric N. Olson.
Cell culture.
Cerebellar granule neurons were obtained from dissociated cerebella of 7- to 8-day-old Wistar rats or C57BL/6 mice as described previously (12). Animals were treated in accordance with the on-site Institutional Animal Care and Use Committee policies as well as those prescribed by the National Institutes of Health. Briefly, cells were plated in basal Eagle's medium with Earle's salts (BME) supplemented with 10% fetal bovine serum, 25 mM KCl, 2 mM glutamine (Gibco-BRL), and 100 μg/ml gentamicin on dishes coated with poly-l-lysine at a density of 1 × 106 cells/well in 24-well dishes, 1.2 × 107 cells/60-mm dish, or 3.0 × 107 cells/100-mm dish. Cytosine arabinofuranoside (10 μM) was added to the culture medium 18 to 22 h after plating to prevent replication of nonneuronal cells. Cultures were maintained for 6 to 7 days prior to experimental treatments. Upon treatment, the cells were rinsed once and then maintained in LK medium (serum-free BME medium, 5 mM KCl) or HK medium (serum-free BME medium, 20 mM KCl). Treatment of cultures with pharmacological inhibitors was initiated at the time of switching the medium to HK or LK medium, unless specified otherwise. Neuronal viability was assayed by the MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay as previously described (9, 22). Briefly, the tetrazolium salt MTT was added to the cultures at a final concentration of 1 mg/ml, and incubation of the cultures was continued in a CO2 incubator for another 30 min at 37°C. The assay was stopped by adding lysis buffer (20% sodium dodecyl sulfate [SDS] in 50% N,N-dimethyl formamide, pH 4.7). The absorbance was measured spectrophotometrically at 570 nm after an overnight incubation at room temperature. The absorbance of a well without cells was used as the background and subtracted from the experimental values. Viability was also quantified by staining chromatin with 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI) as previously described (22, 45).
Adenovirus-mediated overexpression.
An adenoviral expression vector encoding c-Myc-tagged HDRP (Ad-HDRP) and a recombinant adenovirus expressing green fluorescent protein (Ad-GFP) were used to infect 4- to 5-day-old granule neuron cultures at a multiplicity of infection of 50. Briefly, serum-containing medium was removed, saved, and replaced with serum-free HK medium containing 4 μg/ml of hexadimethrin bromide (Sigma) and virus. Infection was allowed to persist for 1 h before the cells were washed once and the original serum-containing medium was added back. Treatments were performed 48 h after infection. Infected neurons were detected by GFP fluorescence or by positive staining for c-myc by immunocytochemistry procedures as described previously (22). Cerebellar granule neurons positive for GFP or c-myc staining were analyzed for viability by DAPI staining. For subcellular localization analysis, images were obtained using a Leica DM IRE2 confocal microscope.
RT-PCR.
RNAs were extracted from cultured neurons by using Trizol (Invitrogen) according to the manufacturer's instructions. RNAs were normalized, and cDNAs were made from 5 μg RNA using the Thermoscript reverse transcriptase PCR (RT-PCR) system (Invitrogen) according to the manufacturer's instructions. PCR was performed with PCR master mix (Promega, Madison, WI). The primers used for PCR amplification were as follows: rat GAPDH forward, 5′-CCATCACCATCTTCCAGGAG-3′; rat GAPDH reverse, 5′-CCTGCTCACCACCTTCTTG-3′; rat HDRP forward, 5′-AACTTGAAGGTGCGGTCCA-3′; rat HDRP reverse, 5′-TTACAAATCCTGGAGCTAAAT-3′; rat HDAC9 forward, 5′-AAATCTATTGAACAACTGAAGCAACCAGGC-3′; rat HDAC9 reverse, 5′-AGCTCATTCCAAATGGTGTCACTGTCCACC-3′; rat β-actin forward, 5′-AGGACTCCTATGTGGGTGACGA-3′; and rat β-actin reverse, 5′-CGTTGCCAATAGTGATGACCTG-3′.
Western blotting.
The culture medium was removed, and the cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in lysis buffer (1% Triton, 20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM sodium EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and one protease inhibitor tablet). Protein concentrations were measured and normalized using the Bradford protein assay reagent (Bio-Rad, Hercules, CA). Following normalization, 40 μg of protein was subjected to Western blotting as previously described (22, 45). Immunoreactivity was examined by enhanced chemiluminescence (Amersham Bioscience, Piscataway, NJ).
Immunoprecipitation.
Cultured cells were treated accordingly and then washed twice with ice-cold PBS followed by lysis in lysis buffer. Lysates were centrifuged for 10 min at 10,000 × g at 4°C. The protein concentrations in supernatant fractions were determined and normalized using the Bradford protein assay reagent (Bio-Rad). Equal amounts of protein were incubated overnight with 1 μg of primary antibody at 4°C. Samples were then incubated with 20 μl protein A/G Plus-agarose (Santa Cruz) for 2 h at 4°C. Bead complexes were pelleted by centrifugation at 1,000 × g for 5 min at 4°C and washed four times with lysis buffer. Pellets were then resuspended in 3× SDS sample buffer, heated for 5 min at 95°C, and subjected to Western blotting.
Antisense oligonucleotide treatment.
Antisense DNA oligonucleotides were designed against rat RNA transcript segments common to both HDAC9 and HDRP. A control antisense oligonucleotide was generated with the same G/C content as the HDAC9-HDRP antisense DNA but with no appreciable homology to any rodent transcripts in the NCBI database. All antisense oligonucleotides were custom synthesized by Operon Biotechnologies (Huntsville, AL). To inhibit degradation, the oligonucleotides were generated with phosphorothioate linkages between the first three and last three nucleotides. The oligonucleotides were conjugated to Texas Red to visualize cells that had taken up the DNA. The sequences for the HDAC9-HDRP- and HDAC9-specific and control antisense oligonucleotides referred to in the manuscript are 5′-GGATCCACCACAGGCATC-3′, 5′-CTACCGTCAGGGAGGCTA-3′, and 5′-GGTCCACACGTCGTAGTC-3′, respectively. Antisense DNA was added directly to the medium of 4-day-old CGN cultures to a final concentration of 1, 2, or 4 μM. Cells were maintained for 48 h, after which the medium was switched to serum-free HK or LK medium containing antisense DNA at 1, 2, or 4 μM. After 24 h, cells were washed twice with ice-cold PBS to remove noninternalized oligonucleotides, and viability was assessed by DAPI staining.
JNK activity assay.
For JNK activity assays, a JNK assay kit from Cell Signaling Technology was used. The assay was performed in accordance with the manufacturer's instructions, with the exception of the reaction incubation time and the ATP concentration. Briefly, culture medium was removed, and cells were washed with ice-cold PBS and lysed with lysis buffer. Lysates were centrifuged for 10 min at 10,000 × g at 4°C. The protein concentrations in supernatant fractions were determined and normalized using the Bradford protein assay reagent (Bio-Rad). Immunoprecipitation with immobilized c-Jun-conjugated beads was performed by adding 20 μl of the beads to normalized lysates. The mixture was incubated overnight at 4°C with gentle agitation. Immune complexes were centrifuged for 30 s at 10,000 × g at 4°C. Pellets were washed twice with lysis buffer and twice with kinase buffer (Cell Signaling). The pellets were then resuspended in 50 μl of kinase buffer containing 150 μM ATP and incubated for 12 min at 30°C. The addition of 25 μl of 3× SDS sample buffer stopped the reaction. Samples were boiled for 5 min and subjected to Western blotting. Phosphorylation of c-Jun at serine 63 was visualized using a phospho-c-Jun antibody (Cell Signaling).
ChIP.
Chromatin immunoprecipitation (ChIP) assays were carried out using a kit from Upstate Cell Signaling Solutions and the protocol provided by the manufacturer. Briefly, the neuronal cultures were fixed by the addition of formaldehyde to a final concentration of 1% and incubated for 10 min at 37°C. The medium was aspirated, and cells were washed twice with ice-cold PBS. Cells were then harvested, pelleted by centrifugation at 800 × g for 5 min (all centrifugation steps in this assay were performed at 4°C), and lysed in 200 μl SDS lysis buffer per 106 cells (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1). Each 200-μl aliquot of lysate was sonicated with 3 pulses of 12 seconds at 30% power. Samples were diluted 1:10 in ChIP dilution buffer (Upstate Cell Signaling Solutions) and precleared with protein A beads containing salmon sperm DNA (Upstate Cell Signaling Solutions). Acetylated histone H3 was then immunoprecipitated and washed with a low-salt wash (Upstate Cell Signaling Solutions), a high-salt wash (Upstate Cell Signaling Solutions), a lithium chloride wash (Upstate Cell Signaling Solutions), and two washes with Tris-EDTA buffer (Upstate Cell Signaling Solutions). The histone complex was eluted by the addition of 250 μl of elution buffer (1% SDS, 0.1 M NaHCO3). The immune complex was spun down at 800 × g for 2 min, and a second cycle of elution was repeated. Histone-DNA cross-links were reversed by adding 20 μl of 5 M NaCl and incubating the mixture at 65°C for 4 h. Protein was degraded by the addition of 10 μl EDTA, 20 μl 1 M Tris-HCl, pH 6.5, and 20 μg of proteinase K followed by incubation for 1 h at 45°C. Samples were then phenol-chloroform extracted (with yeast tRNA as a carrier), and nucleic acids were precipitated with ethanol, washed with 70% ethanol, and subjected to PCR. The primer sequences used to detect the rat c-Jun promoter (3) correspond to a region that extends from the transcription start site and terminates at the −504 position and are as follows: forward, 5′-TGTAACCTCTACTCCCACCCA-3′; and reverse, 5′-TCTGAGTCCTTATCCAGCCTG-3′.
UV light induction of apoptosis in HeLa cells.
HeLa cells were maintained in Dulbecco's modified Eagle's medium containing 10% newborn calf serum, 50 U/ml penicillin, and 50 μg/ml streptomycin in 60-mm dishes. Cells were infected with Ad-HDRP or Ad-GFP, and expression was allowed to persist for 48 h. The medium was removed and replaced with 2 ml of warm PBS. Cells were then irradiated with 20 J/m2 of UV light using a UV cross-linker (Fisher, Hampton, NH). The PBS was aspirated, the original medium was added back, and cells were incubated for 1 h before lysates were harvested. Western blotting was performed, and the membrane was probed with both c-Jun and phospho-JNK antibodies.
In vitro HDRP-JNK binding assay.
To further characterize the interaction between HDRP and JNK1, -2, and -3, binding assays were performed by using full-length HDRP, partial HDRP domains translated in vitro, and bacterially expressed GST-JNK1, -2, and -3 proteins. GST-JNK1, -2, and -3 fusion proteins were expressed in Escherichia coli strain BL21 by adding 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and were purified by being conjugated to glutathione-Sepharose beads (Sigma-Aldrich) according to the manufacturer's instructions. After extensive washing steps, immobilized fusion proteins on glutathione-agarose beads were directly used for in vitro binding assays without elution. In vitro-synthesized HDRP protein and the N and N1 domains were synthesized with [35S]methionine (MP Biomedicals, Inc., Irvine, CA) by using a TNT coupled lysate system (Promega). Five to 10 μg of GST or GST-fused JNK1, -2, and -3 immobilized on glutathione-Sepharose beads was incubated with 5 to 10 μl of 35S-labeled HDRP proteins in binding buffer (200 mM NaCl, 20 mM Tris-Cl, pH 7.6, 1 mM EDTA) for 3 h at 4°C and washed three times with the same buffer. After removal of the washing buffer, protein complexes were eluted by boiling with 20 to 40 μl of 2× SDS sample buffer (100 mM Tris-Cl, pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol, and 200 mM beta-mercaptoethanol) and then separated by SDS-polyacrylamide gel electrophoresis. Separated protein bands were detected by exposing dried gels to BioMax X-ray film (Eastman Kodak, Rochester, NY).
Statistical analysis.
All graphs present mean values for data obtained from two or more separate experiments. Standard deviations were used to determine error bars for experiments with three or more replicates. Statistical significance for all data sets was determined by using the unpaired t test, where P values of <0.05 were deemed significant.
RESULTS
Induction of neuronal apoptosis by treatment with HDAC inhibitors.
Granule neurons harvested from the postnatal rat or mouse cerebellum survive in medium containing depolarizing levels of potassium (HK medium). The lowering of potassium to nondepolarizing levels (LK medium) induces apoptosis (12). In this commonly used paradigm of neuronal apoptosis, commitment to cell death occurs within 6 h of LK treatment, although cell death itself is not evident until about 12 h (2). The survival-promoting effect of HK mimics the well-established need for electrical activity in the survival of several neuronal populations in vivo during nervous system development. In view of the findings of other investigators showing that pharmacological inhibitors of HDACs protect against neurodegeneration in certain in vivo and in vitro experimental paradigms (13, 19, 36), we examined whether LK-induced neuronal apoptosis could be prevented by HDAC inhibition. HDAC inhibitors failed to prevent LK-induced neuronal death (data not shown). On the contrary, HDAC inhibitors blocked the ability of HK medium to maintain neuronal survival. The utilization of three structurally distinct HDAC inhibitors argues against the possibility that the induction of neuronal death was due to an effect other than HDAC inhibition (Fig. 1A). This result is consistent with those of other investigators using cultured cerebellar granule neurons (4, 37). Since it was possible that long-term treatment with HDAC inhibitors was toxic and could thus mask potential neuroprotective efficacy, we treated neuronal cultures with HDAC inhibitors in HK medium for only 6 h, after which the cultures were washed and the medium replaced with HK medium without the inhibitor. While no signs of degeneration were evident at 6 h, the neurons that were treated with the inhibitors died to a similar extent as those that received the drug for the entire 24-h period (Fig. 1B). Substantial cell death was also observed after a mere 4 h of exposure to HDAC inhibitors (data not shown), indicating that commitment to death occurred within 4 to 6 h.
FIG. 1.

Effect of pharmacological inhibition of HDAC activity on neuronal survival. (A) Neuronal cultures were treated with HK medium containing different concentrations of TSA, HDACi, and sodium butyrate (NaB). Survival was determined using the MTT assay 24 h after treatment (means ± standard deviations [SD]; n = 3). All TSA-, HDACi-, and NaB-treated cultures showed a significant decrease in viability compared to HK cultures (P < 0.05). (B) To examine if HDAC inhibitors exhibit toxicity, cultured neurons were treated in HK medium with 5 μM TSA, 50 μM HDACi, or 100 μM NaB for 6 h and then switched to HK medium without inhibitors. Survival was determined by the MTT assay (means ± SD; n = 3). HDAC inhibitor addition resulted in significant reductions in neuronal survival compared to HK cultures (P < 0.05).
Decreased expression of HDRP and HDAC9 during neuronal apoptosis.
Since treatment with HDAC inhibitors in HK medium blocked neuronal survival, it was possible that apoptosis induced by stimuli such as LK treatment was caused by a downregulation of HDAC function. We investigated this possibility by initially performing RT-PCR analysis to examine the expression of class I and class II HDACs in healthy neurons and neurons primed to die by LK treatment. As shown in Fig. 2A, the expression of two class II HDACs, HDAC9 and HDAC5, was reduced by LK treatment. HDAC9 is known to be expressed in two major forms, a form containing an HDAC domain (henceforth referred to as HDAC9) and a truncated form lacking the HDAC domain, called HDRP/MITR (hereafter referred to as HDRP), that is generated as a result of alternative splicing (32, 48). While mRNAs encoding both forms are downregulated by LK treatment, the reduction of HDRP expression occurs more rapidly and to a greater extent than that of HDAC9 (Fig. 2B). This pattern of downregulation was also seen at the protein level (Fig. 2C). We also examined the expression of the various SIRT genes, which comprise the third deacetylase gene family. Although all seven SIRT genes are expressed in neurons, their expression was not significantly changed by LK treatment (Fig. 2D). Also, treatment of neuronal cultures with nicotinamide (doses of 0.1 to 1 mM), which inhibits the activity of SIRTs with a 50% inhibitory concentration of 50 μM, had no effect on neuronal survival in HK or LK medium (data not shown).
FIG. 2.

Expression of HDRP and other deacetylases during neuronal apoptosis. (A, B, and D) RT-PCR analysis was used to determine the RNA expression of indicated genes from cultures treated for 6 h with HK or LK medium. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin were used to demonstrate that similar quantities of sample were used. (C) HDAC9 protein expression in neuronal cultures subjected to time course treatment with HK or LK medium. Lysates were obtained from cultures treated for 0 (untreated), 1, 6, and 12 h with either HK or LK medium. Western blotting was performed, and the resulting membrane was probed with an HDAC9 antibody, followed by an α-tubulin antibody to confirm equal protein loading. HDRP-infected neuronal lysates served as a positive control for HDRP expression, while 3T3 cytoplasmic extract served as a negative control.
We also examined the effect of treatment with HDAC inhibitors on expression of the various HDAC genes. As shown in Fig. 3, treatment with both trichostatin A (TSA) and sodium butyrate (NaB) reduced HDRP expression. Interestingly, the expression of HDAC9 was not discernibly reduced by these inhibitors. Similarly, with the exception of HDAC7 expression, which was reduced by pharmacological HDAC inhibition, the expression of the other HDAC mRNAs was not altered to a significant extent (Fig. 3).
FIG. 3.
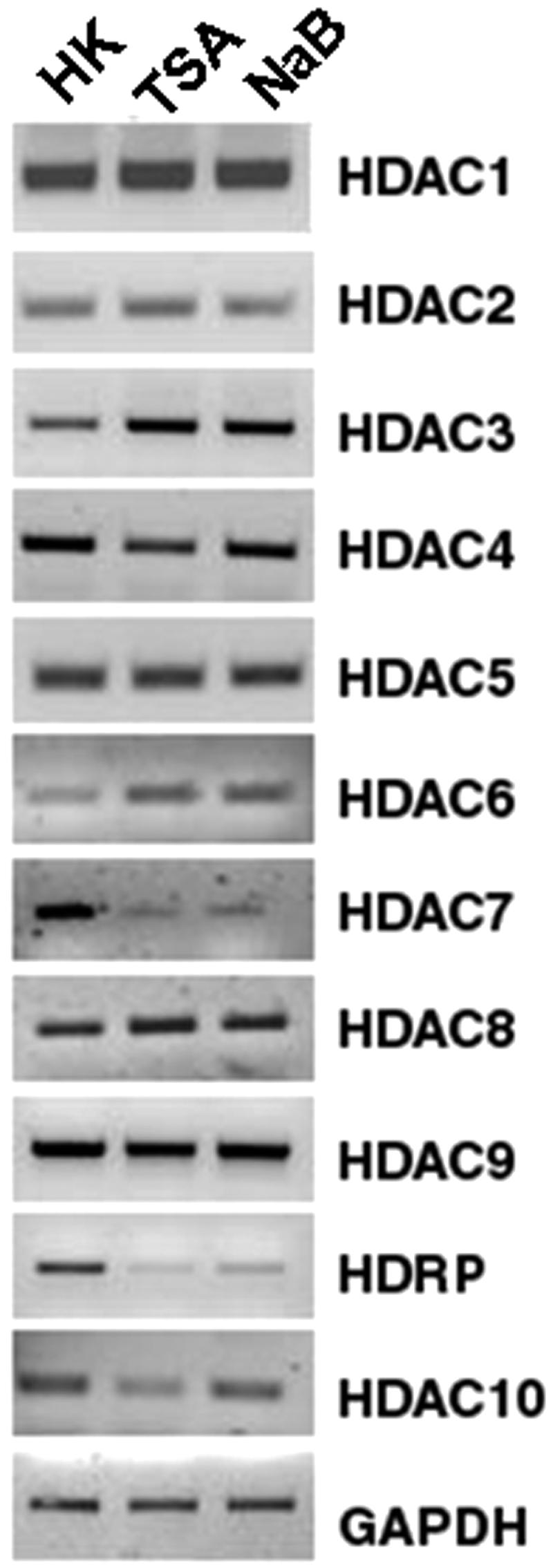
Effects of HDAC inhibition on HDAC mRNAs. Cultured CGNs were treated with HK medium, HK medium with 5 μM TSA, or HK medium with 2 mM NaB for 6 h. RNA levels for HDAC family members were determined using primers specific for individual HDAC members by RT-PCR. The levels of GAPDH were measured to ensure equal sample loading.
HDRP is involved in promoting the survival of cultured neurons.
To determine whether the reduction of HDRP and HDAC9 expression in neurons was correlative with or causative of neuronal death, we overexpressed HDRP in neuronal cells and examined if the presence of HDRP was neuroprotective. We focused initially on HDRP because its downregulation was more robust and because it was a smaller protein lacking an HDAC domain. As shown in Fig. 4A, the forced expression of HDRP using an adenoviral expression vector completely blocked LK-induced cell death, while cultures infected with a GFP-adenovirus construct and treated with LK showed an average viability of 59.4%. Since HDAC5 shares sequence similarity with HDAC9, is also expressed highly in the brain, and is downregulated by LK treatment, we examined whether its forced expression would also be protective. In contrast to HDRP, however, HDAC5 did not display any neuroprotection (N. Majdzadeh and S. R. D'Mello, unpublished observation).
FIG. 4.

Survival of neurons exhibiting altered HDAC9 or HDRP expression. (A) Neuronal cultures were infected with Ad-HDRP (Myc tagged) or Ad-GFP. Cells were then treated with HK or LK medium for 24 h, and the viability of infected cells was examined by immunocytochemistry and DAPI staining (means ± SD; n = 3). *, P > 0.05 for comparison with HDRP-LK. (B) Cerebellar granule neuron cultures were obtained from wild-type (+/+) and HDAC9/HDRP knockout (−/−) mouse littermates and treated for 24 h with either HK or LK medium. The proportions of cells that were apoptotic, as judged by condensed or fragmented nuclei based on DAPI staining, were then determined (means ± SD; n = 3). *, P < 0.05 for comparison with wild-type cells in LK medium; **, P < 0.05 for comparison with +/+ cells in HK medium. (C) HDAC9−/− and +/+ mice were obtained as in panel B and infected with Ad-HDRP or Ad-GFP. Neurons were then treated with HK or LK medium for 24 h, and the viability of infected neurons was assessed by immunocytochemistry.
To confirm that HDRP was involved in maintaining neuronal survival, we cultured cerebellar granule neurons from HDAC9 knockout mice (both forms are missing in the knockout mice) and wild-type littermates. While the HDAC9−/− neurons survived in vitro, switching them to either HK or LK medium resulted in decreased survival, at 90.4 and 45.7%, respectively, compared to neurons from wild-type littermates, at 100% for HK medium and 63.0% for LK medium (Fig. 4B). To verify that the increased vulnerability of HDAC9−/− mice was cell intrinsic and solely due to the absence of HDRP, we reexpressed HDRP in these mutant neurons by infection with Ad-HDRP. As shown in Fig. 4C, forced expression of HDRP abolished the increased susceptibility and protected HDAC9−/− neurons from LK-mediated death.
The higher vulnerability displayed by HDAC9−/− neurons confirms a role for HDRP in promoting neuronal survival. It is likely that compensation by other class II HDAC proteins masks the full role of HDRP in promoting neuronal survival. To examine this issue, we analyzed whether the expression of any other member of the HDAC family was upregulated in HDAC9−/− neurons treated with HK or LK medium. As shown in Fig. 5, the expression profiles of other HDACs remained unaltered, suggesting that compensatory effects by other class II HDACs in HDAC9−/− neurons may be mediated at the level of protein-protein interaction rather than by increased expression of other HDAC family members.
FIG. 5.

Expression of HDAC family members in HDAC9−/− neuronal cultures. The RNA expression of each HDAC family member was analyzed by RT-PCR, using specific primers. GAPDH RNA levels were measured to ensure equal sample loading.
To examine whether forced downregulation of HDRP expression also inhibited neuronal survival, we utilized antisense oligonucleotides. As shown in Fig. 6 (upper panels), treatment with antisense oligonucleotides that reduce the expression of both HDRP and HDAC9 also reduces neuronal viability. The maximum effect of HDAC9/HDRP knockdown was seen at an oligonucleotide concentration of 4 μM, where survival was reduced by 25.3% and 24.3% for HK and LK cultures, respectively. In contrast to HDRP, selective suppression of HDAC9 using an antisense oligonucleotide that does not affect HDRP expression (corresponding to the 3′ end of HDAC9 mRNA, which is missing in HDRP mRNA) does not affect neuronal survival (Fig. 6, lower panels). Thus, HDRP, but not HDAC9, is of importance to neuronal survival.
FIG. 6.

Antisense oligonucleotide-mediated suppression of HDRP and HDAC9 expression. Cultured neurons were pretreated (for 48 h) with antisense or control oligonucleotide DNA (conjugated to the Texas Red fluorochrome) by direct addition to the medium at the indicated concentrations. The culture medium was then switched to HK or LK medium containing antisense oligonucleotides at the indicated concentrations. The graphs show neuronal viability in HK or LK medium, expressed as percentages of the viability with control oligonucleotide. Survival was determined by examining the DAPI staining pattern of Texas Red-positive cells (means ± SD; n = 3). Asterisks indicate statistical significance (P < 0.05) compared with the corresponding control oligonucleotide at the given concentrations. Lysates from cultures treated with control (C) or antisense (AS) oligonucleotide were also analyzed by Western blotting using antibodies against HDAC9/HDRP, HDAC5 (to ensure the specificity of the antisense oligonucleotide), and tubulin (for equality of loading). (Top panels) Results of viability assays and Western blot analysis using an antisense oligonucleotide that binds both HDRP and HDAC9. (Bottom panels) Results using an antisense oligonucleotide specific for HDAC9. No concentration of HDAC9 antisense treatment showed a significant difference in viability compared to the control oligonucleotide.
Neuroprotection by HDRP is mediated in the nucleus.
HDRP lacks the nuclear export signal that has been mapped to the HDAC domains of other class II members (40, 47, 48). Not unexpectedly, therefore, HDRP localizes predominantly to the nucleus in a variety of cell types (47). In neurons maintained in HK medium, however, adenovirally expressed HDRP was abundant in the cytosol (Fig. 7). Upon LK treatment, HDRP was found almost exclusively within the nucleus (Fig. 7). In myoblasts, HDRP and other class II HDACs are phosphorylated by CaMK (28, 30, 31). In the case of HDAC4 and HDAC5, inhibition of CaMK prevents their export from the nucleus (28, 30, 31). It is well established that in neurons, membrane depolarization results in an influx of calcium (Ca2+) through L-type voltage-gated calcium channels, which in turn activates CaMK (15, 39). To test whether the cytoplasmic localization of HDRP in HK-treated neurons was CaMK dependent, we used KN-62, a highly selective pharmacological inhibitor of CaMK. As shown in Fig. 8, in neuronal cultures treated with KN-62, HDRP localized to the nucleus even in HK medium. Interestingly, CaMK inhibition had no effect on HDRP-mediated neuroprotection, although it did reduce HK-mediated neuronal survival (Fig. 8). Since HDRP was in the nucleus in the presence of KN-62 and neuronal viability was still intact, our results suggest that neuroprotection by HDRP does not require cytoplasmic localization and is CaMK independent.
FIG. 7.

Subcellular localization of HDRP. Cultured neurons were infected with Ad-HDRP (c-Myc tag) before replacement of the medium with HK or LK medium. After 24 h of treatment, the localization of adenovirally expressed HDRP was visualized using a c-Myc antibody and a Texas Red-conjugated secondary antibody (red). Chromatin was stained with Syto 13 (green) to visualize nuclei. (A) Representative images of neurons expressing exogenous HDRP in HK and LK medium. (B) Subcellular localization of HDRP quantified in neurons treated with HK and LK medium (means ± SD; n = 3). “N only” refers to the nucleus only, while “N + C” denotes nuclear and cytoplasmic localization. *, P < 0.05 for comparisons between both HK populations and between both LK populations.
FIG. 8.

Effect of Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62 on exogenous HDRP localization. Neuronal cultures were infected with Ad-HDRP (c-Myc tag) and then treated with HK medium or HK medium with 50 μM KN-62 for 24 h. HDRP was visualized with a c-Myc antibody and a Texas Red-conjugated secondary antibody (red). Nuclei were detected using the chromatin stain Syto 13 (green). (A) Representative images of neurons infected with Ad-HDRP and treated with HK medium or HK medium with KN-62. (B) The subcellular localization of exogenous HDRP was tabulated, and results from three independent experiments are shown in the graph (means ± SD). “N only” refers to the nucleus only, while “N + C” denotes nuclear and cytoplasmic localization. *, P < 0.05 for comparison between both HK and KN-62 populations; **, P < 0.05 for comparison between both HK-only populations.
Neuroprotection by HDRP does not require MEK-ERK or PI 3-kinase-Akt signaling.
The best studied antiapoptotic pathway in neurons is the phosphatidylinositol 3-kinase (PI 3-kinase)-Akt pathway. An important event in this pathway is the phosphorylation of Akt, which leads to its activation. Once activated, Akt phosphorylates a number of proapoptotic molecules, including the Bcl-2 protein BAD and glycogen synthase kinase 3 (GSK-3), leading to their inactivation (7). However, neither the reduced phosphorylation of Akt nor the increased phosphorylation of Gsk3β that normally results following LK treatment was affected by forced expression of HDRP (Fig. 9). These results suggest that HDRP does not activate the PI 3-kinase-Akt pathway. Supporting this conclusion is the finding that the pharmacological inhibition of this pathway using the highly selective PI 3-kinase inhibitor wortmannin or the Akt inhibitor ML-9 has no effect on HDRP-mediated neuroprotection (see Fig. S1 in the supplemental material).
FIG. 9.

Signaling mechanisms involved in HDRP-mediated neuroprotection. (A) Effect of HDRP expression on various signaling proteins. Neuronal cultures infected with the Ad-HDRP or Ad-GFP expression vector were treated with HK or LK medium for 3 h, and protein extracts were prepared and subjected to Western blot analysis using different antibodies sequentially on the same membrane. Equal protein loading was confirmed by probing the membrane with α-tubulin antibody. (B) c-Jun expression in HDAC9−/− neuronal cultures during forced expression of HDRP. Neuronal cultures were prepared from HDAC9−/− mice and infected with Ad-HDRP or Ad-GFP. Cells were then treated with HK or LK medium for 3 h. (Top panel) RNAs were extracted from treated neurons, and RT-PCR was performed using primers specific to the c-Jun transcript. Equal sample loading was ensured by examining GAPDH levels. (Bottom panel) Cellular protein was extracted from treated neurons, and Western blotting was performed. The membrane was then probed with antibodies against c-Jun and HDAC9. Equal protein loading was confirmed by probing the membrane with α-tubulin antibody.
Another antiapoptotic signaling pathway is the Raf-MEK-ERK pathway (18). LK treatment caused a reduction in the phosphorylation of MEK and ERK (Fig. 9A). The reduction in MEK or ERK phosphorylation was not prevented by HDRP expression. Also, treatment with two structurally distinct and highly selective MEK inhibitors, U0126 and PD98059 (see Fig. S1 in the supplemental material), had no effect on HDRP-mediated neuroprotection, arguing against the need for the Raf-MEK-ERK signaling pathway.
Ectopic HDRP inhibits c-Jun transcription and phosphorylation via interaction with HDAC1 and JNK.
Several studies have shown that the induction of neuronal apoptosis in vivo or in culture requires the activation of c-Jun (16, 44). Activated c-Jun stimulates its own transcription, leading to its increased expression. As shown in Fig. 9A, forced expression of HDRP prevents the stimulation of c-Jun phosphorylation and expression. We extended our analysis to neuronal cultures prepared from HDAC9−/− mice. The basal levels of c-Jun mRNA and protein were not substantially different in HDAC9-deficient neurons from those in wild-type cultures (data not shown), likely reflecting the relatively small difference in the vulnerabilities of these neurons in culture. LK treatment of these neurons increased c-Jun expression and phosphorylation (Fig. 9B). As observed with wild-type neurons, forced expression of HDRP in HDAC9−/− neurons inhibited the LK-mediated stimulation of c-Jun.
Phosphorylation by c-Jun is mediated by JNK, and the requirement for JNK in neuronal apoptosis is well documented (6, 23, 43). Three JNK proteins are expressed in mammalian cells, namely, JNK1, JNK2, and JNK3 (45, 46). In most cell types, JNK1 is expressed predominantly as a 46-kDa protein. JNK2 and JNK3 exist as 46- and 54-kDa proteins due to alternative splicing at the C terminus. Since c-Jun phosphorylation was inhibited by HDRP, it was likely that HDRP inhibited JNK activity. Western blot analysis confirmed that LK-induced JNK phosphorylation was reduced in Ad-HDRP-infected neurons (Fig. 10A). The reduction in phosphorylation was restricted to the 54-kDa form of JNK, suggesting that JNK2 and/or JNK3 was the target of HDRP inhibition. HDRP-mediated inhibition of JNK was confirmed in activity assays using immunoprecipitated JNK, with c-Jun as a substrate (Fig. 10B). To examine whether the inhibition of c-Jun by HDRP also occurred in nonneuronal cell types, we extended our studies to HeLa cells. As shown in Fig. 10C, the stimulation of c-Jun expression and phosphorylation that normally occurs after exposure to UV radiation was reduced in HeLa cells by HDRP. As observed in apoptotic neurons, HDRP also prevented the induction of JNK phosphorylation in HeLa cells. Once again, reduction was more pronounced with the 54-kDa form of JNK.
FIG. 10.

HDRP expression inhibits JNK phosphorylation and activity. (A) Neurons were infected with Ad-GFP or Ad-HDRP and treated with HK or LK medium for 3 h. Whole-cell lysates were obtained, Western blot analysis was performed using a phospho-JNK (T183/Y185) antibody, and an α-tubulin antibody was used to confirm equal protein loading. (B) JNK activity (upper panel) was assayed in neuronal cultures infected with Ad-HDRP or Ad-GFP and treated with HK or LK medium for 30 min. A truncated c-Jun-GST fusion peptide (c-Jun residues 1 to 89) conjugated to glutathione-Sepharose beads was used to immunoprecipitate JNK and also to act as a substrate in an in vitro kinase reaction using an antibody specific for c-Jun phosphorylated at Ser63. The specificity of JNK activity in the reaction was assessed by using controls in which the kinase reaction was performed in the presence of 20 μM of JNK inhibitor peptide (C1) or with agarose-beads alone (C2; no c-Jun substrate). The membrane was stripped and probed with phospho-JNK (T183/Y185) antibody to determine if active JNK was pulled down in the kinase assay (lower panel). (C) HeLa cells were infected with Ad-HDRP or Ad-GFP and subjected to UV light 48 h later. After exposure to UV light, the cells were incubated for 1 h, lysed, and subjected to Western blotting. The blot was probed with antibodies specific for c-Jun as well as phospho-JNK (T183/Y185). Sample size equality was confirmed using an α-tubulin antibody.
To examine the mechanism by which HDRP regulates JNK activity, we examined whether the two proteins associated physically. As shown in Fig. 11A, HDRP coimmunoprecipitated with JNK in CGNs. Furthermore, HDRP was pulled down by c-Jun-conjugated beads from Ad-HDRP-infected CGNs (Fig. 11B). Since the c-Jun beads pulled down JNK (Fig. 10B), this result shows that HDRP, JNK, and c-Jun are all part of a protein complex. To verify that HDRP interacts with JNK and to identify the form(s) of JNK involved in this interaction, we expressed JNK1, JNK2, and JNK3 as GST fusion proteins and used them in binding assays with in vitro-translated and 35S-labeled HDRP. As shown in Fig. 11C, full-length HDRP interacted with JNK3 but bound to only a modest extent with JNK2. A substantial interaction between JNK1 and HDRP was also observed. Using two deletion mutants of HDRP spanning amino acids 1 to 343 and 175 to 343, we have mapped the JNK interaction site to the region between residues 175 and 343 of HDRP (Fig. 11C). Neither of the two deletion constructs displayed significant binding to JNK1. This finding, along with the cytoplasmic localization of JNK1, suggests that JNK3 is the primary target of HDRP-mediated JNK inhibition.
FIG. 11.

Association of HDRP with JNK, HDAC1, and c-Jun complexes. (A) JNK (JNK1, -2, and -3), HDAC1, and HDRP were immunoprecipitated from Ad-HDRP-infected neurons treated for 3 h in HK or LK medium. Immunoprecipitates were subjected to Western blotting, and the membrane was probed with c-Myc antibody for the detection of exogenous HDRP. A bead-only control (C) was used to confirm the fidelity of the immunoprecipitation. (B) Truncated c-Jun-GST conjugated to glutathione-Sepharose beads was used to immunoprecipitate exogenous HDRP from Ad-HDRP-infected neuronal lysates that were treated with HK or LK medium for 3 h. Western blotting was performed on the immunoprecipitate using a c-Myc antibody to detect exogenous HDRP. The specificity of the interaction was confirmed using glutathione-Sepharose beads in place of the c-Jun beads. (C) For in vitro binding assays, p54 isoforms of JNK1-, JNK2-, and JNK3-GST fusion proteins were conjugated to glutathione-Sepharose beads and incubated with [35S]methionine-labeled HDRP and HDRP N (1-343) and N1 (175-343) domains. Associated proteins were resolved by SDS-polyacrylamide gel electrophoresis followed by autoradiography. 35S-labeled HDRP and the N and N1 domains interacted with immobilized GST-JNK1, -2, and -3 but not with the negative control, glutathione-Sepharose resin (beads). The input lane represents 10% of the [35S]methionine-labeled protein that was added to each reaction and serves as a reference marker.
Besides affecting c-Jun phosphorylation, HDRP also inhibited the LK-induced increase in c-Jun expression (Fig. 9A). It is well established that once activated, c-Jun stimulates its own transcription. Gene transcription is often regulated at the level of histone acetylation at the promoter. To examine if the inhibition of c-Jun expression by HDRP was associated with a reduction of promoter acetylation, we performed ChIP assays. As shown in Fig. 12A, histone H3 acetylation was readily detectable in cultures expressing GFP and treated with HK medium. The extent of acetylation was higher in LK medium, consistent with the higher level of c-Jun gene transcription. Forced HDRP expression reduced the extent of c-Jun promoter-associated acetylation under both LK and HK conditions. Since HDRP has no intrinsic HDAC activity, the inhibition of histone H3 acetylation at the c-Jun promoter is likely to involve another HDAC protein. Indeed, HDRP has been shown to acquire deacetylase activity by recruitment of HDAC1 or HDAC3 (48). We found that HDRP is pulled down with an antibody that immunoprecipitates HDAC1 (Fig. 11A). In contrast, an association between HDRP and HDAC3 could not be detected (data not shown). Consistent with HDAC1 involvement in HDRP-mediated c-Jun promoter repression is the finding that HDAC1 associates with the c-Jun promoter (Fig. 12B). The interaction was increased substantially in LK-treated cultures upon forced expression of HDRP, suggesting that neuroprotection by HDRP is mediated by the recruitment of HDAC1 to the c-Jun promoter, permitting histone deacetylation of the promoter. Since HDRP also interacts with JNK, our results suggest that HDRP, JNK, and HDAC1 are part of a complex. Supporting this conclusion, we found that immunoprecipitation of neuronal lysates with an HDAC1 antibody pulls down JNK (Fig. 12C). Consistent with our previous observation indicating that JNK3 is the target of HDRP, HDAC1 also bound primarily to the 54-kDa form of JNK (Fig. 12C).
FIG. 12.

Histone H3 acetylation and HDAC1 interaction at the c-Jun promoter. (A) ChIP assays were performed with neurons infected with Ad-HDRP or Ad-GFP. Expression persisted for 48 h before cells were treated with HK medium, LK medium, or LK medium with TSA (1 μM) for 3 h. Following fixation of the cultured neurons and sonication of DNA, an acetyl histone H3 antibody was used to immunoprecipitate acetylated chromatin. PCR was then performed using primers that amplify a 504-bp fragment of the c-Jun promoter (top panel). Aliquots of cell lysate were also taken before histone immunoprecipitation, the DNA was purified, and the c-Jun promoter was amplified by PCR and used as DNA input (center panel). A second immunoprecipitation was performed in tandem from the same fixed and sonicated lysates to determine the efficacy of acetyl-H3 being pulled down. Immune complexes were subjected to Western blotting, and the resulting membrane was probed with acetyl-H3 antibody (bottom panel). Rat tail genomic extract was used as a positive control (PC) for PCR. Immunoprecipitation was also performed with a nonhistone binding antibody of the same isotype (NC) to further verify the specificity of the bands. (B) ChIP assays were performed and are displayed as in panel A, with the exception that HDAC1 antibody was used to immunoprecipitate the sonicated chromatin. HDAC1 interaction with the c-Jun promoter was quantified by PCR (top panel). (C) JNK (JNK1, -2, and -3) and HDAC1 were immunoprecipitated using antibodies of rabbit origin from Ad-HDRP-infected or uninfected neurons and treated with HK or LK medium for 3 h. Immunoprecipitates were subjected to Western blotting, and the membrane was probed with a phospho-JNK (T183/Y185) antibody of mouse origin to prevent the detection of IgG heavy and light chains from immunoprecipitated antibodies.
If HDAC1-mediated histone deacetylation at the c-Jun promoter is important for neuroprotection by HDRP, treatment with HDAC inhibitors would reduce the neuroprotective efficacy of HDRP. As shown in Fig. 13, treatment with either TSA or sodium butyrate did inhibit the neuroprotection conferred by HDRP. Interestingly, we have observed that TSA and HDAC inhibitor 1 (HDACi) cause an atypical form of chromatin condensation not seen following neuronal responses to several other forms of apoptotic stimuli (Fig. 13B). Furthermore, the nuclear fragmentation that is a normal feature of apoptotic death did not occur following treatment with either of the HDAC inhibitors. Since the partial pattern of nuclear condensation was not entirely conclusive, we verified our results using another well-established assay of cell viability. The MTT assay confirmed that HDAC inhibitors do indeed prevent HDRP-mediated neuronal survival in LK medium (Fig. 13C).
FIG. 13.

Pharmacological inhibition of HDACs blocks neuroprotection by HDRP. Neuronal cultures that were infected with HDRP were treated with 5 μM TSA or 30 μM HDACi. Viability was quantified 24 h later by visualization of DAPI-stained nuclei (A and B) or by using the MTT assay (C). (A) Quantification using DAPI staining of nuclei. The proportions of partially and fully condensed nuclei were quantified and scored as apoptotic. (B) Atypical appearance of nuclei following treatment with HDAC inhibitors. The images show the appearance of nuclei from cultures infected with Ad-HDRP and then treated in the presence (LK + TSA) or absence (LK) of TSA for 24 h. Note the appearance of healthy (asterisks) and apoptotic (arrows) nuclei in LK-treated cultures. In the presence of TSA, the majority of the nuclei displayed an atypical appearance, in which the nuclei were brighter than healthy nuclei but often only partially condensed. (C) Quantification of cell viability using the MTT assay. The incomplete neuroprotection by Ad-HDRP reflects the successful infection of only ∼30% of the neurons in the culture. LK-GFP control cultures as well as all HDAC inhibitor-treated cultures showed reduced viability compared to LK-treated Ad-HDRP-infected neurons (*, P < 0.05).
DISCUSSION
Although several HDACs are expressed at high levels in the brain, their role in brain function has not been explored. We show here that HDRP is involved in the promotion of neuronal survival. Our conclusion is based on the following findings. HDRP expression is downregulated in apoptotic neurons, and this occurs prior to the time at which these neurons become irreversibly committed to death. Neurons cultured from mice lacking HDRP are more vulnerable to LK-induced apoptosis. Similarly, antisense oligonucleotide-mediated downregulation of HDRP expression induces apoptosis in HK medium and increases the extent of cell death in LK medium. Finally, forced expression of HDRP completely prevents LK-induced neuronal death. This is the first report demonstrating the involvement of HDRP (or any other specific HDAC protein) in promoting the survival of neurons. The human HDAC9 gene is located at 7p21.1, a region implicated in neurological disorders and a variety of cancers, including peripheral nerve sheath tumors (20, 26, 33, 38, 41). In cancers involving chromosomal region 7p, tumor progression is associated with a gain of a genomic region (38), consistent with the possibility that increased HDAC9/HDRP expression might have a prosurvival or antiapoptotic effect. Supporting such a role is the observation that HDAC9 and HDRP expression is upregulated in several cell lines and tumors (32). In the context of neuroprotection, HDRP is likely to be of particular significance. Supporting such a conclusion is our finding that selective suppression of HDAC9 expression has no effect on neuronal survival. Also, neuronal death induced by treatment with HDAC inhibitors reduces HDRP expression without an appreciable effect on the expression of HDAC9.
Results from studies using pharmacological inhibitors of HDACs have provided contradictory conclusions. While the administration of HDAC inhibitors protects against neurodegeneration in Drosophila and mouse models of Huntington's disease, the same inhibitors actively kill certain types of neurons in culture, such as cerebellar granule neurons (5, 13, 35-37). One possibility to reconcile these findings is that while some HDAC members, such as HDRP, have prosurvival effects, other HDAC proteins may be proapoptotic. Our finding that HDRP is a neuroprotective protein, along with reports by other investigators showing that HDAC4 and HDAC5 have proapoptotic effects in neurons, is consistent with such a possibility (1, 27). Because the currently available HDAC inhibitors block the activities of all HDAC proteins, whether these compounds are neuroprotective or neurotoxic could depend, at least in part, on the relative abundances of different HDAC family members in the specific cell type.
HDRP and other class II HDACs have been most extensively studied in the context of muscle differentiation, where they play an inhibitory role by repressing the activity of MEF2 within the nucleus. The phosphorylation of these HDACs by CaMK or protein kinase D results in their dissociation from MEF2, permitting MEF2 to activate the transcription of myogenic genes. In the case of HDAC4 and HDAC5, phosphorylation induces their translocation out of the nucleus. A similar phosphorylation of HDRP only relocalizes it within the nucleus, presumably because it lacks a nuclear export sequence (47). In contrast to the case in muscle cells, adenovirally expressed HDRP is found abundantly in the cytosol in neurons maintained in HK medium but translocates to the nucleus following LK treatment. The mechanism by which HDRP is transported to the cytosol in HK medium is unclear, but it depends on CaMK activity, as inhibition of this kinase restricts HDRP to the nucleus. It has been previously established that HK treatment activates CaMK in neurons and that this activation is required for depolarization-mediated neuronal survival (15). Although preventing its translocation to the cytosol, the inhibition of CaMK does not inhibit the ability of HDRP to maintain neuronal survival. Thus, neuroprotection by HDRP is mediated in the nucleus.
The roles of the PI 3-kinase-Akt and Raf-MEK-ERK pathways in the promotion of neuronal survival are well established. We found that forced expression of HDRP does not activate either of these pathways and that inhibition of these pathways does not reduce the ability of HDRP to provide neuroprotection. How, then, does HDRP prevent LK-induced neuronal death? We found that HDRP inhibits c-Jun, a transcription factor that is necessary for apoptosis in a variety of in vitro and in vivo paradigms of neuronal death (17). Activation of c-Jun in neurons requires its phosphorylation by JNK. We found that HDRP associates with JNK and that JNK activity is reduced when HDRP is overexpressed, explaining the inhibition of c-Jun by HDRP. Exposure of cells to UV radiation is known to stimulate JNK, leading to the induction of c-Jun activity. As observed with LK treatment of neurons, the stimulation of JNK and c-Jun by UV exposure of HeLa cells is also blocked by HDRP. In contrast to JNK1, which is cytoplasmic in neurons, JNK2 and JNK3 translocate to the nucleus, where they phosphorylate c-Jun (10). Since neuroprotection by HDRP occurs in the nucleus, the target of HDRP is thus likely to be JNK2 or JNK3. Consistent with this conclusion is the finding that HDRP inhibits the 54-kDa form of JNK, which is expressed by the JNK2 and JNK3 genes, in neurons and HeLa cells. In vitro binding assays using GST-JNK constructs confirmed the interaction between HDRP and JNK3, but the association with JNK2 was found to be minimal. Taken together, our results indicate that JNK3 is the primary target of HDRP-mediated neuroprotection. It is noteworthy that the loss of JNK3 protects mice against neuronal loss induced by axotomy, ischemic stroke, and glutamate-induced excitotoxicity (21, 34, 46). In contrast, the loss of either JNK1 or JNK2 alone has no serious consequences. JNK3 has therefore been considered to be the most proapoptotic member of the family in the context of neurodegeneration and is currently being targeted for the design of neuroprotective drugs (14, 34).
In addition to c-Jun phosphorylation, transcription of the c-Jun gene is known to be stimulated in neurons primed to undergo apoptosis. Consistent with this increased transcription, histone H3 acetylation of the c-Jun promoter is higher in neurons induced to undergo apoptosis. Forced expression of HDRP inhibits this acetylation. Although HDRP has no intrinsic HDAC activity, other investigators have shown that it can acquire such activity by recruitment of HDAC1 or HDAC3 (48). Consistent with previous reports, we found that HDRP does interact with HDAC1 in neurons. Furthermore, HDAC1 does associate strongly with the c-Jun promoter in neurons expressing HDRP, and treatment with pharmacological inhibitors of HDAC inhibits neuroprotection by HDRP. This result suggests that in addition to the inhibition of JNK activity, deacetylation of the c-Jun promoter via recruitment of HDAC1 contributes to the neuroprotective action of HDRP.
It is known that the induction of c-Jun gene transcription follows phosphorylation and activation of preexisting c-Jun proteins. Once activated, c-Jun binds to its own promoter, stimulating its expression. Thus, the prevention of c-Jun phosphorylation via JNK inhibition is likely to be the primary mechanism by which HDRP protects neurons from apoptosis. Previous studies using pharmacological inhibitors of JNK signaling in cell culture and in vivo models of neurodegeneration have established that cell death is completely prevented by the inhibition of c-Jun phosphorylation (6, 23, 43). We and others have found that pharmacological inhibitors of HDAC induce apoptosis in cerebellar granule neuron cultures (4, 37). In this study, we show that these inhibitors reduce the expression of HDRP in neurons. This is likely to contribute to the neurotoxic effect of the inhibitors. Other investigators have reported that neuronal apoptosis by HDAC inhibitors involves stimulation of E2F-1, a transcription factor with established proapoptotic activity in neurons (5). The expression of E2F target genes, such as those for cyclin E and Apaf-1, is induced in cerebellar granule neurons by pharmacological inhibition of HDACs (5). The relationship between HDRP downregulation and the stimulation of E2F activity is currently unclear and deserves investigation. In addition to blocking HK-mediated neuronal survival, HDAC inhibitors also inhibit HDRP-mediated neuroprotection. This is likely to occur via inhibition of HDAC1, which we have found to associate with the c-Jun promoter.
Our results are consistent with the following model. In neurons treated with HK medium, HDRP associates with JNK3 in the cytosol. Treatment with LK medium results in phosphorylation of JNK3, permitting it to translocate to the nucleus. LK-mediated degradation of HDRP allows the translocated JNK to activate c-Jun via phosphorylation. While not affecting the translocation of JNK3, ectopically expressed HDRP remains associated with the kinase and thus interferes with its ability to phosphorylate c-Jun. While HDRP inhibits the phosphorylation of c-Jun by JNK, HDRP also facilitates the recruitment of HDAC1 to the c-Jun promoter and, through histone deacetylation of the promoter, inhibits c-Jun transcription.
In summary, our studies reveal a neuroprotective function for HDRP in the brain. Finding ways of increasing HDRP expression in the intact brain by pharmacological means or appropriate viral vectors could have therapeutic value in the treatment of neurodegenerative diseases.
Supplementary Material
Acknowledgments
The research described in this report was funded by NIH-NINDS grants NS40408 and NS047201 to S.R.D. This work was also supported by funds from the Department of Defense (DNMD 17-99-1-9566) to S.R.D.
We thank Asligul Yalcin and Megan Kong for preliminary work that contributed to the development of this project.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bolger, T. A., and T. P. Yao. 2005. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 25:9544-9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borodezt, K., and S. R. D'Mello. 1998. Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J. Neurosci. Res. 53:531-541. [DOI] [PubMed] [Google Scholar]
- 3.Borras, E., R. Zaragoza, M. Morante, C. Garcia, A. Gimeno, G. Lopez-Rodas, T. Barber, V. J. Miralles, J. R. Vina, and L. Torres. 2003. In vivo studies of altered expression patterns of p53 and proliferative control genes in chronic vitamin A deficiency and hypervitaminosis. Eur. J. Biochem. 270:1493-1501. [DOI] [PubMed] [Google Scholar]
- 4.Boutillier, A. L., E. Trinh, and J. P. Loeffler. 2002. Constitutive repression of E2F1 transcriptional activity through HDAC proteins is essential for neuronal survival. Ann. N. Y. Acad. Sci. 973:438-442. [DOI] [PubMed] [Google Scholar]
- 5.Boutillier, A. L., E. Trinh, and J. P. Loeffler. 2003. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 84:814-828. [DOI] [PubMed] [Google Scholar]
- 6.Bozyczko-Coyne, D., M. S. Saporito, and R. L. Hudkins. 2002. Targeting the JNK pathway for therapeutic benefit in CNS disease. Curr. Drug Targets CNS Neurol. Disord. 1:31-49. [DOI] [PubMed] [Google Scholar]
- 7.Brunet, A., S. R. Datta, and M. E. Greenberg. 2001. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11:297-305. [DOI] [PubMed] [Google Scholar]
- 8.Buck, S. W., C. M. Gallo, and J. S. Smith. 2004. Diversity in the Sir2 family of protein deacetylases. J. Leukoc. Biol. 75:939-950. [DOI] [PubMed] [Google Scholar]
- 9.Chin, P. C., L. Liu, B. E. Morrison, A. Siddiq, R. R. Ratan, T. Bottiglieri, and S. R. D'Mello. 2004. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J. Neurochem. 90:595-608. [DOI] [PubMed] [Google Scholar]
- 10.Coffey, E. T., G. Smiciene, V. Hongisto, J. Cao, S. Brecht, T. Herdegen, and M. J. Courtney. 2002. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J. Neurosci. 22:4335-4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Ruijter, A. J., A. H. van Gennip, H. N. Caron, S. Kemp, and A. B. van Kuilenburg. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370:737-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Mello, S. R., C. Galli, T. Ciotti, and P. Calissano. 1993. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 90:10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrante, R. J., J. K. Kubilus, J. Lee, H. Ryu, A. Beesen, B. Zucker, K. Smith, N. W. Kowall, R. R. Ratan, R. Luthi-Carter, and S. M. Hersch. 2003. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 23:9418-9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graczyk, P. P., A. Khan, G. S. Bhatia, V. Palmer, D. Medland, H. Numata, H. Oinuma, J. Catchick, A. Dunne, M. Ellis, C. Smales, J. Whitfield, S. J. Neame, B. Shah, D. Wilton, L. Morgan, T. Patel, R. Chung, H. Desmond, J. M. Staddon, N. Sato, and A. Inoue. 2005. The neuroprotective action of JNK3 inhibitors based on the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole scaffold. Bioorg. Med. Chem. Lett. 15:4666-4670. [DOI] [PubMed] [Google Scholar]
- 15.Hack, N., H. Hidaka, M. J. Wakefield, and R. Balazs. 1993. Promotion of granule cell survival by high K+ or excitatory amino acid treatment and Ca2+/calmodulin-dependent protein kinase activity. Neuroscience 57:9-20. [DOI] [PubMed] [Google Scholar]
- 16.Ham, J., C. Babij, J. Whitfield, C. M. Pfarr, D. Lallemand, M. Yaniv, and L. L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron 14:927-939. [DOI] [PubMed] [Google Scholar]
- 17.Ham, J., A. Eilers, J. Whitfield, S. J. Neame, and B. Shah. 2000. c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol. 60:1015-1021. [DOI] [PubMed] [Google Scholar]
- 18.Hetman, M., and A. Gozdz. 2004. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 271:2050-2055. [DOI] [PubMed] [Google Scholar]
- 19.Hockly, E., V. M. Richon, B. Woodman, D. L. Smith, X. Zhou, E. Rosa, K. Sathasivam, S. Ghazi-Noori, A. Mahal, P. A. Lowden, J. S. Steffan, J. L. Marsh, L. M. Thompson, C. M. Lewis, P. A. Marks, and G. P. Bates. 2003. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA 100:2041-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson, D., S. W. Horsley, D. M. Moloney, M. Oldridge, S. R. Twigg, S. Walsh, M. Barrow, P. R. Njolstad, J. Kunz, G. J. Ashworth, S. A. Wall, L. Kearney, and A. O. Wilkie. 1998. A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am. J. Hum. Genet. 63:1282-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keramaris, E., J. L. Vanderluit, M. Bahadori, K. Mousavi, R. J. Davis, R. Flavell, R. S. Slack, and D. S. Park. 2005. c-Jun N-terminal kinase 3 deficiency protects neurons from axotomy-induced death in vivo through mechanisms independent of c-Jun phosphorylation. J. Biol. Chem. 280:1132-1141. [DOI] [PubMed] [Google Scholar]
- 22.Koulich, E., T. Nguyen, K. Johnson, C. Giardina, and S. D'Mello. 2001. NF-kappaB is involved in the survival of cerebellar granule neurons: association of IkappaBbeta [correction of Ikappabeta] phosphorylation with cell survival. J. Neurochem. 76:1188-1198. [DOI] [PubMed] [Google Scholar]
- 23.Kuan, C. Y., and R. E. Burke. 2005. Targeting the JNK signaling pathway for stroke and Parkinson's diseases therapy. Curr. Drug Targets CNS Neurol. Disord. 4:63-67. [DOI] [PubMed] [Google Scholar]
- 24.Langley, B., J. M. Gensert, M. F. Beal, and R. R. Ratan. 2005. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. Drug Targets CNS Neurol. Disord. 4:41-50. [DOI] [PubMed] [Google Scholar]
- 25.Legube, G., and D. Trouche. 2003. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 4:944-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewanda, A. F., E. D. Green, J. Weissenbach, H. Jerald, E. Taylor, M. L. Summar, J. A. Phillips III, M. Cohen, M. Feingold, W. Mouradian, et al. 1994. Evidence that the Saethre-Chotzen syndrome locus lies between D7S664 and D7S507, by genetic analysis and detection of a microdeletion in a patient. Am. J. Hum. Genet. 55:1195-1201. [PMC free article] [PubMed] [Google Scholar]
- 27.Linseman, D. A., C. M. Bartley, S. S. Le, T. A. Laessig, R. J. Bouchard, M. K. Meintzer, M. Li, and K. A. Heidenreich. 2003. Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous Ca(2+)/calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J. Biol. Chem. 278:41472-41481. [DOI] [PubMed] [Google Scholar]
- 28.Lu, J., T. A. McKinsey, R. L. Nicol, and E. N. Olson. 2000. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 97:4070-4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marks, P. A., V. M. Richon, T. Miller, and W. K. Kelly. 2004. Histone deacetylase inhibitors. Adv. Cancer Res. 91:137-168. [DOI] [PubMed] [Google Scholar]
- 30.McKinsey, T. A., C. L. Zhang, J. Lu, and E. N. Olson. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKinsey, T. A., C. L. Zhang, and E. N. Olson. 2000. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 97:14400-14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrie, K., F. Guidez, L. Howell, L. Healy, S. Waxman, M. Greaves, and A. Zelent. 2003. The histone deacetylase 9 gene encodes multiple protein isoforms. J. Biol. Chem. 278:16059-16072. [DOI] [PubMed] [Google Scholar]
- 33.Powlesland, R. M., A. K. Charles, K. T. Malik, P. A. Reynolds, S. Pires, M. Boavida, and K. W. Brown. 2000. Loss of heterozygosity at 7p in Wilms' tumour development. Br. J. Cancer 82:323-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resnick, L., and M. Fennell. 2004. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov. Today 9:932-939. [DOI] [PubMed] [Google Scholar]
- 35.Rouaux, C., N. Jokic, C. Mbebi, S. Boutillier, J. P. Loeffler, and A. L. Boutillier. 2003. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 22:6537-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryu, H., J. Lee, B. A. Olofsson, A. Mwidau, A. Dedeoglu, M. Escudero, E. Flemington, J. Azizkhan-Clifford, R. J. Ferrante, and R. R. Ratan. 2003. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. USA 100:4281-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salminen, A., T. Tapiola, P. Korhonen, and T. Suuronen. 1998. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res. Mol. Brain Res. 61:203-206. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt, H., H. Taubert, P. Wurl, M. Kappler, H. Lange, F. Bartel, M. Bache, H. J. Holzhausen, and R. Hinze. 2002. Gains of 12q are the most frequent genomic imbalances in adult fibrosarcoma and are correlated with a poor outcome. Genes Chromosomes Cancer 34:69-77. [DOI] [PubMed] [Google Scholar]
- 39.See, V., A. L. Boutillier, H. Bito, and J. P. Loeffler. 2001. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 15:134-144. [DOI] [PubMed] [Google Scholar]
- 40.Sparrow, D. B., E. A. Miska, E. Langley, S. Reynaud-Deonauth, S. Kotecha, N. Towers, G. Spohr, T. Kouzarides, and T. J. Mohun. 1999. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 18:5085-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stankiewicz, P., H. Thiele, C. Baldermann, A. Kruger, I. Giannakudis, S. Dorr, N. Werner, J. Kunz, G. A. Rappold, and I. Hansmann. 2001. Phenotypic findings due to trisomy 7p15.3-pter including the TWIST locus. Am. J. Med. Genet. 103:56-62. [DOI] [PubMed] [Google Scholar]
- 42.Verdin, E., F. Dequiedt, and H. G. Kasler. 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19:286-293. [DOI] [PubMed] [Google Scholar]
- 43.Wang, L. H., C. G. Besirli, and E. M. Johnson, Jr. 2004. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu. Rev. Pharmacol. Toxicol. 44:451-474. [DOI] [PubMed] [Google Scholar]
- 44.Watson, A., A. Eilers, D. Lallemand, J. Kyriakis, L. L. Rubin, and J. Ham. 1998. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J. Neurosci. 18:751-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yalcin, A., E. Koulich, S. Mohamed, L. Liu, and S. R. D'Mello. 2003. Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-kappaB. J. Neurochem. 84:397-408. [DOI] [PubMed] [Google Scholar]
- 46.Yang, D. D., C. Y. Kuan, A. J. Whitmarsh, M. Rincon, T. S. Zheng, R. J. Davis, P. Rakic, and R. A. Flavell. 1997. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865-870. [DOI] [PubMed] [Google Scholar]
- 47.Zhang, C. L., T. A. McKinsey, and E. N. Olson. 2001. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 98:7354-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou, X., V. M. Richon, R. A. Rifkind, and P. A. Marks. 2000. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 97:1056-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.