

Abstract
Neurons have ion channels that are directly gated by voltage, ligands and temperature but not by light. Using structure-based design, we have developed a new chemical gate that confers light sensitivity to an ion channel. The gate includes a functional group for selective conjugation to an engineered K+ channel, a pore blocker and a photoisomerizable azobenzene. Long-wavelength light drives the azobenzene moiety into its extended trans configuration, allowing the blocker to reach the pore. Short-wavelength light generates the shorter cis configuration, retracting the blocker and allowing conduction. Exogenous expression of these channels in rat hippocampal neurons, followed by chemical modification with the photoswitchable gate, enables different wavelengths of light to switch action potential firing on and off. These synthetic photoisomerizable azobenzene-regulated K+ (SPARK) channels allow rapid, precise and reversible control over neuronal firing, with potential applications for dissecting neural circuits and controlling activity downstream from sites of neural damage or degeneration.
Natural photoreceptive proteins, such as rhodopsin, have a covalently attached chromophore that directly activates the protein on exposure to light. Several strategies have been used to enable light regulation of proteins that are not intrinsically light sensitive. Light can be used indirectly to activate receptor proteins, for example, by making a ligand available from a caged precursor1. Light can also be used to directly photoisomerize a synthetic molecule that is covalently attached to a protein, thereby imposing conformational changes2,3. We have combined these ideas by synthesizing a photoisomerizable tether that attaches a specific ligand on a protein near its normal binding site. Photoswitching of the tethered ligand rapidly regulates its ability to reach its binding site, thereby switching the protein on and off without causing unnatural conformational changes. Photoswitchable tethered ligands can be agonists (for example for nicotinic acetylcholine receptors4), antagonists, or other regulators of protein function. We have applied this general design principle to an ion channel, where the chosen ligand is a blocker of the channel's pore.
Our approach is particularly valuable for controlling neuronal firing because ion channels are the basis of neuronal signaling. Many techniques exist for controlling neural activity, but they all have considerable limitations. Traditional electrical and chemical methods require invasive electrodes or chemical delivery systems that cannot control patterns of activity in densely packed neural tissue. Optical techniques utilizing caged neurotransmitters5,6 are less invasive and can be more precise, but reversal of the effects of the uncaged transmitter is limited by its diffusion kinetics. Recently, the exogenous expression of genes encoding ion channels has been used to influence electrical activity in specific neurons7-9, but the onset and reversal of gene expression is slow. A modification of this technique, in which a receptor from one species is introduced into another that lacks a natural ligand, dramatically improves temporal control10,11, but this approach still relies on a diffusible ligand whose time course of removal limits reversibility. Finally, photic regulation has been conferred on neurons by introducing a rhodopsin-based signal transduction cascade12. This technique requires coordinated exogenous expression of three different genes and produces light responses that can be slow in onset and offset and variable in different neurons, possibly because the nature of the native ion channels that are regulated by the cascade can vary between neurons.
We have developed light-activated ion channels as an alternative strategy for controlling neuronal activity. Like other optical methods, light-activated channels allow rapid, remote and noninvasive control. Because the light-activated gate is covalently linked to the ion channel, and because the ion channel is integral to the neuronal cell membrane, control over individual neurons is spatially accurate and does not rely on a diffusible ligand. In addition, the gate can be reversibly photoswitched, allowing recurrent control of neural activity.
RESULTS
Engineering and testing the light-activated channel
As a starting point for engineering a light-activated channel, we used the Shaker K+ channel because of the availability of structural and molecular information13,14. Voltage-gated K+ channels, including Shaker, are blocked by the binding of quaternary ammonium ions, such as tetraethylammonium (TEA), to a site in the pore-lining domain15,16. Amino acid Glu422 is estimated to be 15–18 Å from the TEA binding site17-19. When a cysteine is substituted at position Glu422, tethering of a series of cysteine-reactive compounds that contain quaternary ammonium to this position shows that the degree of block is critically dependent on tether length, with a 5-Å difference in length making the distinction between effective and ineffective block17. We used this information in designing and synthesizing a photoswitchable blocker that can be tethered to the outside of modified Shaker channels. The molecule, MAL-AZO-QA, consists of a maleimide (MAL) for cysteine tethering and a quaternary ammonium group to block the channel, with an azo group between (Fig. 1a). The rigid azo moiety shortens by approximately 7 Å when photoisomerized from the trans to the cis configuration20. We reasoned that coupling MAL-AZO-QA to a cysteine introduced at residue 422 (mutant E422C) would block channels when the compound is in the long trans form, whereas photoconversion to the cis configuration would make the tether too short to permit block (Fig. 1b). Hence, the tethering of MAL-AZO-QA to Shaker should introduce a new extracellular gate that can be opened and closed with appropriate wavelengths of light.
Figure 1.

Photoisomerization of MAL-AZO-QA gates ionic currents through modified Shaker channels. (a) The rigid core of MAL-AZO-QA (between the α carbons flanking the azo moiety) changes by about 7 Å upon photoisomerization. (b) MAL-AZO-QA blocks ion flow in the trans configuration but is too short to block effectively after photoisomerization to the cis configuration. Diagram shows a model of the inner helices of the Shaker K+ channel, derived from the crystal structure of the bacterial K+ channel MthK31, with the dimensions of MAL-AZO-QA drawn roughly to scale.
We first tested the effects of MAL-AZO-QA on Shaker channels expressed in Xenopus laevis oocytes. To observe the time course of channel modification, MAL-AZO-QA was applied to the extracellular surface of the channels in outside-out membrane patches. MAL-AZO-QA application reduced the voltage-gated Shaker current by >60% over 4 min (Fig. 2a), but the limited survival time of excised patches made it difficult to assess the full magnitude of block. Channel block developed slowly and persisted after washout (Fig. 2b), consistent with covalent attachment to the channels. Subsequent exposure to ultraviolet light partly relieved channel block and exposure to visible light restored the block (Fig. 2b). In contrast, light had no effect on channels in patches that had not been treated with MAL-AZO-QA (data not shown).
Figure 2.

Photocontrol of MAL-AZO-QA-modified Shaker channels in X. laevis oocytes. (a) Raw Shaker K+ current traces recorded from an outside-out patch before and after treatment with MAL-AZO-QA. The top trace in each panel shows the current before MAL-AZO-QA application. Bottom traces represent current after 4-min application of 10 μM MAL-AZO-QA and 2-min washout (left trace), after 1-min exposure to ultraviolet (UV; 380 nm) light (middle trace) and after 1-min exposure to visible (Vis.; 500 nm) light (right trace). The patch was held at −90 mV and currents were elicited by 100-ms steps to −20 mV at 1 Hz. (b) K+ current amplitudes from the same outside-out patch during perfusion with MAL-AZO-QA, during washout, and during alternating illumination with 380 and 500 nm light. (c) Inside-out patch from an oocyte treated with 100 μM MAL-AZO-QA for 30 min. The patch shows a large Shaker current in 380 nm light and almost complete block in 500 nm light. Pulse protocol same as above, except pulse duration was 30 ms. (d) Current block in the dark follows a biexponential time course with τ1 = 0.49 min and τ2 = 4.79 min.
To achieve more complete block of the channels, we treated intact oocytes with a higher concentration of MAL-AZO-QA for 30 min and then recorded from inside-out patches (Fig. 2c). In this situation, ultraviolet light unblocked as much as 1 nA of current, visible light reblocked the channels almost completely and both effects nearly reached steady state within 5 s under standard epifluorescence illumination. With steady ultraviolet illumination, the channels remained unblocked. In the dark, however, unblocked channels slowly (>5 min) returned to the blocked state (Fig. 2d), consistent with thermal relaxation of the azo moiety to the more stable trans configuration in the absence of light20. Current block in the dark followed a biexponential time course (Fig. 2d), suggesting that a second process was involved. This may be a decrease in slow inactivation as the channels become reblocked by quaternary ammonium21.
Spectral sensitivity
Which wavelengths are best for opening and closing MAL-AZO-QA-modified channels? To address this question, we first measured the absorbance spectra of MAL-AZO-QA in solution as a glutathione adduct (Fig. 3a). The trans configuration of MAL-AZO-QA has a large absorbance peak at 360 nm and a small shoulder at about 440 nm, as reported for other azo derivatives20. Maximal photoisomerization to the cis configuration considerably decreased the 360-nm peak and slightly increased absorbance between 440 and 540 nm. Although the spectra indicate the wavelengths of maximum absorbance for the trans and cis isomers, the spectral overlap between isomers suggests that these may not be the optimal wavelengths for maximal photoconversion. In addition, coupling of MAL-AZO-QA to the channel protein could affect the absorbance spectra. Finally, the spectral properties of the illumination system used in the experiment (in our case a xenon lamp) will determine the relative intensities of different wavelengths. We therefore determined the optimal wavelengths empirically by measuring the action spectrum of each isomer (Fig. 3b-d).
Figure 3.

Absorbance and action spectra of MAL-AZO-QA. (a) The ultraviolet and visible light spectrum of a MAL-AZO-QA-glutathione adduct (10 μM) in oocyte bath solution. To maximize the trans and cis isomers, the solution was exposed to visible and ultraviolet light, respectively, for 3 min. To generate the adduct, MAL-AZO-QA (1 M) was treated with reduced glutathione (1.5 M) for 12 h at 21 °C. (b) Unblocking of Shaker channels at different wavelengths. Currents are from an inside-out patch alternately exposed to various wavelengths between 300 and 480 nm to unblock the channels, and to 500 nm light to reblock the channels. (c) Reblocking of Shaker channels at different wavelengths. The time course of blocking at various wavelengths of visible light. Each trial is preceded by 1-min irradiation at 380 nm to unblock the channels. Traces are superimposed for comparison. Normalized current amplitudes were measured at 0.2 min after onset of blocking. (d) Action spectra for unblocking (left curve) and blocking (right curve) of Shaker K+ channels. Unblock (left axis): Current unblocked at each wavelength divided by current at 380 nm (n = 3–8 patches per wavelength). Currents were compared within each patch. Block (right axis): Fraction of normalized current blocked at 0.2 min after illumination with visible light (n = 2–7 patches per wavelength).
To determine the wavelength that results in maximal recovery of K+ currents, inside-out patches were initially exposed to long-wavelength light (500 nm) for at least 1 min to maximize occupancy of the blocked state. The patch was then irradiated with a discrete wavelength between 300 and 480 nm for 1 min, and the peak current at steady state was measured to determine the degree of unblock (Fig. 3b). The resulting action spectrum shows that 380 nm is most effective in unblocking MAL-AZO-QA-modified Shaker channels (Fig. 3d). The action spectrum for channel unblock presumably reflects the steady-state ratio of trans and cis isomers (the photostationary state) at each wavelength.
To determine the complementary action spectrum for channel reblocking, patches were exposed to 380 nm for 1 min to maximize occupancy of the cis state. Subsequent exposure to discrete wavelengths between 420 and 600 nm caused reblocking of the channels at different rates (examples are shown in Fig. 3c). In this case, rate of reblocking is the most relevant parameter, because many wavelengths will eventually result in complete blocking, as MAL-AZO-QA reaches its thermodynamically more stable trans configuration. Thus we measured the degree of blocking at a fixed time (0.2 min) for each wavelength. The broad peak of this action spectrum (Fig. 3d) indicates that wavelengths from 460 to 500 nm cause the fastest reblocking of channels.
Expression and use in neurons
We next tested whether light-activated channels could be used to control neuronal excitability. First, we wanted to modify the voltage dependence of the channel, so that the photoswitch is the primary regulator of gating. Normally, Shaker-type K+ channels make only a minor contribution to the membrane conductance at typical resting potentials (−40 to −70 mV). The channels also display voltage-dependent inactivation, further limiting their contribution. We therefore introduced mutations to eliminate rapid inactivation (Δ6–46; ref. 22), to reduce slow inactivation (T449V; ref. 23) and to shift voltage-dependent activation to hyperpolarized potentials (L366A; ref. 24), as confirmed by expression in oocytes (data not shown). Expression of voltage-gated K+ channels with these modifications should result in a high resting conductance for K+ and silencing of spontaneous activity.
This multiply mutated Shaker channel was expressed in cultured hippocampal neurons, which were subsequently treated with MAL-AZO-QA for 15 min in the dark, followed by thorough washout. Current clamp recordings from transfected pyramidal cells, identified by coexpression of green fluorescent protein (GFP), showed that exposure to 390-nm light silenced spontaneous action potentials within 3 s and exposure to 500-nm light restored activity, also within seconds (Fig. 4a). Similar results were obtained in five cells. Activity could also be restored simply by leaving neurons in the dark after silencing (data not shown), but the onset was slow (>30 s), in accord with the slow reblocking of ionic current observed in oocyte patches in the dark. Hence, a 5-s pulse of 390-nm light should produce relatively sustained silencing. Prolonged depolarizing current steps caused repetitive firing in 500-nm light (Fig. 4b, left). In contrast, in 390-nm light the same steps elicited rapidly accommodating responses (usually a single action potential, even with depolarization well above threshold; Fig. 4b, right). On average, 390-nm light decreased the number of action potentials elicited by a depolarizing step by 79% (n = 4; Fig. 4c).
Figure 4.
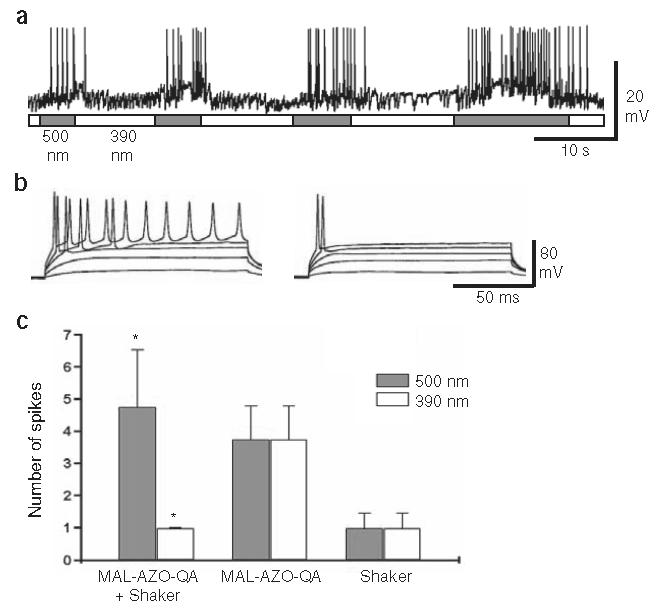
Expression of light-activated channels confers light sensitivity on hippocampal pyramidal neurons. (a) Spontaneous action potentials are silenced and revived by exposure to 390- and 500-nm light, respectively. A neuron that was transfected with the multiply mutated Shaker channel was treated for 15 min with MAL-AZO-QA before recording. The frequency of spontaneous synaptic potentials generated by untransfected presynaptic neurons is not affected by light. (b) Depolarizing current steps elicit repetitive firing in 500-nm light (left) but only single action potentials in 390-nm light (right). Neurons were held under current clamp at about −55 mV and were depolarized to about −15 mV. (c) Summary of repetitive firing data. Number of spikes resulting from a suprathreshold depolarization to −15 mV is significantly modulated by light in the multiply mutated Shaker-transfected neurons treated with MAL-AZO-QA (*P < 0.01). Neurons expressing the channel without MAL-AZO-QA treatment or treated with MAL-AZO-QA without channel expression were unaffected by light.
Light had no effect on MAL-AZO-QA-treated neurons that expressed GFP without the multiply mutated Shaker channels (n = 4), nor on channel-transfected neurons without MAL-AZO-QA treatment (n = 5; Fig. 4c). Hence, it seems that native K+ channels are not susceptible to light-regulated block by tethered MAL-AZOQA, even though many of these channels can be blocked by TEA.
The observation that only Shaker-transfected neurons are light sensitive indicates that MAL-AZO-QA may selectively attach to the introduced cysteine, and the reaction is facilitated by the high effective local concentration of the cysteine-reactive maleimide when the quaternary ammonium binds to the pore25. Nonselective attachment of MAL-AZO-QA to extracellular cysteines on other membrane proteins may have no detectable effects on cellular electrophysiology, because other channels and receptors are unlikely to have a TEA-binding site positioned at the appropriate distance from a modifiable cysteine.
DISCUSSION
Coupling MAL-AZO-QA to the outside of Shaker adds a new light-switchable gate to the channel without compromising its normal voltage-dependent gating mechanism. By combining synthetic chemistry and protein mutagenesis, we have re-engineered the Shaker channel, adding an artificial control mechanism that is orthogonal to the natural one provided by evolution. This strategy may be extended to ionotropic or metabotropic receptors by incorporating specific receptor agonists or antagonists. Two conditions must be met for light-switchable ligands to alter the function of channels or receptors: (i) a given target protein must have a binding site for the ligand component of the molecule, and (ii) that site must be a precise distance from the covalent attachment site on the protein. MAL-AZO-QA attaches to a specific cysteine, but it might be possible to use other attachment strategies, including site-specific recognition sequences26.
In the absence of MAL-AZO-QA, overexpression of the mutant Shaker channel may alter the normal electrical activity of hippocampal neurons. One would expect channel overexpression to hyperpolarize the membrane potential and suppress action potential firing, especially given the mutations that shift activation and reduce inactivation. Overexpression of Shaker channels can, however, result in a compensatory change in the expression of genes encoding native ion channels, causing hyperexcitability of neurons27. In addition, overexpressed Shaker channels could form heteromultimers with native channel subunits, but this should occur slowly, as the turnover rate of native K+ channel subunits probably takes days. None of these perturbations should interfere with the use of light in regulating the activity of the MAL-AZO-QA-modified channels and ultimately in regulating neuronal firing. The procedure used here could, however, be modified to limit undesired effects of channel overexpression. For example, MAL-AZO-QA could be introduced at the onset of channel expression, ensuring that the channels are continually blocked, so that compensation is less likely to occur. Mutations resulting in milder changes in Shaker channel gating might also allow light regulation of firing without perturbing basal activity.
Site-directed functionalization of Shaker channels with MAL-AZO-QA introduces a new and permanent photoswitched gate. The power of this technique lies in its spatial and temporal accuracy, its noninvasiveness and its reversibility. The spatial resolution of channel activation should be limited only by the optics of the experimental system, and the kinetics of the response is limited only by the intensity of the light source.
Because these channels can control the firing of action potentials, we propose the name SPARK (synthetic photoisomerizable azobenzene-regulated K+) channels. Irradiation of SPARK channels may be used to control neuronal populations, individual neurons, or even parts of neurons, such as axonal or dendritic branches. SPARK channels have the potential to transform the experimental analysis of neuronal circuitry not only in vitro but also in vivo. Shaker-type K+ channels can be readily expressed at high density in mammalian neurons using standard gene transfection techniques including lipofection28, viral infection29 and transgenic gene expression27. Amphipathic molecules similar to MAL-AZO-QA, such as voltage-sensitive dyes30, penetrate deeply into neural tissue and label neuronal membranes. These features may enable the widespread use of SPARK channels, even deep in neural tissue. In addition to their use experimentally, SPARK channels may provide an effective means for controlling the activity of specific neurons downstream from sites of neural damage or degeneration. A particularly intriguing possibility is to use these channels to restore light-regulated activity in healthy retinal neurons after degeneration of rods and cones, the native photoreceptors. Light-regulated molecular machines, such as the SPARK channel, may also have applications in fields other than neurobiology, such as nanotechnology, bioelectronics and material sciences.
METHODS
Synthesis of MAL-AZO-QA
MAL-AZO-QA was synthesized in a two-step coupling procedure from commercially available 4,4′-azodianiline and the respective acid chlorides of the maleimide and quaternary ammonium components (M.B., K.B., E.I., R.H.K. and D.T., unpublished data).
Patch recordings from oocytes
X. laevis oocytes were injected with 12.5–100 ng of mRNA encoding Shaker H4, with the following mutations: Δ6–46, E422C and T449V. We found that the effects of light on channel activity were largest for the TEA–binding site mutant T449V as compared with the wild-type channel (Thr449) and two other mutants (T449Y, T449F). Devitillenized oocytes were recorded from 2–10 d postinjection using standard patch-clamp methods. For outside-out patches, glass patch pipettes (2–4 MΩ) were filled with a solution containing 100 mM KCl, 10 mM HEPES, 0.1 mM CaCl2, 0.5 mM MgCl2 and 10 mM EGTA, whereas the bath contained 10 mM KCl, 90 mM NaCl, 10 mM HEPES, 0.1 mM CaCl2, 0.5 mM MgCl2 and 10 mM EGTA. For inside-out patches, patch pipettes and bath both contained 100 mM KCl, 10 mM HEPES, 0.1 mM CaCl2, 0.5 mM MgCl2 and 10 mM EGTA. The pH of all solutions was 7.1. Solid MAL-AZO-QA was dissolved as a concentrated stock solution in DMSO and stored at −20°C until the day of use. Stocks were diluted into oocyte bath solution to final concentrations of 10 μM or 100 μM. The concentration of DMSO in the bath did not exceed 0.1%. Pulse protocols and measurements were carried out with pCLAMP software, a DigiData 1200 series interface and a PC-505 amplifier (Warner Instruments). Samples were taken at 10 kHz, and the data were filtered at 1 kHz. Patches were held at −90 mV, pulsed to −100 mV for 60 ms to monitor leak and pulsed to −20 mV for 30 to 100 ms at 1 Hz to elicit Shaker currents. The peak of the Shaker currents was measured to minimize the contribution of slow inactivation. Patches with a leak conductance >100 pS were not included in the analysis.
Illumination of patches was achieved with a TILL Photonics Polychrome II monochrometer (Applied Scientific Instrumentation, Inc.) containing a 75-W xenon short arc lamp with an output of 250–690 nm, a quartz fiber optic cable and an epifluorescence condenser with an achromatic lens. Discrete wavelengths of light (±10 nm) were focused on patches through a quartz coverslip with a Fluor 20×, 0.5 n.a. objective lens (Nikon). Output intensity was measured for wavelengths between 300 and 600 nm. The measured output intensities for wavelengths between 340 and 600 nm ranged from 0.324 × 10−8 to 6.23 × 10−8 W/cm2.
Recordings from hippocampal neurons
Primary dissociated hippocampal cultures were prepared from embryonic day (E) 18–19 Sprague-Dawley rat embryos and were grown on glass coverslips in serum-containing medium in 7% CO2 in air at 37 °C. All animal care and experimental protocols were approved by the Animal Care and Use Committee at UC Berkeley. Cells were transfected with GFP along with the modified Shaker channel described above with additional L366A and V454L mutations. Transfections with Lipofectamine 2000 (Invitrogen) were carried out at 12–14 days in vitro. About 10% of the cultured neurons were GFP positive at 2–3 d after transfection. Coverslips containing the neurons were treated for 15 min with 300 μM MAL-AZO-QA at 37 °C in an extracellular recording solution containing 135 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 5 mM HEPES, 2.5 mM CaCl2 and 10 mM glucose at pH 7.4. Patch pipettes (7–10 MΩ) were filled with 10 mM NaCl, 135 mM K-gluconate, 10 mM HEPES, 2 mM MgCl2, 2 mM Mg-ATP and 1 mM EGTA at pH 7.4. After washout of MAL-AZO-QA with extracellular solution, the membrane potential was recorded at room temperature under whole-cell current clamp with an AXOPATCH 200A amplifier (Axon Instruments) and was filtered at 10 kHz. Initial recordings were made at the resting potential to evaluate the effects of light on spontaneous activity. In experiments where we wanted to quantify the effect of light on firing (such as Fig. 4b), cultures were treated with CNQX (1 μM) and bicuculline (10 μM) to silence synaptic activity. Baseline currents were adjusted to set the membrane potential at −50 mV before depolarizing current steps ranging from 0.01 to 0.03 nA were applied to evoke action potentials. Cells were irradiated with a Lambda-LS illuminator containing a 125-W xenon arc lamp (Sutter Instruments Company) equipped with narrow-bandpass (±10 nm) filters through a Fluor 20×, 0.5 n.a. objective lens (Nikon). Variability among data is expressed as mean ± s.e.m., unless noted otherwise.
ACKNOWLEDGMENTS
We thank S. Ahituv for technical support, R. Fredrick and J. Harvey for contributing to the chemical synthesis, I. Hafez and C. Nam for help with cell culture, F. Tombola for advice and C. Luetje for comments on the manuscript. This work was supported by a Fight-for-Sight grant to R.H.K. and funds from the Lawrence Berkeley National Laboratory to D.T. K.B. was supported by a Howard Hughes Medical Institute predoctoral fellowship.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Nerbonne JM. Caged compounds: tools for illuminating neuronal responses and connections. Curr. Opin. Neurobiol. 1996;6:379–386. doi: 10.1016/s0959-4388(96)80123-1. [DOI] [PubMed] [Google Scholar]
- 2.James DA, Burns DC, Woolley GA. Kinetic characterization of ribonuclease S mutants containing photoisomerizable phenylazophenylalanine residues. Protein Eng. 2001;14:983–991. doi: 10.1093/protein/14.12.983. [DOI] [PubMed] [Google Scholar]
- 3.Flint DG, Kumita JR, Smart OS, Woolley GA. Using an azobenzene cross-linker to either increase or decrease peptide helix content upon trans-to-cis photoisomerization. Chem. Biol. 2002;9:391–397. doi: 10.1016/s1074-5521(02)00109-6. [DOI] [PubMed] [Google Scholar]
- 4.Lester HA, Krouse ME, Nass MM, Wassermann NH, Erlanger BF. A covalently bound photoisomerizable agonist: comparison with reversibly bound agonists at Electrophorus electroplaques. J. Gen. Physiol. 1980;75:207–232. doi: 10.1085/jgp.75.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz LC, Dalva MB. Scanning laser photostimulation: a new approach for analyzing brain circuits. J. Neurosci. Methods. 1994;54:205–218. doi: 10.1016/0165-0270(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 6.Callaway EM. Caged neurotransmitters. Shedding light on neural circuits. Curr. Biol. 1994;4:1010–1012. doi: 10.1016/s0960-9822(00)00228-1. [DOI] [PubMed] [Google Scholar]
- 7.Nitabach M, Blau J, Holmes T. Electrical silencing of Drosophila pacemaker neurons stops the free-running circadian clock. Cell. 2002;109:485–495. doi: 10.1016/s0092-8674(02)00737-7. [DOI] [PubMed] [Google Scholar]
- 8.Johns D, Marx R, Mains R, O'Rourke B, Marban E. Inducible genetic suppression of neuronal excitability. J. Neurosci. 1999;19:1691–1697. doi: 10.1523/JNEUROSCI.19-05-01691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White B, et al. Targeted attenuation of electrical activity in Drosophila using a genetically modified K+ channel. Neuron. 2001;31:699–711. doi: 10.1016/s0896-6273(01)00415-9. [DOI] [PubMed] [Google Scholar]
- 10.Lechner H, Lein E, Callaway E. A genetic method for selective and quickly reversible silencing of mammalian neurons. J. Neurosci. 2002;22:5287–5290. doi: 10.1523/JNEUROSCI.22-13-05287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slimko E, McKinney S, Anderson D, Davidson N, Lester H. Selective electrical silencing of mammalian neurons in vitro by the use of invertebrate ligand-gated chloride channels. J. Neurosci. 2002;22:7373–7379. doi: 10.1523/JNEUROSCI.22-17-07373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zemelman BV, Lee GA, Ng M, Miesenbock G. Selective photostimulation of genetically chARGed neurons. Neuron. 2002;33:15–22. doi: 10.1016/s0896-6273(01)00574-8. [DOI] [PubMed] [Google Scholar]
- 13.Sigworth F. Voltage gating of ion channels. Q. Rev. Biophys. 1994;27:1–40. doi: 10.1017/s0033583500002894. [DOI] [PubMed] [Google Scholar]
- 14.Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- 15.MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- 16.Heginbotham L, MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- 17.Blaustein R, Cole P, Williams C, Miller C. Tethered blockers as molecular ‘tape measures’ for a voltage-gated K+ channel. Nat. Struct. Biol. 2000;7:309–311. doi: 10.1038/74076. [DOI] [PubMed] [Google Scholar]
- 18.Doyle D, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, et al. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 20.Knoll H. Photoisomerism of azobenzenes. In: Horspool W, Lenci F, editors. CRC Handbook of Organic Photochemistry and Photobiology. 2 CRC Press; Boca Raton, Florida, USA: 2004. pp. 89.1–89.16. [Google Scholar]
- 21.Choi K, Aldrich R, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshi T, Zagotta W, Aldrich R. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Barneo J, Hoshi T, Heinemann S, Aldrich R. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 24.Lopez G, Jan Y, Jan L. Hydrophobic substitution mutations in the S4 sequence alter voltage-dependent gating in Shaker K+ channels. Neuron. 1991;7:327–336. doi: 10.1016/0896-6273(91)90271-z. [DOI] [PubMed] [Google Scholar]
- 25.Blaustein R. Kinetics of tethering quaternary ammonium compounds to K+ channels. J. Gen. Physiol. 2002;120:203–216. doi: 10.1085/jgp.20028613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 27.Sutherland ML, et al. Overexpression of a Shaker-type potassium channel in mammalian central nervous system dysregulates native potassium channel gene expression. Proc. Natl. Acad. Sci. USA. 1999;96:2451–2455. doi: 10.1073/pnas.96.5.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu C, Jan YN, Jan LY. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. 2003;301:646–649. doi: 10.1126/science.1086998. [DOI] [PubMed] [Google Scholar]
- 29.Karschin A, Aiyar J, Gouin A, Davidson N, Lester HA. K+ channel expression in primary cell cultures mediated by vaccinia virus. FEBS Lett. 1991;278:229–233. doi: 10.1016/0014-5793(91)80123-k. [DOI] [PubMed] [Google Scholar]
- 30.Djurisic M, et al. Optical monitoring of neural activity using voltage-sensitive dyes. Methods Enzymol. 2003;361:423–451. doi: 10.1016/s0076-6879(03)61022-0. [DOI] [PubMed] [Google Scholar]
- 31.Jiang Y, et al. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]