

Summary
Replication protein A (RPA) is a eukaryotic single-stranded DNA-binding protein consisting of three subunits of 70-kDa, 32-kDa, and 14-kDa (RPA70, RPA32, RPA14, respectively). It is a protein essential for most cellular DNA metabolic pathways. Checkpoint proteins Rad9, Rad1 and Hus1 form a clamp-like complex which plays a central role in the DNA damage-induced checkpoint response. In this report, we presented the evidence that Rad9-Rad1-Hus1 complex directly interacted with RPA in human cells, and this interaction was mediated by the binding of Rad9 protein to both RPA70 and RPA32 subunits. In addition, the cellular interaction of Rad9-Rad1-Hus1 with RPA or hyperphosphorylated RPA was stimulated by UV irradiation or camptothecin treatment in a dose dependent manner. Such treatments also resulted in the co-localization of the nuclear foci formed with the two complexes. Consistently, knockdown of the RPA expression in cells by the small interference RNA (siRNA) blocked the DNA damage-dependent chromatin association of Rad9-Rad1-Hus1, and also inhibited the Rad9-Rad1-Hus1 complex formation. Taken together, our results suggest that Rad9-Rad1-Hus1 and RPA complexes collaboratively function in DNA damage responses, and that the RPA may serve as a regulator for the activity of Rad9-Rad1-Hus1 complex in the cellular checkpoint network.
Introduction
The maintenance of genomic stability relies on the accurate duplication of the genome and continuous monitoring of its integrity. To accomplish this, cells have evolved complex surveillance mechanisms, termed DNA damage cell cycle checkpoints, in response to DNA damage and replication stress. The checkpoint signaling cascades consist of damage sensors, signal transducers, mediators and effectors that, if activated, eventually inhibit cell cycle progression to stabilize stalled replication forks, and to promote DNA repair or trigger apoptosis (Kastan et al., 2004; Zhou et al., 2000). In mammalian cells, the ATR and ATM proteins, which belong to phosphatidylinositol 3-kinase-related kinase (PIKK) family kinases, and the Rad9-Rad1-Hus1 (9-1-1)/Rad17-Rfc2-5 checkpoint complex have been suggested to be involved in damage recognition and signaling (Rouse et al., 2002; Zhou et al., 2000). The ATM kinase seems to be activated primarily following generation of double-stranded DNA breaks (DSBs), whereas ATR kinase is critical for cellular responses to a variety of DNA damage including DSB. When activated, these protein kinases eventually phosphorylate and modulate the cellular activities of downstream key effectors in DNA damage responses (e.g. Chk1 and Chk2, for ATR and ATM, respectively) (Abraham, 2001; Bartek et al., 2004). The Rad17-Rfc2-5/9-1-1 complex appears to be translocated to the sites of DNA damage independently of the ATR and ATM, but may also be essential for activation of the downstream kinase of ATR (i.e. Chk1) and initiation of the checkpoint responses (Bao et al., 2004; Lowndes et al., 2000; O’Connell et al., 2000; Zou et al., 2002). The 9-1-1 complex is a ring-shape protein complex which as the checkpoint counterpart, resembles the replication clamp PCNA, while the Rad17-Rfc2-5 complex shares structural similarity to the replication clamp loader RFC (Burtelow et al., 2001; Caspari et al., 2000; Parrilla-Castellar et al., 2004; Venclovas et al., 2000). Biochemical studies have demonstrated that the 9-1-1 checkpoint complex can be loaded onto the primed DNA in vitro by Rad17-Rfc2-5 complex and also recruited to the DNA damage sites in vivo by Rad17 (Bermudez et al., 2003; Lindsey-Boltz et al., 2001; Zou et al., 2002). The molecular details of the Rad9-Rad1-Hus1 system, however, remain to be defined.
Replication protein A (RPA) is a ssDNA binding heterotrimeric protein composed of 70-kDa, 32-kDa, and 14-kDa subunits (referred thereafter as RPA70, RPA32, RPA14, respectively) (Binz et al., 2004; Bochkarev et al., 2002; Bochkarev and Bochkarev, 2004; Shell et al., 2005). RPA was initially identified as a factor essential for in vitro SV40 DNA replication (Wold, 1997). RPA represents the major cellular ssDNA binding protein in eukaryotic cells, and participates in almost all aspects of cellular DNA transactions. Through its strong affinity for ssDNA, RPA accumulates on ssDNA filaments generated during DNA repair processes and/or by stalled replication forks, which constitute critical intermediates for multiple DNA metabolic pathways (Iftode et al., 1999). RPA also interacts with numerous proteins involved in DNA replication, recombination, and repair, and its roles in these pathways have been well documented (Binz et al., 2004; Liu et al., 2005).
The evidence for the implication of RPA involvement in the regulation of DNA damage-induced cell cycle checkpoints has recently emerged. In both budding and fission yeasts, several mutations in RPA caused the hypersensitivity of cells to DNA damaging agents, and defective G1/S and intra-S checkpoints, and prevented phosphorylation of downstream targets of ATR/ATM kinases (Binz et al., 2004; Lee et al., 1998; Longhese et al., 1996; Pellicioli et al, 2001). Using Xenopus oocytes system, it has been shown that RPA was necessary for suppression of DNA synthesis in response to DNA strand breaks and immunodepletion of RPA abrogated an aphidiocolin-induced DNA replication checkpoint (Costanzo et al., 2003; You et al., 2002). Similarly, in mammalian cells, the chromatin association and nuclear foci formation of ATR after exposure to genotoxic agents required RPA (Dart et al., 2004; Zou et al, 2003b). The involvement of RPA in checkpoint signaling was further demonstrated by the recent studies, showing that ATRIP, the interacting partner of ATR, recognized RPA-coated ssDNA, and that the ATR-mediated phosphorylation of its kinase effectors Chk1 and Rad17 is dependent on the presence of RPA (Zou et al, 2003a).
RPA-coated ssDNA is also an important part of the intermediate structures recognized by the Rad17-Rfc2-5 complex, which facilitates the recruitment of the Rad9-Rad1-Hus1 complex to the gapped and primed DNA structures in vitro (Zou et al, 2003b). However, mechanistic details of the molecular relationship between Rad9-Rad1-Hus1 and RPA are unclear. Since the Rad9-Rad1-Hus1 complex, once loaded, forms a sliding clamp on DNA substrate (Ellison et al., 2003), and RPA as a ssDNA binding protein has a strong affinity for ssDNA, we postulated that these two complexes may interact with each other at the sites of DNA damage. Here we report that the Rad9-Rad1-Hus1 complex and RPA did directly interact with one another and the interaction was mediated by Rad9 which bound to both RPA70 and RPA32 subunits. Treatment with DNA damage agents enhanced the interaction and also induced apparent co-localization of nuclear foci of the two complexes. Remarkably, silencing of the cellular RPA expression by small interfering RNA (siRNA) diminished the 9-1-1 complex association to chromatin induced by DNA damage. Our results suggest that RPA may directly recruit 9-1-1 complex to the sites of DNA damage in vivo to elicit cell cycle checkpoint responses.
Results
Interactions of 9-1-1 and RPA complexes
To probe the potential association of RPA with 9-1-1 complex, we performed co-immunoprecipitation experiments using total cell lysates prepared from human HeLa cells. Western blotting analysis of the immunoprecipitates with anti-RPA70 and anti-RA32 antibodies revealed the presence of RPA in the anti-Rad9, Rad1 and Hus1 immunoprecipitates (Fig. 1A, lanes 2-4). In the reciprocal experiments, RPA70 antibody was able to co-immunoprecipitate Rad1 and Hus1 (Fig. 1 B), suggesting that these proteins may interact with each other either directly or indirectly in cells. The immunoprecipitation of Rad9 with RPA70 antibody was not successful due to the interference of IgG heavy chain (data not shown). As controls, normal rabbit IgG did not immunoprecipitate RPA, Rad1 or Hus1 (Fig. 1A, lane 5, Fig. 1B, lane 3), indicating that the co-immunoprecipitation of RPA with 9-1-1 complex was not due to nonspecific antibody binding.
Figure 1.

Interaction of RPA with Rad9-Rad1-Hus1 complex. (A): Total cell lysates prepared from exponentially growing HeLa cells were used for co-immunoprecipitation (IP) assays with anti-Rad9 (lane 2), anti-Rad1 (lane 3) and anti-Hus1 (lane 4) or normal rabbit IgG (lane 5) as described in Materials and methods. Proteins from the immunoprecipitates were detected by Western blotting using anti-RPA70 antibody (upper panel) or anti-RPA32 antibody (lower panel). As control, 10% of the total volumes of the whole cellular lysates used for the co-IP reactions were also included (Input, lane 1). IgH is the IgG heavy chain. (B): Total cell lysates were subjected to the co-IP assays with anti-RPA70 antibody and the immunoprecipitated proteins were detected using anti-Rad1 (upper panel) or anti-Hus1 antibody (lower panel). (C): Cell lysates were either treated with 100 μg/mL DNase I (Invitrogen) for 20 min at 37 °C (lane 2) or 50 μg/mL ethidium bromide (EtBr) on ice for 30 min (lane 3) or mock-treated (lane 1). Cell lysates were then subjected to co-IP assays with anti-Rad9 antibody and detected by anti-RPA32 antibody (lower panel). 10% of treated cell lysates were also included as controls (upper panel). Data are representative of three independent experiments.
Since both RPA and 9-1-1 complex are chromatin-bound proteins in cells (Burtelow et al, 2000; Iftode et al., 1999), we considered the possibility that the interaction between RPA and 9-1-1 complex was mediated by the independent binding of both protein complexes to DNA. Nevertheless, pretreatment of cell lysates with DNase I or ethidium bromide (EtBr) prior to co-immunoprecipitation did not affect the interaction of RPA with Rad9 (Fig. 1C), indicating that the observed interactions were probably not mediated via DNA-protein interactions. In spite of this, it remains possible that other proteins may mediate the interaction between RPA and 9-1-1 complex because both complexes possess a variety of interacting partners. To test this possibility and to further characterize the interaction, we carried out the following experiments. After co-immunoprecipitation reaction with anti-Rad9 antibodies, the immunoprecipitates were incubated with increasing ionic strength of buffer to remove endogenous Rad9-binding proteins. As revealed in Fig. 2A, incubation with 0.6 M of NaCl eliminated almost all the co-immunoprecipitated RPA, and also disrupted the 9-1-1 complex (data not shown). Then the purified recombinant RPA complex or individual RPA subunits were supplemented and further incubated in RPA binding buffer to allow for the interactions with the immunoprecipitated Rad9. Under these conditions, the apparent interactions of Rad9 with RPA complex (Fig. 2B), and both RPA70 (Fig. 2D, lanes 2 and 3) and RPA32/14 (Fig. 2C) subunits were observed. These results suggest that RPA and Rad9 may interact directly with one another as the high concentration of salt could readily disrupt most cellular protein-protein interactions. The same experiments were also carried out to investigate the interactions of Rad1 and Hus1 with RPA, respectively. However, no significant association was observed between Rad1 and recombinant RPA trimer (Fig. 2E, lanes 3 and 4) or any individual RPA subunits (Fig. 2D, lanes 4 and 5, Fig. 2E, lanes 5 and 6). We confirmed that Rad1 was still present in anti-Rad1 immunoprecipitates after 0.6 M salt washing (data not shown), indicating that the lack of interaction between Rad1 and RPA was not due to the loss of Rad1 in reaction system. Likewise, no interactions between Hus1 and recombinant RPA were observed either (data not shown). Together, these data demonstrated the constitutive association of RPA with the 9-1-1 checkpoint complex in human cells, which is mediated by the direct interaction of Rad9 with both RPA70 and RPA32/14 subunits of RPA.
Figure 2.
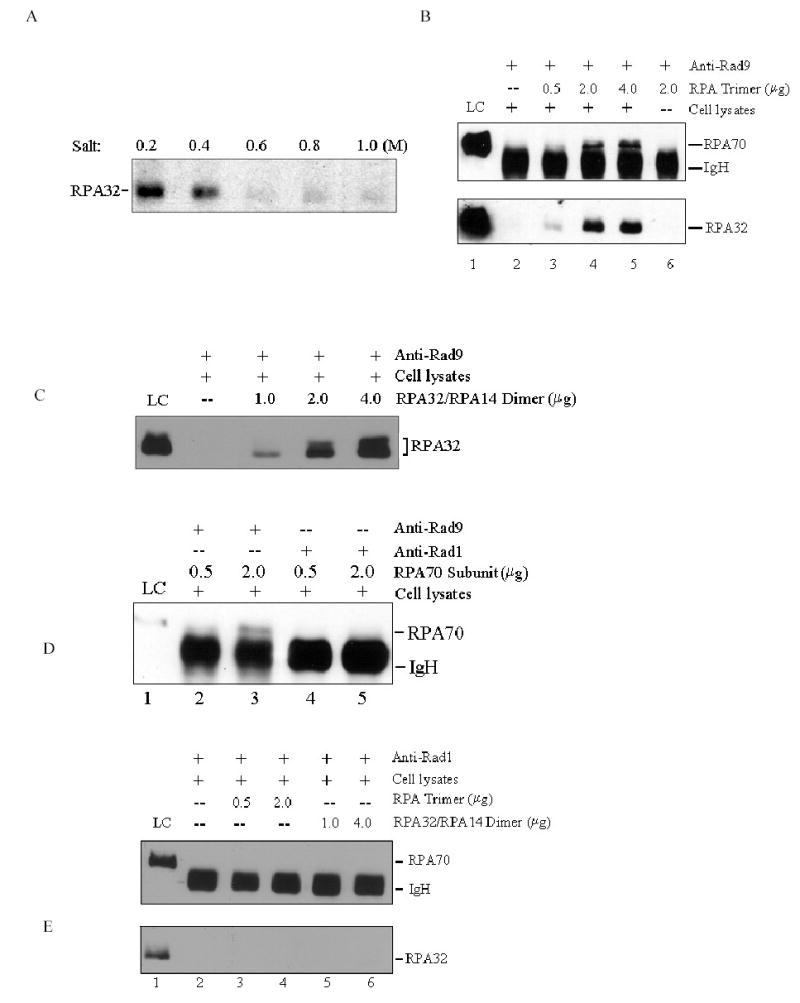
Interaction of Rad9-Rad1-Hus1 complex with RPA is mediated by Rad9. (A): Total cellular lysates were incubated with anti-Rad9 antibodies for 10–14 h, followed by 1-h incubation with protein A/G-agarose beads. After centrifugation, the immunoprecipitates were washed three times with PBS containing 0.05% Nonidet P-40, and further incubated with increasing salt concentrations of buffer (15 mM Tris-Cl, pH 7.5, 0.2~1.0 M NaCl, 0.1% NP-40) for 30 min at 4 °C. After washing, the remaining bound proteins were detected by anti-RPA32 antibody. (B): The co-IP reactions were performed as in Fig. 2A, and the immunoprecipitates were then incubated with the buffer containing 0.6 M NaCl. After centrifugation and washing, purified RPA was added and further incubated in 500 μL RPA binding buffer (40 mM HEPES-KOH, pH 7.5, 75 mM KCl, 8 mM MgCl2, 1 mM DTT, 5% glycerol and 100 μg/mL BSA, 0.1% NP-40) for 4-6 h. The bound proteins were detected by Western blotting with anti-RPA70 and anti-RPA32 antibodies, respectively. (C): The experiments were conducted similarly as described in Fig. 2B, except that RPA32/RPA14 subunit was finally added. (D): The experiments were conducted similarly as described in Fig. 2B, except that anti-Rad9 antibody (lanes 2 and 3) and anti-Rad1 antibody (lanes 4 and 5) were used for co-IP reactions and RPA70 subunit was finally added. (E): The experiments were conducted similarly as described in Fig. 2B, but anti-Rad1 antibody was used for co-IP reactions, and either RPA trimer (lanes 3 and 4) or RPA32/RPA14 subunit (lanes 5 and 6) was finally added. 10% of purified protein used for final binding assays was loaded as control (LC: loading control). Data are representative of two independent experiments.
DNA damage stimulates RPA and 9-1-1 complex interaction
Previous studies showed that both RPA and Rad9 checkpoint complex translocated to nucleus in response to a variety of DNA-damaging agents (Burtelow et al, 2000; Zou et al., 2003a). We speculated that both complexes are probably also localized to the sites of DNA damage and their interactions may be enhanced upon DNA damage to amplify the checkpoint signals. As shown in Fig. 3A, in agreement with the previous reports, the nuclear translocations of RPA and 9-1-1 complexes were triggered by UV irradiation. As expected, the amount of RPA that co-immunoprecipitated with Rad9 increased in a UV-dose dependent manner (Fig. 3B). In particular, Rad9 was able to efficiently interact with hyperphosphorylated RPA (Fig. 3B middle panel, indicated by arrows), which may have biological significance because hyperphosphorylated RPA were located predominantly within nucleus, and in synchronized S-phase cells ~85% of chromatin-bound RPA was hyperphosphorylated (Robison et al., 2004). The similar results were also obtained with an antibody that specifically recognizes the phosphorylated serine 4 and serine 8 of hyperphosphorylated RPA32 (Fig. 3B lower panel). The treatment of cells with CPT, a topoisomerase inhibitor, stimulated the Hus1 and Rad1 interactions with RPA and the hyperphosphorylated RPA (Figs. 3C and D).
Figure 3.
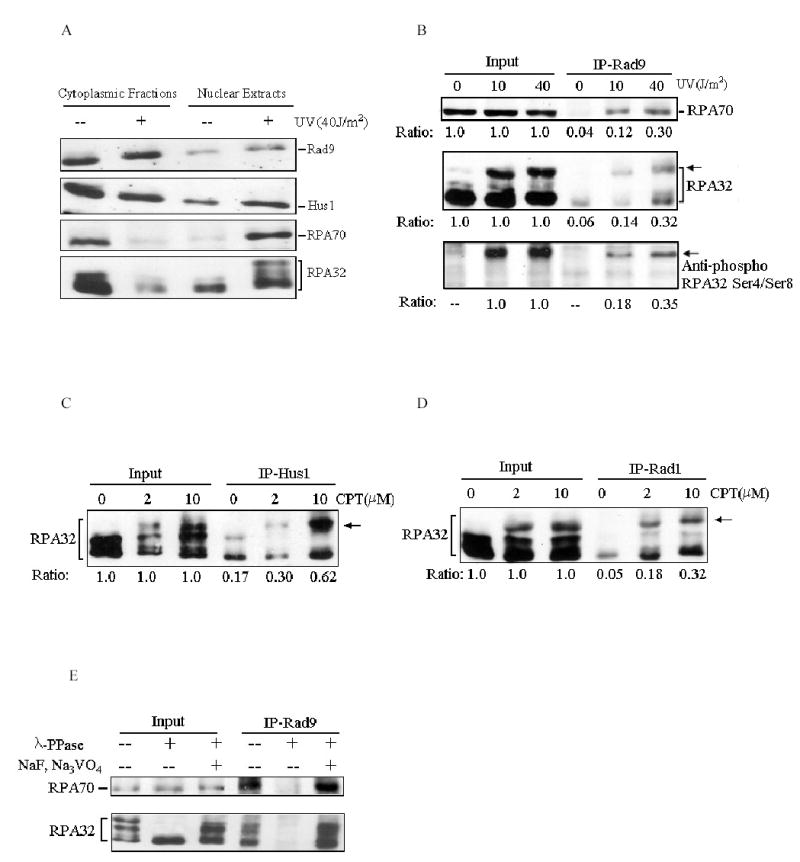
DNA damage stimulates Rad9 checkpoint complex interaction with RPA. (A): Cells were treated with indicated doses of UV irradiation followed by a 2-h recovery. Nuclear extracts were prepared and separated on SDS-PAGE gels, and then probed with anti-Rad9, anti-Rad1, anti-RPA70 and anti-RPA32 antibodies, respectively. (B): Cells were treated with indicated doses of UV and total cell lysates were used for co-IP assays with anti-Rad9 antibody. The immunoprecipitates were then analyzed with anti-RPA70 (upper panel), anti-RPA32 (middle panel) and anti-phospho-RPA32 Ser4/Ser8 (lower panel) antibodies, respectively. The arrows indicate the hyperphosphorylated RPA. The relative quantities of proteins immunoprecipitated by anti-Rad9 antibody were estimated by densitometry and further normalized based on input, which were designated as 1.0. (C) and (D): Cells were treated with indicated doses of CPT, and then the total cell lysates were used for co-IP assays with anti-Hus1(C) or anti-Rad1 (D) antibodies. The immunoprecipitates were detected with anti-RPA32 antibody. (E): Whole cellular lysates were prepared from 40 J/m2 UV-irradiated cells. Prior to co-IP assays, the lysates were either mock treated (−/−), or treated with 2000 units of λ-phosphatase (λ-PPase) (New England Biolabs) for 30 min at 30 °C in the absence (−/+) or presence (+/+) of 10 mM of NaF and 1 mM Na3VO4. Treated cell lysates were then subjected for co-IP reactions with anti-Rad9 antibody. The bound proteins were then detected by Western blotting using anti-RPA32 antibody. Data are representative of at least three independent experiments.
In addition to the phosphorylation of RPA in response to DNA damage, Rad9 and Rad1 have also been reported to undergo complex phosphoryl modifications upon DNA damage and such modifications may modulate the 9-1-1 checkpoint complex formation (Burtelow et al, 2000; Roos-Mattjus et al., 2003; Yoshida et al., 2002, 2003; Volkmer et al., 1999). We therefore examined whether the protein phosphorylation played a role in the RPA/Rad9 complex interaction. To this end, whole cell lyastes from 40 J/m2 UV-treated cells were pre-incubated with lambda phosphatase (λ-PPase) before co-immunoprecipitation assays. As shown in Fig. 3E, the pre-treatment with λ-PPase almost completely abrogated the ability of anti-Rad9 antibodies to co-immunoprecipitate RPA. The addition of sodium fluoride and sodium orthovanadate (NaF, Na3VO4), known inhibitors of λ-PPase, preserved the Rad9/RPA interaction, (Fig. 3E), indicating that the observed disruption of the Rad9/RPA interaction occurred indeed as the consequence of protein de-phosphorylation. Taken together, these results support that RPA and Rad9 checkpoint complex collaboratively participate in the DNA damage checkpoint pathway and the protein phosphorylation may modulate this process, although the exact protein(s) involved remain to be identified.
Co-localization of RPA with Rad9 checkpoint complex after DNA damage
Based on above observations, we investigated the potential co-localization of RPA and Rad9 checkpoint complex into the same nuclear foci after DNA damage. Thus, the immunofluorescent staining analysis was conducted. As shown in Fig. 4A (subpanels B and C), RPA and Rad9 in the mock-treated cells appeared to be homogenously distributed throughout the nucleus. Upon exposure to CPT or UV irradiation, a clear redistribution of RPA and Rad9 proteins to form discrete nuclear foci occurred (Fig. 4A, subpanels F, J and G, K). Some extent of co-localization of these foci was observed, as indicated in Fig. 4A, subpanels H and L (yellow). Such limited degree of co-localization was probably reflecting the fact that both RPA and Rad9 are able to interact with numerous protein partners and function in multiple biological pathways in cells. We also examined the co-localization of hyperphosphorylated RPA with Rad9 checkpoint complex. In control cells, there was no substantial staining with the phospho-specific RPA antibody over the background, while the staining of Rad1 was diffusive (Fig. 4B, subpanels B and C). After treatment of cells with CPT or UV, hyperphosphorylated-RPA aggregated into discrete nuclear foci which also displayed a moderate co-localization with Rad1 foci (Fig. 4B, subpanels F-H and J-L), comparable to the staining with the antibody that recognized all forms of RPA. These results together with those from co-immunoprecipitation experiments suggest that in response to genotoxic stress, both RPA and 9-1-1 complex translocated to the sites of DNA damage or damage-induced intermediate structures where they interacted with each other to activate DNA damage checkpoint signaling.
Figure 4.

Co-localization of RPA with 9-1-1 complex. Cells were treated with 40 J/m2 UV irradiation or 10 μM CPT. After extraction of cytoplasmic proteins with PBS containing 0.5% NP-40, cells were fixed and incubated with primary and secondary antibodies, and visualized with fluorescent microscopy. (A): Cells were stained with anti-Rad9 antibody (green, subpanels B, F, and J) and anti-RPA32 antibody (red, subpanels C, G, and K). Subpanels D, H and L are the merged images of the anti-Rad9 and anti-RPA32 stained cells. Subpanels A, E and I are the DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei. (B): Cells were stained with anti-Rad1 antibody (red, subpanels B, F, and J) and anti-phospho-RPA32 Ser4/Ser8 antibody (green, subpanels C, G, and K). Subpanels D, H and L are the merged images of the anti-Rad1 and anti-phospho-RPA32 Ser4/Ser8 stained cells.
RPA is required for DNA damage-induced chromatin association of Rad9 checkpoint complex
We next sought to address the question of whether RPA is required for 9-1-1 complex to be recruited to the sites of DNA damage. For this purpose, we employed the technique of small interfering RNA (siRNA)-mediated gene repression to knockdown the expression of RPA in cells. Transfection of HeLa cells with siRNA oligonucleotides complementary to RPA70 or RPA32 message RNA resulted in more than 85% reduction of cellular RPA expression level as compared to the cells transfected with siRNA targeting GFP (Figure 5A). Furthermore, expression levels of XPA and Rad52, two repair proteins involved in nucleotide excision repair and DSB repair respectively, remained unaffected by either RPA70 or RPA32 siRNA transfection, illustrating the specificity of this approach and the siRNA sequences used. Interestingly, the siRPA70 transfection also caused a coordinate down-regulation of RPA32, and vice versa, suggesting that the RPA heterotrimer formation is likely an important determinant for the stability of RPA subunits in cells. The transfected cells were then treated with UV irradiation or CPT, and the chromatin-bound fractions were isolated and immunoblotted with anti-Rad9, Hus1 and Rad1 antibodies, respectively. The results in Figs. 5B and 5C revealed that either the RPA70 or RPA32 siRNA transfection significantly inhibited the DNA damage-induced 9-1-1 complex association to the chromatin. In contrast, transfection with siGFP had no effect on Rad9 complex accumulation on chromatin. Since RPA interacted specifically with Rad9 in the 9-1-1 complex, we further examined effects of the RPA deficiency on Rad9 complex assembly. In siGFP transfected cells, UV irradiation strongly induced Rad9 interaction with Hus1 (~ 4.5 fold), which, however, was reduced to ~1.8 fold after siRPA32 transfection (Fig. 5D). Based on these observations, we conclude that RPA is required for the DNA damage-induced recruitment of Rad9 checkpoint complex to chromatin as well as the facilitation of Rad9 complex formation in nucleus.
Figure 5.
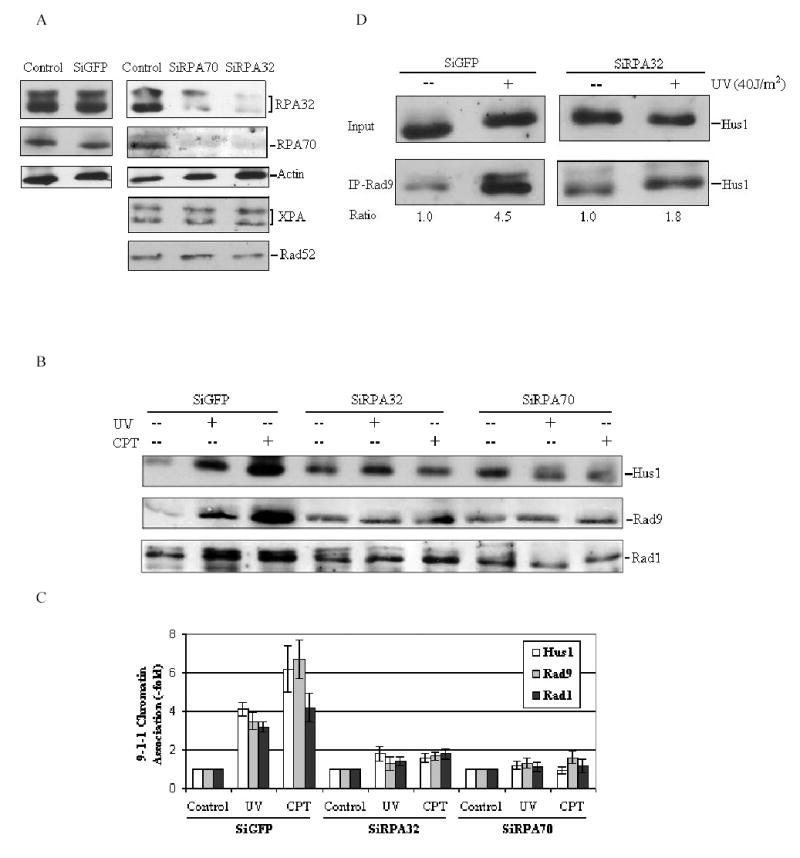
Knockdown of RPA by siRNA affects 9-1-1 complex accumulation on chromatin following DNA damage. (A): HeLa cells were transfected with siRPA70, siRPA32 or siGFP as described in Materials and methods. Controls were treated with transfection reagents only. Total cell lysates were harvested 72 h after transfection, and probed with indicated antibodies, respectively. (B): HeLa cells were transfected with indicated siRNA, and then cells were treated with 40 J/m2 UV irradiation or 10μM CPT at 72h after transfection. The chromatin-bound fractions were isolated as described in materials and methods and immunoblotted with anti-Rad9, anti-Rad1 and anti-Hus1 antibodies, respectively. (C): The data obtained from panel (B) were quantified using Fuji Image Gauge 3.46, and normalized to the controls (as the value of 1) and are the mean ± SD of three independent experiments. (D): HeLa cells were transfected with siRPA32 or siGFP, and then treated with 40 J/m2 UV irradiation 72 h after transfection. The nuclear extracts were prepared and used for co-IP assays with anti-Rad9 antibody. The bound protein was detected with anti-Hus1 antibody. The quantity of Hus1 immunoprecipitated by anti-Rad9 antibody was normalized to input.
Discussion
RPA is one of the most abundant proteins in eukaryotic cells and interacts with a wide variety of protein partners required for DNA replication, repair and recombination, suggesting that RPA is a versatile factor in the cellular DNA metabolic processes (Binz et al., 2004; Iftode et al., 1999). Recent evidences suggested the participation of RPA in DNA damage checkpoint pathway, especially as an upstream regulator in the checkpoint network (Rouse et al., 2002; Zou et al., 2003a; Zou et al., 2003b). In this study, we demonstrated that RPA directly interacted with 9-1-1 checkpoint complex in human cells and thus offered a novel insight into the role of RPA in the DNA damage-induced checkpoint responses.
Although the recent in vitro studies and the work in yeast have suggested RPA as a critical mediator in the recruitment of 9-1-1 complex by Rad17-Rfc2-5 clamp loader to the sites of DNA damage (Ellison et al., 2003; Zou et al., 2003b), the exact role of RPA in this process, particularly in mammalian cells, remains unclear. Analogous to its replicative counterpart PCNA, 9-1-1 complex is also a sliding clamp that slides on DNA substrate once loaded (Ellison et al., 2003). The RPA-mediated recruitment of 9-1-1 complex by Rad17-Rfc2-5 clamp loader therefore may be attributed to (i) the ability of RPA to physically prevent 9-1-1 clamp from sliding off the DNA ends, (ii) the involvement of specific protein-protein interaction, or both. Our results showed that RPA and 9-1-1 complex did interact with each other in human cells, implying that RPA may facilitate the loading of 9-1-1 to chromatin in vivo through the direct interaction with 9-1-1 complex. This is evidenced by the fact that the DNA damage-induced 9-1-1 complex association with chromatin was drastically attenuated by silencing the RPA expression. Moreover, in response to DNA damage, both RPA and 9-1-1 complex redistributed into nucleus, and their interactions and nuclear co-localization were significantly stimulated, supporting that RPA and 9-1-1 complex work cooperatively to activate checkpoint signaling. These observations could be attributed to two different possible mechanisms. First, RPA, with its strong affinity to ssDNA, binds to the ssDNA intermediate structures produced by DNA repair processes or stalled replication forks, and then recruits 9-1-1 complex directly to DNA damage sites. Upon localization to damage site, 9-1-1 complex may therefore be loaded onto primer/template junctions by Rad17-Rfc2-5 clamp loader and form a sliding clamp. The interaction of RPA with 9-1-1 complex may represent a critical step in the initiation of checkpoint signaling. In support of this, the yeast RPA was unable to substitute for human RPA to stimulate loading of 9-1-1 complex to DNA substrates by Rad17-Rfc2-5 clamp loader in vitro probably due to the deficient protein-protein interactions (Ellison et al., 2003). Alternatively, however, RPA may play only a facilitating role in the recruitment of 9-1-1 complex by Rad17-Rfc-2-5. It has been reported that loss of Rad17 significantly abolishes DNA damage-induced recruitment of 9-1-1 complex to the chromatin (Zou et al., 2002). Thus it is also possible that cellular knockdown of RPA may disrupt the Rad17-dependent 9-1-1 loading.
In addition to association with Rad1 and Hus1 to form a heterotrimeric complex that functions as a damage sensor in checkpoint pathway, Rad9 has been shown to interact with a number of other cellular proteins such as androgen receptor, antiapoptotic proteins Bcl-2 and Bcl-xL, kinase PKC δ and carbamoyl phosphate synthetase/aspartate transcarbamoylase/dihydroorotase (Komatsu et al., 2000; Lindsey-Boltz et al., 2004; Wang et al., 2004; Yoshida et al., 2003). Here we demonstrated that the interaction of 9-1-1 complex with RPA is also meditated by Rad9, which interact with both RPA70 and RPA32 subunits of RPA. Interestingly, the loading of 9-1-1 complex onto DNA substrate by Rad17-Rfc2-5 clamp loader is also mainly mediated by the interaction of Rad9 with Rad17 (Bermudez et al., 2003). However, Rad17 was not converted into extraction-resistant form in response to DNA damage (Burtelow et al., 2000) and could not associate with the chromatin-bound fraction of 9-1-1 complex (Rauen et al., 2000), suggesting that Rad17 complex may loosely and transiently interact with 9-1-1 complex and chromatin. This is in contrast with our observations that RPA and 9-1-1 complex translocated into nucleus simultaneously following DNA damage and the interaction and co-localization were significantly induced. Together these data strongly suggest that RPA works cooperatively with Rad17 complex and plays a role in recruiting Rad9-Rad1-Hus1 checkpoint complex in response to DNA damage.
Numerous checkpoint proteins are phosphorylated in response to DNA damage and the phosphorylation may modulate protein-protein interactions and the protein functions in checkpoint pathways (Sun et al., 1998; Yaffe et al., 2001; Yu et al., 2003). We found that hyperphosphorylated RPA was able to efficiently interact and co-localize with 9-1-1 checkpoint complex upon DNA damage. It has been reported that hyperphosphorylated RPA was defective in support of in vitro DNA replication and failed to associate with replication centers in vivo, while had no effect on nucleotide excision repair activity (Carty et al., 1994; Pan et al., 1995; Vassin et al., 2004). Our present results indicate that hyperphosphorylated RPA is also active in 9-1-1 checkpoint signaling pathway. Furthermore, protein phosphorylation appears to be essential for the efficient interaction between RPA and 9-1-1 complex as de-phosphorylation of whole cell lysates almost completely abrogated Rad9 interaction with RPA. Nevertheless, the exact mechanisms by which the protein phosphorylation modulates RPA-Rad9 complex interaction is out of the scope of this study, and need to be examined in future.
Materials and methods
Cell culture and treatments
HeLa cells were obtained from American Type Culture Collection and maintained at 37 °C and 5% CO2 in Dubelco’s Modified Eagles Medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. For UV exposure, cells were irradiated with various doses of UV using a UV crosslinker at a dose rate of 0.5 J/m2/s and further incubated for 2 h at 37 °C before harvesting. For CPT treatments, cells were incubated with indicated doses of CPT for 3 h before cell lysates were prepared.
Subcellular fractionation
The cellular protein fractionation was performed essentially as described (Zou et al., 2002) with following modifications. To prepare whole cell extracts, cells were lysed in solution A (50 mM Tris-HCl, pH7.8, 420 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 0.34 M sucrose, 10% glycerol, 1 mM Na3VO4, 10 mM NaF and β-glycerophosphate, 1 mM PMSF, and protease inhibitor cocktail). Lysates were cleared by centrifugation and protein concentration was determined by Bradford assay (Bio-Rad). For nuclear extracts preparation, cells were first lysed in buffer B (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, protease and phosphatase inhibitors). Cytoplasmic proteins were separated from nuclei by low-speed centrifugation (1300g for 4 min). Isolated nuclei were washed once with solution B and then further lysed in solution A as above. To prepare chromatin-bound proteins, isolated nuclei were lysed in solution C (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT) to release soluble nuclear proteins and then further lysed in solution A as above.
Western blotting
Cell lysates and immunoprecipitates were separated on SDS-polyacrylamide gels and transferred to PVDF membrane. The membranes were blocked with TBST buffer containing 5% powered milk and probed using following primary antibodies: anti-RPA32, anti-Rad1 (Kamiya), and anti-RPA70, anti-Rad9, anti-Hus1 (Santa Cruz). The membranes were then incubated with horseradish peroxidase-linked secondary anti-mouse antibodies and bound antibodies were visualized using ECL chemiluminescent method.
Co-immunoprecipitation assays
Cell lysates were diluted with dilution buffer (15 mM Tris-HCl, pH7.8, 1 mM EDTA, 10% glycerol, protease and phosphatase inhibitors), and incubated with 4 μg of rabbit anti-Rad9, anti-Hus1, goat anti-RPA70 or mouse anti-Rad1 antibodies, respectively, for 10–14 h at 4 °C with end-over-end mixing. Then protein A/G-agarose beads were added and the reaction mixtures were further mixed for 1 h at 4 °C. The immunoprecipitates were separated from supernatant by centrifugation and washed with PBS containing 0.05% Nonidet P-40. Proteins were extracted from the agarose beads by boiling in 1× SDS gel loading buffer and resolved on 12% SDS- polyacrylamide gels.
RPA purification
Recombinant human RPA p70 and p32/p14 complex was expressed in E. coli BL21(DE3)-RP cells and purified as previously described (Yang et al., 2002) with the following modifications. Following immobilization of RPA on a chitin column (New England Biolabs) the protein was washed with 1.5M NaSCN HI buffer and RPA70 subunit purified from the flow-through by FPLC using a Superdex 200 HR size exclusion column (Amersham). RPA p32/p14 complex was eluted from the chitin column by cleavage of the fusion protein with 30mM DTT and further purified by FPLC.
Immunofluorescence
Cells were grown on 18 mm coverslips overnight prior to treatment. After treatment, cells were extracted with PBS containing 0.5% NP-40, fixed with 100% methanol, and blocked in PBS containing 15% FBS. Primary antibody dilutions used are as follows: rabbit anti-Rad9 1:500, mouse anti-RPA32 1:1000, rabbit anti-phospho-RPA32 Ser4/Ser8 1:2000 (Bethyl Laboratories, Inc.), mouse anti-Rad1 1:250. Secondary antibody dilutions are as follows: anti-rabbit Alexa Fluor 488 1:250 and anti-mouse Alexa Fluor 568 1:250 (Molecular Probes). Images were captured with a Nikon inverted fluorescent microscope with attached CCD camera at 100 X magnification and processed using Photoshop 6.0 (Adobe) software.
Small interfering RNA (siRNA) transfections
SiRNA transfection experiments were carried out using TransIT-TKO Transfection Reagent (Mirus) essentially following the manufacture’s instructions. Transfection-ready siRNA duplexes were purchased from Ambion Inc. SiRNA sequences used in this study were: RPA70:5′-AACUGGUUGACGAAAGUGGUG-3′ and RPA32:5′-GGCUCCAACCAACAUUGUU-3′.
Acknowledgments
We thank Drs. Antonio E. Rusinol and Krishna Singh for their assistance in immunofluorescence technique. We also thank Dr. Deling Yin for helpful suggestions on siRNA experiments.
Footnotes
This study was supported by NCI grant CA86927 (to Y.Z.)
References
- Abraham RT. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Bao S, Lu T, Wang X, Zheng H, Wang LE, Wei Q, Hittelman WN, Li L. Oncogene. 2004;23:5586–5593. doi: 10.1038/sj.onc.1207753. [DOI] [PubMed] [Google Scholar]
- Barr SM, Leung CG, Chang EE, Cimprich KA. Curr Biol. 2003;13:1047–1051. doi: 10.1016/s0960-9822(03)00376-2. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz SK, Sheehan AM, Wold MS. DNA Repair (Amst) 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Bochkarev A, Bochkarev E. Curr Opin Struc Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Bochkarev E, Korolev S, Lees-Miller SP, Bochkarev A. EMBO J. 2002;21:1855–1863. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtelow MA, Kaufmann SH, Karnitz LM. J Biol Chem. 2000;275:26343–26348. doi: 10.1074/jbc.M001244200. [DOI] [PubMed] [Google Scholar]
- Burtelow MA, Roos-Mattjus PM, Rauen M, Babendure JR, Karnitz LM. J Biol Chem. 2001;276:25903–25909. doi: 10.1074/jbc.M102946200. [DOI] [PubMed] [Google Scholar]
- Carty MP, Zernik-Kobak M, McGrath S, Dixon K. EMBO J. 1994;13:2114–2123. doi: 10.1002/j.1460-2075.1994.tb06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T, Dahlen M, Kanter-Smoler G, Lindsay HD, Hofmann K, Papadimitriou K, Sunnerhagen P, Carr AM. Mol Cell Biol. 2000;20:1254–1262. doi: 10.1128/mcb.20.4.1254-1262.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Dart DA, Adams KE, Akerman I, Lakin ND. J Biol Chem. 2004;279:16433–16440. doi: 10.1074/jbc.M314212200. [DOI] [PubMed] [Google Scholar]
- Ellison V, Stillman B. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftode C, Daniely Y, Borowiec JA. Crit Rev Biochem Mol Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Komatsu K, Miyashita T, Hang H, Hopkins KM, Zheng W, Cuddeback S, Yamada M, Lieberman HB, Wang HG. Nature Cell Biol. 2000;2:1–6. doi: 10.1038/71316. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- Lindsey-Boltz LA, Bermudez VP, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 2001;98:11236–11241. doi: 10.1073/pnas.201373498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey-Boltz LA, Wauson EM, Graves LM, Sancar A. Nucleic Acids Res. 2004;32:4524–30. doi: 10.1093/nar/gkh789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yang Z, Utzat CD, Liu Y, Geacintov NE, Basu AK, Zou Y. Biochem J. 2005;385:519–26. doi: 10.1042/BJ20041151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Neecke H, Paciotti V, Lucchini G, Plevani P. Nucleic Acids Res. 1996;24:3533–3537. doi: 10.1093/nar/24.18.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes NF, Murguia JR. Curr Opin Genet Dev. 2000;10:17–25. doi: 10.1016/s0959-437x(99)00050-7. [DOI] [PubMed] [Google Scholar]
- O’Connell MJ, Walworth NC, Carr AM. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/s0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- Pan ZQ, Park CH, Amin AA, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 1995;92:4636–4640. doi: 10.1073/pnas.92.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrilla-Castellar ER, Arlander SJ, Karnitz L. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- Rauen M, Burtelow MA, Dufault VM, Karnitz LM. J Biol Chem. 2000;275:29767–29771. doi: 10.1074/jbc.M005782200. [DOI] [PubMed] [Google Scholar]
- Robison JG, Elliott J, Dixon K, Oakley GG. J Biol Chem. 2004;279:34802–34810. doi: 10.1074/jbc.M404750200. [DOI] [PubMed] [Google Scholar]
- Roos-Mattjus P, Hopkins KM, Oestreich AJ, Vroman BT, Johnson KL, Naylor S, Lieberman HB, Karnitz LM. J Biol Chem. 2003;278:24428–24437. doi: 10.1074/jbc.M301544200. [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- Shell SM, Hess S, Kvaratskhelia M, Zou Y. Biochemistry. 2005;44:971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnerhagen P, Carr AM. Mol Cell Biol. 2000;20:1254–1262. doi: 10.1128/mcb.20.4.1254-1262.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS, Stern DF. Science. 1998;281:272–274. doi: 10.1126/science.281.5374.272. [DOI] [PubMed] [Google Scholar]
- Vassin VM, Wold MS, Borowiec JA. Mol Cell Biol. 2004;24:1930–1943. doi: 10.1128/MCB.24.5.1930-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venclovas C, Thelen MP. Nucleic Acids Res. 2000;28:2481–2493. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmer E, Karnitz LM. J Biol Chem. 1999;274:567–570. doi: 10.1074/jbc.274.2.567. [DOI] [PubMed] [Google Scholar]
- Wang L, Hsu CL, Ni J, Wang PH, Yeh S, Keng P, Chang C. Mol Cell Biol. 2004;24:2202–2213. doi: 10.1128/MCB.24.5.2202-2213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Elia AE. Curr Opin Cell Biol. 2001;13:131–138. doi: 10.1016/s0955-0674(00)00189-7. [DOI] [PubMed] [Google Scholar]
- Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Biochemistry. 2002;41:13012–13020. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Komatsu K, Wang HG, Kufe D. Mol Cell Biol. 2002;22:3292–3300. doi: 10.1128/MCB.22.10.3292-3300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Wang HG, Miki Y, Kufe D. EMBO J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Kong L, Newport J. J Biol Chem. 2002;277:27088–27093. doi: 10.1074/jbc.M204120200. [DOI] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Science. 2003a;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, Elledge SJ. Proc Natl Acad Sci USA. 2003b;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]