

Abstract
The precise regulation of protein activity is fundamental to life. The allosteric control of an active site by a remote regulatory binding site is a mechanism of regulation found across protein classes, from enzymes to motors to signaling proteins. We describe a general approach for manipulating allosteric control using synthetic optical switches. Our strategy is exemplified by a ligand-gated ion channel of central importance in neuroscience, the ionotropic glutamate receptor (iGluR). Using structure-based design, we have modified its ubiquitous clamshell-type ligand-binding domain to develop a light-activated channel, which we call LiGluR. An agonist is covalently tethered to the protein through an azobenzene moiety, which functions as the optical switch. The agonist is reversibly presented to the binding site upon photoisomerization, initiating clamshell domain closure and concomitant channel gating. Photoswitching occurs on a millisecond timescale, with channel conductances that reflect the photostationary state of the azobenzene at a given wavelength. Our device has potential uses not only in biology but also in bioelectronics and nanotechnology.
Many proteins function like molecular machines that undergo mechanical movements in response to input signals. These signals can consist of changes in voltage, membrane tension, temperature or, most commonly, ligand concentration. Ligands provide information about events in the external world or about the energetic or biosynthetic state of the cell. They can be as small as a proton or as large as a whole protein. In allostery, ligand binding induces a structural change of a sensor domain, which propagates to a functional domain of the protein and alters its behavior. Such conformational control can operate over long distances, crossing a membrane or passing from one protein to another in a complex.
Reengineering of nanoscopic protein machines to contain artificial control elements would be a major benefit for biology and technology. Optical switches would be especially powerful, as they could be activated remotely with precise temporal and spatial control1,2. A simple design strategy would be to modify a protein by attaching a synthetic ligand whose binding ability could be altered by light. These ligands tethered via an optical switch could function in two ways. They could conditionally block the active site of an enzyme or the pore of a channel without inducing major conformational changes in the protein, or they could reversibly present an agonist to an allosteric binding site and conditionally trigger the normal conformational changes of activation (Fig. 1a).
Figure 1.
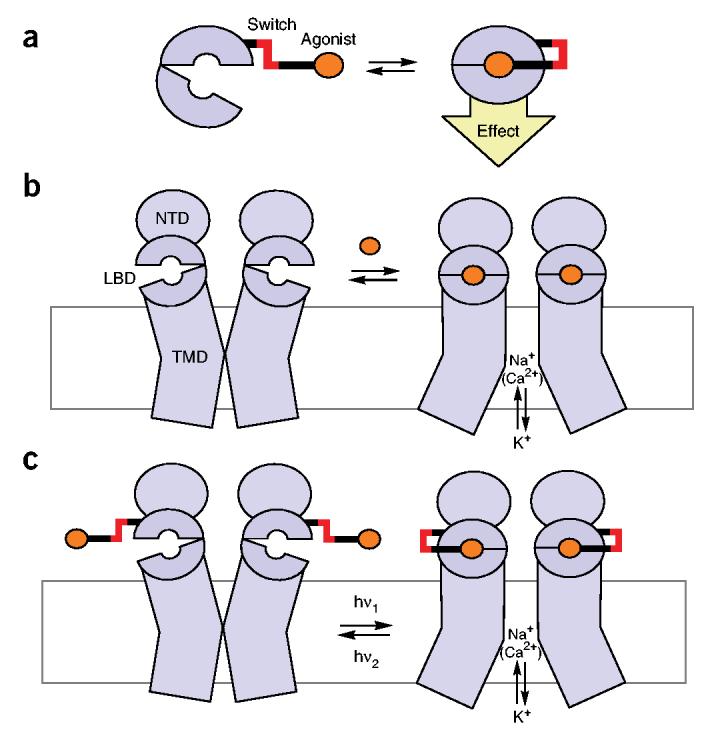
Design of an allosteric photoswitch. (a) An agonist (orange) is tethered to an LBD through an optical switch (red) via linkers (black). In one state of the switch, the ligand cannot reach the binding pocket, whereas in the other state, the ligand docks and stabilizes the activated (closed) conformation of the LBD. (b) Schematic representation of the operating mode of iGluRs. Binding of an agonist (orange) stabilizes the activated (closed) conformation of the LBD and allosterically opens the pore, allowing flow of Na+, Ca2+ and K+. NBD: N-terminal domain; TMD: transmembrane domain. (c) The principle of LiGluR. Reversible optical switching of a tethered agonist on the LBD opens and closes the pore.
An early version of an optically controlled system was reported in 1980 (ref. 3) in the form of a nicotinic acetylcholine receptor that could be photoactivated via a tethered choline analogue. This work lay dormant for many years because of the difficulty of generalizing the approach to other proteins in the absence of crystal structures and modern approaches to protein engineering. Recently, success was reported for a light-gated ‘nanovalve’, in which a photoisomerizable moiety actuates a nonselective, large-pore, mechanosensitive channel from bacteria4. Although attractive, it is not clear whether this approach could work on smaller-gauge, selective pores, or how it could be extended to other classes of proteins.
We recently demonstrated a synthetic light gate for a potassium channel that works in cells. The light gate consists of a pore blocker attached site-specifically to the protein through a photoswitchable azobenzene linker5. The potassium channel is an attractive target for several reasons. The pore is symmetric and is very accessible, located in the center of a wide platform that can seat objects as large as peptide toxins. However, these features of the potassium channel are unusual. Most proteins have active sites or allosteric sites that bind ligands much more intimately, at higher affinity, and asymmetrically, via multiple spatially precise interactions in deep crevices. Furthermore, in allosteric sites, domain closure around the ligand is key to the regulatory signal. Thus, a generalizable method is still needed to control function in the broad spectrum of proteins that bind ligands in deep sites.
With this in mind, we chose as our next target for optical control the ubiquitous ‘venus flytrap’ or ‘clamshell’ ligand-binding domain that is homologous to periplasmatic binding proteins6. Ligand-binding domains of this type represent an ancient module for allosteric activation and are present in pharmacologically important receptors, including ion channels7 and G protein–coupled receptors8. Using structure-based design, we have engineered an ionotropic glutamate receptor (iGluR) that can be turned on and off by irradiation with different wavelengths of light. Our device, termed LiGluR, thus functions as a light- and ligand-gated ion channel and can be used to rapidly and briefly inject current into cells with light.
The iGluR family members mediate the major excitatory currents in the central nervous system9. Structurally, they are tetrameric protein assemblies with subunits consisting of an extracellular N-terminal domain (NTD), an extracellular ligand binding domain (LBD) and a transmembrane domain (TMD) that forms the pore (Fig. 1b)10. The LBD closes like a clamshell as it binds the agonist glutamate. This reversible binding and closure is allosterically coupled, in an unknown way, to the opening of the pore. The detailed structures of the LBD of several iGluRs in their apo state or in complex with agonists (such as glutamate, kainate, AMPA or domoic acid) have been solved by X-ray crystallography11-13. These structures provide a vivid picture of how the LBD closes when the agonist binds.
Our approach to engineering LiGluR was to site-specifically attach a tethered analogue of glutamate containing a photoisomerizable azobenzene moiety to a ‘lip’ of the LBD clamshell (Fig. 1c). In one state of the azobenzene, the LBD would not bind the tethered agonist and therefore would remain open. Only after isomerization would the tether present the agonist to the binding site and thus effect closure. Overall, the reversible switching of an azobenzene would allosterically trigger the opening and closing of the entire ion channel, mediated by the clamshell-like movement of the LBD.
We now report the successful implementation of this strategy. Using a rational, stepwise approach, we have synthesized a suitable tethered agonist and identified an effective point of attachment to create a light-gated ionotropic glutamate receptor.
RESULTS
Synthesis and evaluation of a tether model
Our design of a tethered agonist was based both on extensive structure-activity relationship analyses that have been performed on iGluR agonists14,15 and, notably, on the X-ray structure of the LBD of iGluR6 in complex with the agonist (2S,4R)-4-methyl glutamate (1; Fig. 2a,c)12. It is evident from this structure that the ligand-bound form of the clamshell, although closed, features a narrow ‘exit channel’. Our hope was that the exit channel would enable a tether appended to an agonist to protrude and reach an attachment site at the surface of the protein, while still permitting the clamshell to close over the agonist and activate the channel.
Figure 2.
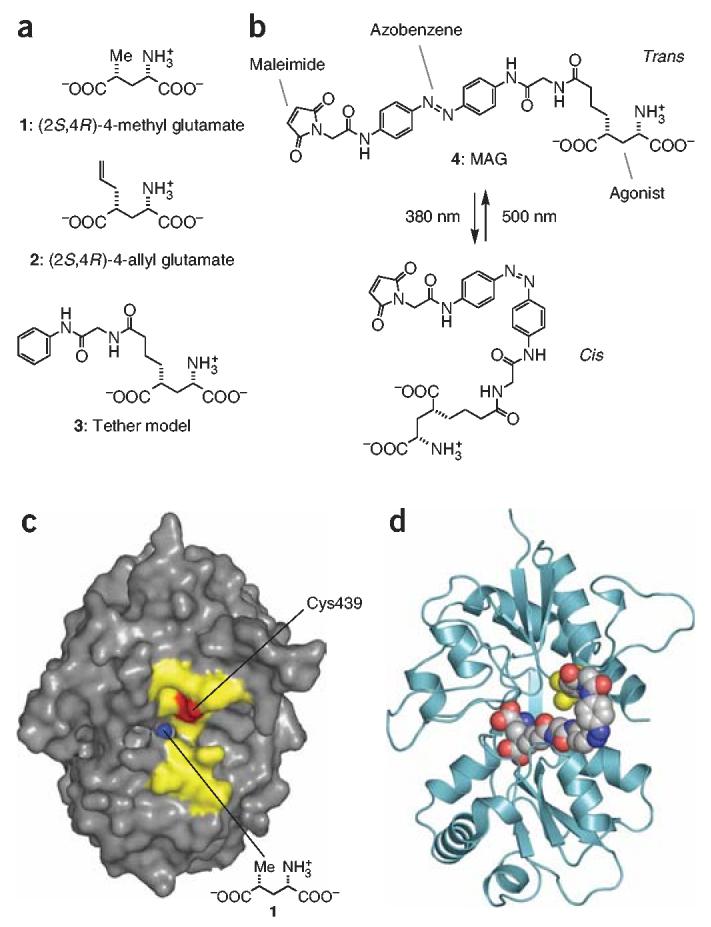
Structures and fit of photoswitched agonist and iGluR6 LBD. (a) Chemical structure of the iGluR6 agonists (2S,4R)-4-methyl glutamate 1; (2S,4R)-4-allyl glutamate 2; and tether model 3. (b) Structure of MAG 4 in its trans state (dark and 500 nm) and cis state (380 nm). (c) View looking into the ‘mouth’ of iGluR6 LBD in complex with 1 (ref. 12). Residues on clamshell ‘lips’ that were individually mutated to cysteine are highlighted in yellow. Position 439 is shown in red. The methyl group of 1 can be seen in blue at the bottom of the ‘exit channel’. (d) Docking model of MAG in the cis state attached at L439C (yellow) and bound to the activated (closed) conformation of the LBD.
To explore the feasibility of this idea, we first synthesized a ‘tether model’ (3; Fig. 2a). This compound is in essence an alkylated version of glutamate and resembles the known iGluR6 agonist (2S,4R)-4-allyl glutamate (2; Fig. 2a)15. We have extended the allyl side chain of this compound to include a moiety that mimics half of an azobenzene. This partial tether should, in principle, be long enough to project out of the exit channel and thus serve to determine if a full-length azobenzene tether would impede LBD activation.
To assay LBD activation, we took advantage of the calcium permeability of iGluR6 (ref. 16). We expressed iGluR6 in HEK293 cells, loaded the cells with the fluorescent calcium indicator FURA-2-AM17 and exposed them to various concentrations of agonist 2 or the tether model 3 to quantify receptor activation (Fig. 3). Tether model 3 evoked large responses (Fig. 3b). Allyl glutamate 2 had an EC50 of 18 μM, whereas model 3 showed an EC50 of 180 μM (Fig. 3d). The maximal response of tether model 3 was similar to that evoked by saturating glutamate but was ∼30% lower than that of agonist 2, indicating that the side chain may interfere with clamshell closure to a minor degree (Fig. 3d). These results suggest that tethering a glutamate analogue is possible while maintaining effective agonism. The loss in apparent affinity due to the side chain of model 3 should be compensated for by the high effective local concentration of a tethered ligand on its short leash.
Figure 3.

Calcium imaging of iGluR6 activity. (a) Superimposed bright-field (gray) and enhanced yellow fluorescent protein (EYFP; green) images of HEK293 cells transfected with both iGluR6 and EYFP. (b) Calcium image (350/380 nm) of FURA-2-AM–loaded cells during perfusion of 300 μM glutamate. Red and blue correspond to high and low Ca2+ concentrations, respectively. (c) Simultaneous Ca2+ concentration traces from individual cells in response to indicated concentrations of the tether model 3. (d) Dose-response curves from Ca2+ traces as in c. Higher concentrations of model 3 activate iGluR6 to levels similar to those resulting from saturating concentrations of glutamate (1 mM). Responses were normalized with respect to saturating concentrations of glutamate. Tether model 3 has an EC50 of 180 μM, compared with the higher-affinity molecule 2 on which it was based, which has an EC50 of 18 μM. (e) MAG confers light sensitivity to iGluR6-L439C–expressing cells, as seen by reversible increases in intracellular Ca2+ concentration at 380 nm and decreases at 500 nm. MAG does not confer light sensitivity to wild-type iGluR6-expressing cells (WT). Agonism by free MAG is transient and reverses upon washout for both iGluR6-L439C and wild-type iGluR6. IGluR6-L439C retains ability to be activated by free glutamate after MAG 4 conjugation. Note that Ca2+ concentration was not measured during irradiation at 380 or 500 nm or during conjugation.
Design and synthesis of a tethered agonist
After evaluation of the stereochemistry and synthetic accessibility of several candidates, we decided to focus on a tethered agonist that we call MAG (4; Fig. 2b). This compound features a cysteine-reactive maleimide (‘M’), an azobenzene photoswitch (‘A’), and a glutamate head group (‘G’). As the iGluR X-ray structure on which our design was based provides only a snapshot of a flexible protein, a certain amount of conformational flexibility was also built into MAG by adding freely rotatable bonds to the linker. The UV-visible spectra of the cis and trans isomers of soluble MAG are typical of azobenzenes (data not shown). MAG was prepared by multistep synthesis featuring a Grubbs olefin metathesis, several amide couplings and an intricate sequence of protective group manipulations (Scheme 1). The tether model 3 was prepared along similar lines (Supplementary Methods online).
Scheme 1.
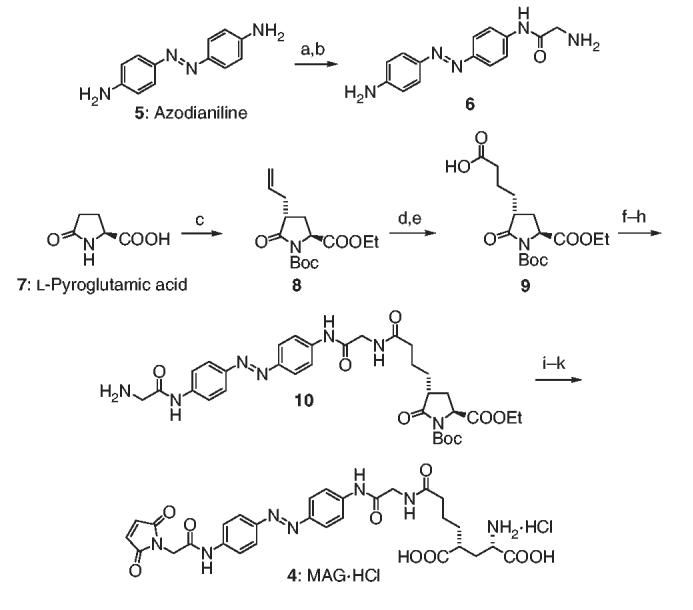
Total synthesis of MAG 4. Reagents and conditions: (a) Boc-Gly-OH, EDCI, HOBt, DIPEA (66%); (b) TFA, CH2Cl2 (98%); (c) see ref. 30; (d) acrylic acid, 5% Grubbs' second-generation catalyst (92%); (e) H2, Pd/C, MeOH (97%); (f) 6, EDCI, HOBt, DIPEA (97%); (g) Fmoc-Gly-OH, (COCl)2, DMF; (h) piperidine, DMF (43%, over two steps); (i) 1.0 M LiOH H2O/THF, 0 °C (80%); (j) N-methoxycarbonylmaleimide, NaHCO3, THF/H2O (71%) and (k) HCl-saturated EtOAc (87%). Boc, t-butoxycarbonyl; CH2Cl2, dichloromethane; (COCl)2, oxalyl chloride; DIPEA, diisopropylethylamine; DMF, N,N-dimethylformamide; EDCI, N-ethyl-N′-(3-dimethyldiaminopropyl)-carbodiimide HCl; EtOAc, ethyl acetate; Fmoc, 9-fluorenylmethoxycarbonyl; HOBt, 1-hydroxybenzotriazole hydrate; MeOH, methanol; Pd/C, palladium on carbon; TFA, trifluoroacetic acid; THF, tetrahydrofuran.
Cysteine screening
In parallel to our synthetic work, we prepared a series of single cysteine mutants of iGluR6 by site-directed mutagenesis. The positions were chosen to form a perimeter around the exit channel, close to where the maleimide end of the tether was predicted to stick out (Fig. 2c). Ca2+ imaging was used to search for cysteine mutants that would provide optical activation after covalent attachment of MAG. Although Ca2+ imaging has shortcomings for this application (slow kinetics and illumination at wavelengths that are absorbed by azobenzene, preventing continuous imaging that can follow the kinetics of the response), this assay enabled us to rapidly test 11 attachment positions and find three with clear responses to light, in which Ca2+ concentration increased at 380 nm and declined back to basal levels at 500 nm. The recordings were done in the presence of concanavalin A to prevent desensitization. Of the three, the version of the receptor with a cysteine at 439 (iGluR-L439C) had the largest responses (Fig. 3e). Because the rise in free cytoplasmic Ca2+ concentration depends not only on influx through iGluRs but also on Ca2+ buffering and pumping, we turned for further characterization to whole-cell patch clamping to measure the kinetics of channel gating directly and to obtain quantitative measures of activation efficiency.
Electrophysiological evaluation
Whole-cell patch clamping showed that iGluR-L439C conjugated with MAG (LiGluR) was activated both by free glutamate and by illumination (Fig. 4). The photostationary cis/trans ratio of azobenzenes depended on the wavelength, with maximum cis state occupancy typically observed at ∼380 nm and maximum trans state occupancy observed at ∼500 nm1,18. We therefore illuminated at 500 nm (to favor the inactive trans form) and tested illumination at wavelengths that ranged from 280 to 460 nm (to photoisomerize to the active cis form). The shortest test wavelengths evoked no response, the intermediate wavelengths evoked substantial inward currents and the longer wavelengths had smaller responses. The largest current was at 380 nm, agreeing with peak photoisomerization of free azobenzene to the cis form. To examine the opposite transition, we maximally activated the receptor with 380-nm illumination and tested wavelengths between 400 and 600 nm. Receptors were most efficiently turned off at 500 nm, agreeing with the peak photoisomerization of free azobenzene to the trans form. Thus, switching between 380 and 500 nm gave a maximal dynamic range for the optical control, and the degree of protein activation was precisely controlled at intermediate wavelengths.
Figure 4.
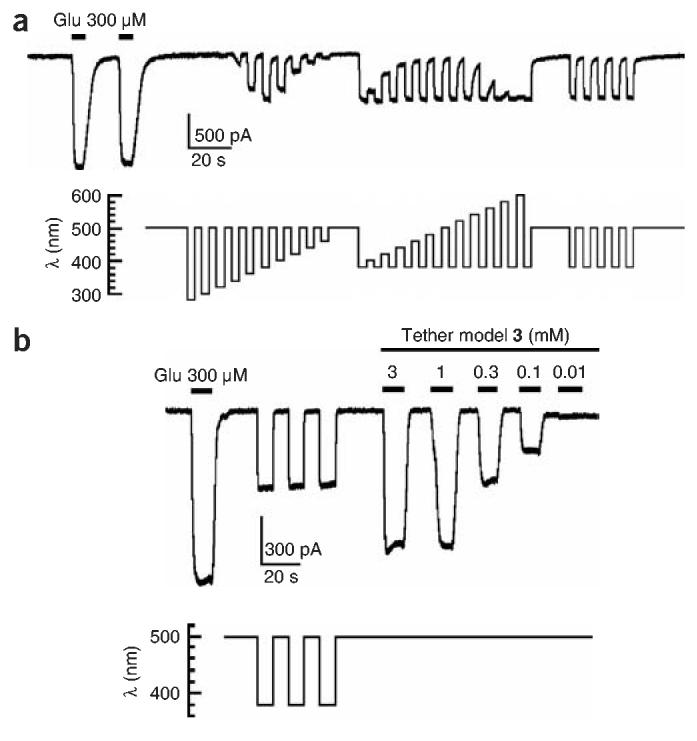
Whole-cell patch-clamp current recordings from HEK293 cells expressing iGluR6-L439C after conjugation of MAG. (a) Inward currents (downward deflections, carried mainly by Na+ influx) in response to glutamate are preserved in LiGluR. Irradiation with short wavelengths of light (280–480 nm, in 20-nm increments) results in maximal activation at 380 nm. Irradiation with long wavelengths of light (400–600 nm, in 20-nm increments) leads to maximal deactivation at 500 nm. Alternation between 380- and 500-nm illumination evokes highly reproducible responses. (b) Patch-clamp traces comparing the effects on LiGluR of a saturating concentration of glutamate, optical switching and the titration of the tether model 3. Saturating responses elicited by 3 are slightly higher than those elicited by 380-nm irradiation, suggesting geometric constraints that prevent the LBD from fully closing on MAG.
Notably, the photocurrents were fully reversible and highly reproducible. Repeated switching between 380 and 500 nm evoked responses of similar amplitude over a period of more than 30 min, consistent with the resistance of azobenzenes to bleaching and demonstrating that the system is robust. Even with weak illumination from a standard fluorescent lamp, attenuated by passage through a monochromator and fiber guide, the receptor turned on and off rapidly (τon-380 nm = 115 ± 3 ms and τoff-500 nm = 92.3 ± 0.3 ms; mean ± s.e.m., n = 3) at a power of 12.4 W m−2 (irradiance at 500 nm).
LiGluR was turned on and off with light but was also activated by freely diffusible glutamate (Fig. 4). The currents generated by irradiation were smaller than currents evoked by saturating concentrations of glutamate (300 μM) and by saturating concentrations of tether model 3 (≥1 mM). This could be due to incomplete labeling, but we consider that unlikely, because increased exposure (in either concentration or time) to MAG during the conjugation period did not change the size of the optical response. Alternatively, MAG may permit only partial closure of the LBD. Incomplete closure of the ligand-binding domain has been previously linked to partial agonism in the related iGluR2 channel19.
The efficient activation of iGluR6-L439C by MAG can be explained by a model that shows cis-MAG docked into the glutamate-binding site of the closed (activated) conformation of the LBD (Fig. 2d). The linker between the glutamate head group and the azobenzene moiety can comfortably protrude through the exit channel, with the azobenzene almost completely exposed to solvent.
DISCUSSION
In the dark, azobenzenes thermally isomerize to the lower-energy trans state18. To obtain optical control of protein function but avoid the need for constant illumination, it is therefore preferable for the optically switched channel to be turned off in the trans state, as we have shown here for LiGluR. The structure of the LBD clamshells and the geometry of azobenzene photoisomers are perfectly suited for this arrangement. The LBDs have deep and narrow cavities without a straight pathway from the sites of covalent attachment to the binding site. As a result, when the trans form of the azobenzene extends the tether, it cannot point into the binding pocket, even if the flexibility of the linkers is taken into account (Figs. 1 and 2). Photoisomerization to the cis form bends the tether and permits the agonist to turn into the mouth of the clamshell, reach the binding site and turn the protein on.
In principle, the approach we have used here should also work for tethered antagonists. In such a case it may be preferable to obtain ligand binding with a trans-azobenzene so that the engineered receptor can function only during timed bouts of illumination. This reverse arrangement should be possible for LBDs that have a tethering site that has an unobstructed, straight pathway to the binding pocket.
As clamshell-type LBDs are widespread in nature, our system could be extended to other proteins homologous to periplasmic binding proteins, such as the lac repressor20, the metabotropic glutamate receptors (mGluRs)21, NMDA receptors22, GABAB receptors, bacterial quorum sensors23 and pheromone receptors8. Detailed X-ray structures and extensive structure-activity studies of agonists and antagonists also exist for other classes of LBDs. In each of these cases, the appeal of the approach is not only that the proteins can be turned on and off under remote control but also that the color of the illumination can be chosen to set the degree of activation.
The development of light-activated ion channels could have a major impact on neurobiology, and the reengineering of nature's molecular machines could also provide valuable devices for biosensing24,25, bioelectronics26 and nanotechnology27. Our synthetic azobenzene-regulated K+-channel (SPARK) functions, in essence, as a nanoscale photo- and field-effect transistor3. LiGluR, on the other hand, can be activated by light and soluble agonists. As the SPARK and LiGluR channels are each regulated by two different input signals, they literally function as logic gates (AND and OR gates, respectively). Future investigations will show whether these nanoscale devices can be interfaced with macroscopic systems.
METHODS
Synthetic protocols
See Supplementary Methods online.
Site directed mutagenesis
Cysteine point mutations were introduced to the iGluR6 DNA, containing glutamine at the position 621 RNA editing site16 using the QuikChange site-directed mutagenesis kit (Stratagene). The following PCR profile was used: one cycle (95 °C for 30 s); 20 cycles (95 °C for 30 s, 55 °C for 1 min, 68 °C for 12 min). The forward and reverse oligonucleotide sequences designed for the L439C mutant were 5′-GATTGTTACCACCATTTGCGAAGAACCGTATGTTCTG-3′ and 5′-CAGAACATACGGTTCTTCGCAAAATGGTGGTAACAATC -3′, respectively.
Cell culture and transfection
HEK293 cells were plated at approximately 3 × 106 cells ml−1 on poly-l-lysine–coated glass coverslips (Deutsche Spiegelglas, Carolina Biological) and were maintained in DMEM with 5% fetal bovine serum, 0.2 mg ml−1 streptomycin and 200 U ml−1 penicillin at 37 °C. Cells were transiently transfected with various plasmids using Lipofectamine 2000 (Invitrogen). The amount of total transfected iGluR6 DNA and enhanced yellow fluorescent protein (EYFP) DNA per well was fixed at 4 μg and 200 ng, respectively. All recordings were carried out 36 to 48 h after transfection.
Attachment of MAG
To conjugate MAG to cysteine mutants of iGluR6, the compound was diluted to 10–100 μM (final concentration 0.5–5% DMSO) in the HEK cell control solution, and the cells were incubated in the dark for 15–30 min.
Calcium imaging
Cells were washed in PBS and loaded with 5 μM FURA-2-AM (Molecular Probes) for 30 min. Changes of [Ca2+]i in individual cells were measured as intracellular Fura2 fluorescence intensity using mercury arc lamp illumination. We alternated excitation with band-pass filters of 350 nm and 380 nm for 66 ms at 5–20 s intervals and detected emission at 545 nm17. Fluorescence was monitored on an inverted microscope system (Nikon). Images were captured with a charge-coupled device (CCD) camera using the Imaging Workbench software (INDEC Biosystems), which was also used to irradiate the cells at 380 and 500 nm for 1–2 min in order to produce photoisomerization of MAG. Measurements were performed in a control solution (in mM): 135 NaCl, 5.4 KCl, 0.9 MgCl2, 1.8 CaCl2, 10 HEPES and 10 glucose, pH 7.6, containing 300 mg l−1 concanavalin A type IV (Sigma) to block desensitization28,29.l-Glutamate was applied as reported in text and figures. The results are representative data from multiple cells in at least two independent cultures.
Whole-cell patch clamping
Patch clamp recordings were carried out using an Axopatch 200A amplifier in the whole-cell mode. Cell voltage was held at −60 mV. Pipettes had resistances of 4–8 MΩ and were filled with a solution containing (in mM) 145 CsCl, 5 EGTA, 0.5 CaCl2, 1.0 MgCl2 and 10 HEPES, pH 7.2. Illumination was applied using a TILL Photonics Polychrome II monochromator through a 60 ×/1.2-W objective (power output: 12.4 W m−2; irradiance: 500 nm, as measured with a Newport optical power meter). Data was recorded with pClamp software (Axon Instruments), which was also used to control the monochromator.
Accession codes
Protein Data Bank: iGlu R6-methyl glutamate, 1SD3.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Partin for the iGluR6 cDNA and for advice, T. Machen for guidance on calcium imaging and S. Szobota for participation in initial imaging experiments. P.G. was supported by postdoctoral fellowships from the Generalitat de Catalunya (Nanotechnology Program), Ministerio de Educación y Ciencia (Spain) and the Human Frontier Science Program. R.N. was supported by a postdoctoral fellowship from the Japan Society for the Promotion of Science. This work was supported by a Laboratory Directed Research Development Award from the Lawrence Berkeley National Laboratory and by a grant from the Human Frontier Science Program. D.T. thanks Eli Lilly, Astra Zeneca, Glaxo Smith Kline, Amgen and Merck & Co. for Young Investigator Awards.
Footnotes
Note: Supplementary information is available on the Nature Chemical Biology website.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Feringa BL, editor. Molecular Switches. Wiley-VCH, Weinheim; Germany: 2001. [Google Scholar]
- 2.Goeldner M, Givens R. Dynamic Studies in Biology. Wiley-VCH, Weinheim; Germany: 2005. [Google Scholar]
- 3.Lester HA, Krouse ME, Nass MM, Wassermann NH, Erlanger BF. Covalently bound photoisomerizable agonist - Comparison with reversibly bound agonists at electrophorus electroplaques. J. Gen. Physiol. 1980;75:207–232. doi: 10.1085/jgp.75.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kocer A, Walko M, Meijberg W, Feringa BL. A light-actuated nanovalve derived from a channel protein. Science. 2005;309:755–758. doi: 10.1126/science.1114760. [DOI] [PubMed] [Google Scholar]
- 5.Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Light-activated ion channels for remote control of neuronal firing. Nat. Neurosci. 2004;7:1381–1386. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paas Y. The macro- and microarchitectures of the ligand-binding domain of glutamate receptors. Trends Neurosci. 1998;21:117–125. doi: 10.1016/s0166-2236(97)01184-3. [DOI] [PubMed] [Google Scholar]
- 7.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 8.Pin JP, Galvez T, Prezeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 2003;98:325–354. doi: 10.1016/s0163-7258(03)00038-x. [DOI] [PubMed] [Google Scholar]
- 9.Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. 4 McGraw-Hill; New York: 2000. [Google Scholar]
- 10.Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Glutamate receptor gating. Crit. Rev. Neurobiol. 2004;16:187. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-Sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 12.Mayer ML. Crystal structures of the GluR5 and GluR6 ligand binding cores: Molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539–552. doi: 10.1016/j.neuron.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 13.Nanao MH, Green T, Stern-Bach Y, Heinemann SF, Choe S. Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc. Natl. Acad. Sci. USA. 2005;102:1708–1713. doi: 10.1073/pnas.0409573102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johansen TN, Greenwood JR, Frydenvang K, Madsen U, Krogsgaard-Larsen P. Stereostructure-activity studies on agonists, at the AMPA and kainate subtypes of ionotropic glutamate receptors. Chirality. 2003;15:167–179. doi: 10.1002/chir.10177. [DOI] [PubMed] [Google Scholar]
- 15.Pedregal C, et al. 4-alkyl- and 4-cinnamylglutamic acid analogues are potent GluR5 kainate receptor agonists. J. Med. Chem. 2000;43:1958–1968. doi: 10.1021/jm9911682. [DOI] [PubMed] [Google Scholar]
- 16.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2+ permeability in both TM1 and TM2 of high-affinity kainate receptor channels - diversity by RNA editing. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 17.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca-2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Knoll H. In: CRC Handbook of Organic Photochemistry and Photobiology. Horspool W, Lenci F, editors. Vol. 89. CRC, Boca Raton; Florida: 2004. pp. 1–89. [Google Scholar]
- 19.Jin RS, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- 20.Kercher MA, Lu P, Lewis M. Lac repressor operator complex. Curr. Opin. Struct. Biol. 1997;7:76–85. doi: 10.1016/s0959-440x(97)80010-3. [DOI] [PubMed] [Google Scholar]
- 21.Kunishima N, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, et al. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 24.Bayley H, Jayasinghe L. Functional engineered channels and pores. Mol. Membr. Biol. 2004;21:209–220. doi: 10.1080/09687680410001716853. [DOI] [PubMed] [Google Scholar]
- 25.Dwyer MA, Hellinga HW. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Willner I, Willner B. Molecular and biomolecular optoelectronics. Pure Appl. Chem. 2001;73:535–542. [Google Scholar]
- 27.Balzani ACV, Venturi M. Molecular Devices and Machines: A Journey Into the Nanoworld. Wiley-VCH, Weinheim; Germany: 2003. [Google Scholar]
- 28.Wilding TJ, Huettner JE. Activation and desensitization of hippocampal kainate receptors. J. Neurosci. 1997;17:2713–2721. doi: 10.1523/JNEUROSCI.17-08-02713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin-A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 30.Ezquerra J, et al. Stereoselective reactions of lithium enolates derived from N-Boc protected pyroglutamic esters. Tetrahedron. 1993;49:8665–8678. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.