

Abstract
The nuclear zinc-finger protein encoded by the hindsight (hnt) locus regulates several cellular processes in Drosophila epithelia, including the Jun N-terminal kinase (JNK) signaling pathway and actin polymerization. Defects in these molecular pathways may underlie the abnormal cellular interactions, loss of epithelial integrity, and apoptosis that occurs in hnt mutants, in turn causing failure of morphogenetic processes such as germ band retraction and dorsal closure in the embryo. To define the genetic pathways regulated by hnt, 124 deficiencies on the second and third chromosomes and 14 duplications on the second chromosome were assayed for dose-sensitive modification of a temperature-sensitive rough eye phenotype caused by the viable allele, hntpeb; 29 interacting regions were identified. Subsequently, 438 P-element-induced lethal mutations mapping to these regions and 12 candidate genes were tested for genetic interaction, leading to identification of 63 dominant modifier loci. A subset of the identified mutants also dominantly modify hnt308-induced embryonic lethality and thus represent general rather than tissue-specific interactors. General interactors include loci encoding transcription factors, actin-binding proteins, signal transduction proteins, and components of the extracellular matrix. Expression of several interactors was assessed in hnt mutant tissue. Five genes—apontic (apt), Delta (Dl), decapentaplegic (dpp), karst (kst), and puckered (puc)—are regulated tissue autonomously and, thus, may be direct transcriptional targets of HNT. Three of these genes—apt, Dl, and dpp—are also regulated nonautonomously in adjacent non-HNT-expressing tissues. The expression of several additional interactors—viking (vkg), Cg25, and laminin-α (LanA)—is affected only in a nonautonomous manner.
DURING development, tissues and organs are formed through dynamic cell shape changes and movements that are orchestrated in time and space (reviewed by Gumbiner 1996; Geiger et al. 2001). Data gathered from both vertebrate and invertebrate systems have implicated several cell surface, cytoskeletal, and extracellular matrix (ECM) molecules in the establishment and maintenance of cell architecture, cell movement, and tissue integrity during morphogenesis (reviewed by Gumbiner 1996; Lauffenburger and Horwitz 1996; Wilk et al. 2004). However, to date, there has been little analysis of genetic regulatory hierarchies that control the expression and function of these molecules in specific tissues.
Previous analyses have shown that the Drosophila hindsight (hnt) gene encodes a nuclear zinc-finger protein found in several epithelia during development (Yip et al. 1997). These include the extraembryonic amnioserosa, the midgut and tracheae of the embryo, and the photoreceptor cells of the developing adult retina (Yip et al. 1997; Lamka and Lipshitz 1999; Wilk et al. 2000; Reed et al. 2001; Pickup et al. 2002). HNT expression in these epithelia regulates several local and global morphogenetic processes. For example, the expression of HNT in the amnioserosa is required for embryonic germ band retraction (Yip et al. 1997; Lamka and Lipshitz 1999). HNT also plays an important role in embryonic dorsal closure by downregulating JNK signaling in the amnioserosa, thus enabling assembly of the F actin-based purse string in the adjacent, leading edge epidermal cells (Reed et al. 2001). During tracheal development, tertiary branching fails (Wilk et al. 2000) and, during eye morphogenesis, the shape of individual photoreceptor cells is often abnormal (Pickup et al. 2002) in hnt mutant tissue. During eye development, HNT function is required for the accumulation of F actin in the apical tip of photoreceptor precursor cells in the ommatidial clusters, as well as in the developing rhabdomere during the pupal period (Pickup et al. 2002).
HNT has also been shown to be essential for maintenance of epithelial tissue integrity. While hnt mutant tracheae undergo normal specification, invagination, and primary and secondary branching, at later embryonic stages the epithelial tubes lose their integrity (Wilk et al. 2000). Similarly, in hnt mutant embryos the amnioserosa falls apart prematurely leading to defects in germ band retraction and dorsal closure (B. H. Reed and H. D. Lipshitz, unpublished observations; see Reed et al. 2004). In hnt mutant eye tissue, the developing retinal epithelium breaks down at the midpupal stages (Pickup et al. 2002).
Thus, hnt has all of the hallmarks of a regulatory gene, which functions in specific epithelia to control processes that are required for morphogenesis. However, direct transcriptional targets of HNT as well as genetic pathways that are regulated by HNT remain largely undefined. Here we carry out a series of genetic modifier screens aimed at identifying loci that genetically interact with hnt. Two different hnt hypomorphic alleles—one a viable eye-specific allele (hntpeb; Pickup et al. 2002), the other a leaky embryonic lethal allele (hnt308; Reed et al. 2001)—were used to produce sensitized genetic backgrounds in which we could identify dominant modifier loci. Over 60 interactors were identified, including genes encoding transcription factors and cytoskeletal, signal transduction, and ECM components. Expression of a subset of the interactors was assayed in hnt mutant tissue. These analyses showed that several genes (dpp, puc, kst, apt, and Dl) are regulated tissue autonomously by HNT in embryo and/or eye tissue. Expression of three of these (dpp, apt, and Dl) as well as expression of several additional genes (vkg, Cg25C, and LanA) is also affected nonautonomously in hnt mutants.
MATERIALS AND METHODS
Drosophila mutants and lines:
Most deficiencies, duplications, mutations, P-element lethal lines, and enhancer trap lines were obtained from the Bloomington Drosophila Stock Center and are described in FlyBase (http://flybase.bio.indiana.edu/). In(2LR)lt616-LBR29 is a duplication from 60C to 60E (Reed 1992); Df(1)rb1 has been previously described (Wilk et al. 2000). hnt mutants included hntXE81 (described in Yip et al. 1997), hnt1142 (described in Wilk et al. 2000), hnt308 (described in Reed et al. 2001), and hntpeb (described in Yip et al. 1997; Pickup et al. 2002). LanA3A1, LanA4A8, vkg177, and Cgc25C234 (from N. McGinnis, University of California, San Diego) are described in Gellon et al. (1997). To visualize the embryonic tracheal system, the trachealess enhancer trap 1-eve-1 was utilized (described in Wilk et al. 1996).
Screen for chromosomal regions that dominantly interact with hntpeb:
hntpeb virgin females were crossed to males bearing either a deficiency or a duplication in trans to a dominantly marked balancer chromosome. Crosses were maintained at 29°, the restrictive temperature at which hntpeb shows a rough eye phenotype (Pickup et al. 2002). A total of 124 deficiency lines (Df) from the “deficiency kit” for the second and third chromosome were tested, along with 14 duplications (Dp) covering most of the second chromosome. hntpeb/Y; Balancer/+ progeny were compared to the hntpeb/Y; Df or Dp/+ sibs (these sibs were identified by their lack of the dominantly marked balancer). We evaluated and compared the roughness of the eyes between these two groups (∼10 pairs of flies). Any consistent difference (enhancement or suppression of the hntpeb rough eye phenotype) was noted and interactors were retested for confirmation.
Screen for loci that dominantly interact with hntpeb:
Crosses similar to those described above were also used to identify individual loci that exhibit dominant interactions with hntpeb. The P-element lethal lines tested mapped to the regions identified by the first screen and included lines with elements mapping close to, but outside of, the rearrangement breakpoints (this was done to take into account the uncertainties in the cytological breakpoints; Figure 3). In addition to P-element lines, a dozen other mutations were tested (see Table 2). Any interaction was confirmed by performing at least two independent crosses. Where possible, additional alleles of the same gene were tested for modification of the rough eye phenotype (see Table 2).
Figure 3.—

Schematic of P-element lines that genetically interact with hntpeb. (A) Chromosome 2. (B) Chromosome 3. All the P-element lethal stocks that mapped to any of the interacting regions defined by deficiencies or duplications were tested for modification of the hntpeb rough eye phenotype. A vertical line marks each fly line that was tested. A grayscale vertical line underneath represents any modification to the hntpeb rough eye phenotype. The grayscale code and the schematic representation of deficiencies and duplications are the same as in Figure 2. The name of the gene mutated by an interacting P element is shown. Larger font represents genes where at least two alleles genetically interact with hntpeb. Details are given in Table 2.
TABLE 2.
Loci that genetically interact withhntpeb
| Locus (cytology) | Allele | Genetic interaction (expected direction) |
Molecular identity of gene product |
|---|---|---|---|
| Components of the cytoskeleton | |||
| chickadee (chic) (26A9–B1) | 11 | Su (+) | Profilin; actin polymerization/depolymerization |
| 01320 | Su (+) | ||
| 221 | Su (+) | ||
| k13321 | E (−) | ||
| cactusa(35F9–11) | 4 | Su (−) | Transcription factor; cytoplasmic sequestration of Dorsal |
| 1 | No interaction | ||
| Dynamitin (Dmn) (44F6–8) | k16109 | Su (+) | Dynactin motor; microtubule-based movement |
| RhoAa(52E4) | J3.8 | Su (NR) | Rho small monomeric GTPase |
| E3.10 | No interaction | ||
| karst (kst) (63C5–D1) | 01318 | Su (+) | βHeavy-spectrin; actin binding, microtubule binding |
| rolling pebbles (rols) (68F1) | 08232 | E (−) | Component of the cytoplasm; involved in myoblast fusion |
| Extracellular matrix component | |||
| viking (vkg) (25C1) | 01209 | Su (+) | Type IV collagen α2 chain |
| k00236 | Su (+) | ||
| k07138 | Su (+) | ||
| k16721 | Su (+) | ||
| k16502 | Su (+) | ||
| 177-27 | Su (+) | ||
| Cg25C (25C1–2) | k00405 | Su (+) | Type IV collagen α1 chain |
| 234-9 | Su (+) | ||
| Components of signal transduction pathways EGFR signaling pathway | |||
| echinoid (ed) (24D2–4) | k01102 | Su (+) | Contains immunoglobulin domains |
| MESK2 (57E6–9) | k0019 | Su (−) | Suppressor of KSR2; alpha/beta-hydrolase domains |
| EgfRa(57E9–F1) | f1 | E (+) | Epidermal growth factor receptor; protein tyrosine kinase |
| TGFβ/Dpp signaling pathway | |||
| thickveins (tkv) (25C9–D1) | k16713 | Su (+) | Protein kinase; involved in dorsal closure and tracheal system development |
| 09415 | No interaction | ||
| baboon (babo) (44F12–45A1) | k16912 | Su (+) | Type I TGFβ receptor; serine/threonine kinase |
| 32 | No interaction | ||
| JNK signaling pathway | |||
| anterior opena (aop) (22D1) | 1 | Su (NR) | RNA polymerase II transcription factor; transcriptional repressor |
|
Jun related antigen (Jra)a (46E4–5) |
1 | Su (+) | Transcription factor bZIP; Jun related |
| puckered (puc) (84E10–13) | A251.1f3 | Su (−) | Protein tyrosine phosphatase; Jun kinase (JNK) phosphatase |
| Notch signaling pathway | |||
| l(2)44DEa (44D3–6) | k10313 | Su (+) | Acetate-CoA ligase; interacts with l(1)Sc and N |
| 05847 | Su (+) | ||
| Delta (Dl) (92A1–2) | 05151 | E (+) | Notch receptor ligand |
| X | E (+) | ||
| 9P | E (+) | ||
| Other signaling pathways | |||
| plexus (px) (58E3–8) | k08316 | Su (−) | Localized to the nucleoplasm; interacts genetically with Delta, rho, and EGFR |
| k08134 | Su (−) | ||
|
Atypical protein kinase C (aPKC) (51D7–8) |
k06403 | Su (−) | Atypical protein kinase C; mutants affect epithelial apical-basal polarity; associates with Bazooka; expressed apically in tracheae and other epithelia, not in amnioserosa |
| Nucleic acid binding | |||
| vrille (vri) (25D4–5) | k05901 | Su (+) | Transcription factor; bZIP; expressed in amnioserosa, tracheae, eye, and other tissues |
| eIF-4a (26B1–2) | 02439 | Su (+) | RNA helicase; translation initiation factor; expressed ubiquitously in embryos |
| k14518 | Su (+) | ||
| k01501 | No interaction | ||
| dachshund (dac) (36A2) | P | Su (−) | Transcription factor; expressed in CNS and eye disc |
| domino (dom) (57D4–8) | k08108 | Su (−) | Transcription factor; helicase; involved in cell proliferation; expressed in hemocytes and other tissues |
|
defective proventriculus (dve) (58D1–2) |
01738 | Su (−) | Transcription factor; homeodomain |
| k06515 | Su (−) | ||
| apontic (apt) (59F1–2) | k15608 | Su (−) | Transcription factor; expressed in amnioserosa, tracheal system, and other tissues; mutations affect the larval tracheal system and the embryonic heart |
| 09049 | Su (−) | ||
| 03041 | Su (−) | ||
| 06369 | Su (−) | ||
| retained (retn) (59F2–3) | 02535 | Su (−) | DNA-binding protein; expressed in the amnioserosa and the brain |
| slow border cells (slbo) | ry7 | S (+) | Transcription factor; bZIP; required for border cell migration; expressed in border follicle cells, embryonic foregut, midgut, and epidermis |
| ry8 | Su (+) | ||
| 01310 | Su (+) | ||
| reptin (rept) (76A3–4) | 06945 | Su (+) | DNA binding; helicase |
| osa (90C1–2) | 00090 | E (+) | DNA binding; expressed in the eye disc morphogenetic furrow |
| Avr1 | E (+) | ||
| glassa(91A3) | 1 | E (+) | C2H2 zinc-finger transcription factor; eye photoreceptor development |
| 2 | E (+) | ||
| 3 | No interaction | ||
| Localized to cell membranes | |||
| turtle (tutl) (24E1–4) | k14703 | Su (+) | Contains immunoglobulin domains; flight behavior |
| 01081 | No interaction | ||
| lamin (lam) (25E6–F1) | k11511 | Su (+) | Nuclear membrane protein; involved in nuclear envelope reassembly; mutations affect cytoplasmic extensions for terminal cells of the tracheal system |
| 04643 | No interaction | ||
| heixuedian (heix) (35F7–8) | k11403 | Su (−) | Plasma membrane component; integral membrane protein |
| 1 | Su (−) | ||
| Rya-r44F (44F3–8) | k04913 | Su (+) | Ryanodine receptor; caffeine-sensitive calcium release channel; localized to the ER membrane |
| Miscellaneous | |||
| Pdsw (23F3) | k10101 | Su (+) | NADH dehydrogenase |
| Sec61α (26D7–8) | k04917 | Su (+) | Protein transporter |
| Coprox (27C6–8) | k10617 | Su (−) | Coproporphyrinogen oxidase |
| Cyclin E (CycE) (35D4) | 05206 | Su (−) | G1/S specific cyclin |
| k05007 | No interaction | ||
| Sec61β (51B6) | k03307 | Su (−) | Protein transporter; component of the translocon |
| 07214 | Su (−) | ||
|
Proteasome p44.5 subunit (Rpn6) (51C1) |
k00103 | Su (−) | Involved in proteolysis; component of the proteasome regulatory particle |
| bellwether (blw) (59B2) | k00212 | Su (−) | Hydrogen transporting ATP synthase |
| 03972 | Su (−) | ||
| 1 | No interaction | ||
| Thiolase (60A5–7) | k09828 | Su (−) | Acetyl CoA acyltransferase |
| 00628 | No interaction | ||
| non-stop (not) (75D4) | 02069 | Su (NR) | Ubiquitin protease; axonal target recognition |
| eRF1 (77B4–5) | neo28 | Su (+) | Translation release factor involved in termination of protein synthesis |
| Unknown or novel | |||
| l(2)k10001 (25B8–9) | k10001 | Su (+) | Unknown |
| l(2)k00605 (27A1–2) | k00605 | Su (−) | Unknown |
| l(2)k10113 (27F4–6) | k10113 | Su (−) | Unknown |
| l(2)k13905 (36A10–11) | k13905 | Su (−) | Unknown |
| l(2)s1878 (44D5–6) | s1878 | Su (+) | Unknown |
| l(2)00297 (47A13–14) | 00297 | Su (+) | Unknown |
| l(2)k15826 (47C3–4) | k15826 | Su (+) | Unknown; homology to a transtyretin-like protein (BLAST) |
| fs(2)neo12 (48C) | 1 | Su (+) | Unknown |
| unchained (49D1–50D1) | k15501 | Su (+) | Novel; mutations affect the chordotonal organ |
| charlatan (51E2) | 02064 | Su (−) | Novel; transcription factor domains; mutations affect the chordotonal organ and the PNS |
| k04218 | No interaction | ||
| l(2)03605 (57F8–10) | 03605 | Su (−) | Unknown |
| l(2)k13211 (58D6–7) | k13211 | Su (−) | Unknown |
| l(2)k06617 (58D6–7) | k06617 | Su (−) | Unknown |
| l(2)k00611 (58F4–5) | k00611 | Su (−) | Unknown; transcription factor domains |
| l(3)L3930 (75C5–6) | L3930 | Su (+) | Unknown |
| ms(3)neo94 (77B–C) | 1 | Su (+) | Unknown |
| l(3)10615 (85D16) | 10615 | E (+) | Unknown |
| l(3)neo51 (92A) | 1 | Su (−) | Unknown |
All loci are listed that modify the rough eye phenotype exhibited by hntpeb at 29°, including all the P-element lines and other types of mutations. The table is organized by “functional classes.” Su, suppressor; E, enhancer; NR, not relevant (the gene does not map to an interacting region). Plus indicates that the corresponding deficiency showed the same result; minus indicates that it did not show the same result.
Additional, candidate gene not within an interacting region defined in Table 1.
Most of the identified mutations mapped to the second chromosome and behaved as moderate dominant suppressors. One possible explanation for this bias is a difference in genetic background between P-element lines on the second compared to the third chromosome. For a subset of the second-chromosome loci, we therefore tested additional alleles induced on distinct genetic backgrounds for interactions with hntpeb (see Table 2). In 64% of the cases (14 of 22) more than one allele interacted with hntpeb. Moreover, hundreds of second-chromosome P-element mutations that had been induced on the same genetic background as those exhibiting moderate suppression did not exhibit any dominant genetic interaction with hntpeb. We therefore conclude that most of the interactions are real and that in each case the mutation in the identified modifier gene itself, and not the genetic background, is likely to be responsible for the observed interaction.
Confirmation of genetic interactions utilizing hnt308:
Virgin hnt308/FM7z females were crossed to balanced mutant males carrying a mutation in the gene to be tested. Embryos from these crosses were collected on grape juice agar plates, aligned in groups of 50 on fresh agar plates, aged for >24 hr at 25° and scored for embryonic lethality. In most cases, the percentage of embryonic lethality was compared to a control cross that was identical except for the absence of the mutation on the autosome (i.e., with the same balancers). Exceptions were chickadee (chic), puckered1 (puc1), and RhoA (see below). Embryonic lethality was calculated by counting dead (brown eggs with cuticle) and unfertilized eggs (white and undeveloped) and hatched embryos (empty cuticle case). The embryonic lethality was (brown embryos/n), where n was the total number of aligned embryos minus the unfertilized eggs. Embryonic lethality for each mutant was normalized to the lethality observed in control crosses. Most lines were crossed to hnt308/FM7 female virgins. chic221 and chic01320 were crossed to hnt308/FM6 female virgins. The chic control was hnt308/FM6 virgin females crossed to w1118/Y males. The control cross for puc1 and RhoA used hnt308/FM7 female virgins crossed to Oregon-R males (as described in Reed et al. 2001). Embryonic lethality among specific controls was as follows: CyO, 0.125 ± 0.08; TM3, 0.161 ± 0.05; TM1, 0.122; TM6B, 0.146; and chic control, 0.08. The following embryonic lethalities, normalized to control values of 1.0, were calculated and used to create Figure 4: (viking) vkg01209/CyO, 0.575; vkg177/CyO, 0.95; vkgk16721/CyO, 0.16; vkgk00236/CyO, 0.17; vkgk07138/CyO, 0.17; vkgk16502/CyO, 0.13; Cg25Ck00405/CyO, 0.93; Cg25C234-9/CyO, 0.067; (Laminin A) LanA3A1/TM1 Me, 0.088; LanA4A8/TM1 Me, 0.088; (karst) kst01318/TM3 Sb, 0.22; kst1/TM6B, 0.17; kst2/TM6B, 0.15; (turtle) tutlk14703/CyO, 0.086; (thickveins) tkvk16713/CyO, 0.09; (heixuedian) heixk11403/CyO, 0.1; (Delta) Dl05151/TM3 Sb, 0.067; (slow border cells) slbory8/CyO, 0.11; pucA251.1f3/TM3 Sb, 0.148; Df(3L)kto2/TM6B,Tb1, 0.038; chic221/CyO, 0.11; and chic01320/CyO, 0.104.
Figure 4.—

Genetic interactions with hnt308. Fly lines that were crossed to hnt308 virgin females are shown, organized by their biological function. Embryonic lethality was scored for the progeny of all crosses. The data are normalized to the appropriate control, and controls are all normalized to one. The light gray horizontal bar represents the normalized control plus or minus the normalized error bars. The number of embryos scored (N) and the P-values for the χ2 test are shown. Statistically significant interactions are shaded as in Figures 2 and 3. NS, no significant interaction; (*), P = 0.05–0.1; *, P = 0.025–0.05; **, P = 0.01–0.025; ***, P ≪ 0.005.
The statistical significance of the results was determined using the χ2 test (Dixon and Massey 1957). To calculate the χ2, we used the results from the balancer control crosses to generate the expected frequencies and the results from the testcrosses with candidate mutations to generate the observed frequencies. We considered a P-value of <0.05 to be significant. If the percentage of embryonic lethality increased or decreased significantly when the mutation was present, the mutation is listed as a dominant enhancer or a suppressor of hnt308 embryonic lethality, respectively.
Test for molecular regulation by HNT:
Embryos from the following candidate enhancer trap lines were stained with anti-β-galactosidase antibody: Dl05151/TM3, chicK13321/Cyo, chic35A/Cyo, chic13E/Cyo, chicRM1/Cyo, chic11/Cyo, kst01318/TM3, vriK05901/CyO, pucA251.1F3/TM3, dpp10638/CyO, aptK15608/CyO, apt03041/CyO, and RhoAK02107b/CyO. If expression was detected in either the tracheal system or the amnioserosa, expression was assayed in hnt mutants as follows. Virgin hntXE81/FM7z females were crossed to males from the following enhancer trap lines: Dl05151/TM3, pucA251.1F3/TM3, dpp10638/CyO, tkvK16713/CyO, apt03041/CyO, and Rho1K02107b/CyO. Overnight embryo collections from these crosses were immunostained for β-galactosidase to determine if there was any difference in staining between hntXE81 mutant embryos with the enhancer trap and their wild-type sibs that only carried the enhancer trap (the ftz-lacZ marker on the FM7z balancer chromosome distinguished them from the hnt embryos).
Standard protocols were used to generate FLP-induced hnt clones in the eye disc (Xu and Rubin 1993). The FRT line w1118 P{w+mC=piM}5A P{w+mC=piM}10D P{ry+t7.2=neoFRT}18A and the FLP recombinase stock w1118; MKRS, P{ry+t7.2=hsFLP}86E/TM6B Tb1 were obtained from the Bloomington Drosophila Stock Center. Eye discs were dissected from third instar larvae of the genotype 182piM FRT/hntXE81FRT:Dl05151/FLP, 182piM FRT/hntXE81FRT: kst01318/FLP, or 182piM FRT/hntEH704aFRT:FLP/+ and immunostained with α-HNT (to identify hnt patches) and either α-β-galactosidase (for Dl-lacZ and kst-lacZ) or α-Apontic antibody, respectively, to determine whether the hnt mutant area shows any difference in staining for the candidate gene product.
Immunostaining and microscopy:
Staining was carried out using standard procedures with the following antibodies: mouse monoclonal anti-Drosophila collagen type IV (from L. I. Fessler, University of California, Los Angeles; 1:70 dilution); rabbit anti-Drosophila laminin [from L. I. Fessler; used at 1:700 dilution as described in Fessler et al. (1987)]; rabbit anti-β-galactosidase (Cappel, Malvern, PA; 1:1000 dilution); chicken anti-β-galactosidase (ab-cam; 1:1000 dilution); guinea pig anti-tracheal lumen 55 [from B. Shilo; used at 1:150 dilution as described in Reichman-Fried et al. (1994)]; rabbit anti-Apontic/Tracheae defective [APT; from R. Schuh, Max Planck Institute; used at 1:30 dilution as in Eulenberg and Schuh (1997)]; mouse monoclonal anti-HNT, used at 1:20 dilution as described in Yip et al. (1997). Double staining for laminin and tracheal lumen as well as double staining for TDF and HNT was performed as previously described (Wilk et al. 2000). HRP-secondary antibodies were used for light microscopy (Jackson, West Grove, PA; 1:300 dilution); rhodamine and FITC-conjugated secondary antibodies were used for confocal analyses (Jackson; 1:300 dilution).
Light microscopy was carried out using a Zeiss Axioplan 2 imaging microscope. Images were captured with a Spot digital camera (Diagnostic Instruments) and Spot software or with a Zeiss AxioCam digital camera and AxioVision 3.1 software. Confocal analyses were conducted using a Zeiss inverted microscope with LSM 510 software. Images were processed with PhotoShop (Adobe) and Illustrator software (Adobe).
RESULTS
Identification of autosomal regions that exhibit dominant genetic interactions with hntpeb:
To identify chromosomal regions that genetically interact with hnt, we performed a genetic screen for dose-dependent modifiers of the temperature-sensitive rough eye phenotype exhibited by the viable allele, hntpeb (Figure 1; Yip et al. 1997; Pickup et al. 2002). We tested 58 deficiencies on the second chromosome and 64 on the third chromosome that, respectively, remove a total of ∼84% and ∼78% of the loci on these chromosomes (Figure 2). In addition we used 14 duplications that cover ∼84% of the second chromosome (Figure 2). hntpeb males carrying one copy of the deficiency or the duplication were compared to sibling hntpeb males carrying a balancer chromosome (for details, see materials and methods). Dominant genetic modifiers of hntpeb were identified on the basis of a consistent and reproducible alteration in eye roughness. Twenty-nine deficiencies or duplications consistently modified the hntpeb rough eye phenotype (∼21% of the lines tested; see example in Figure 1C): 17 were suppressors and 12 were enhancers, representing 19 different regions (8 on the second and 11 on the third chromosome; see Figures 2 and 3; Table 1).
Figure 1.—

Dose-dependent modification of the hntpeb rough eye phenotype is shown in scanning electron micrographs of adult eyes. (A) Eye from a wild-type male fly. (B) Eye from an hntpeb male fly raised at the restrictive temperature, showing a rough eye due to disorganization of facets (modified from Pickup et al. 2002). (C) Eye from an hntpeb/Y; Df(3L)kto2/+ fly raised at the restrictive temperature showing suppression of the hntpeb/Y eye phenotype. (D) Eye from hntpeb/Y; Dl9P/+ fly raised at the restrictive temperature showing enhancement of the hntpeb/Y eye phenotype.
Figure 2.—

Dose-dependent modification of hntpeb by autosomal deficiencies and duplications. The schematic represents all the regions in the fly chromosome that were found to genetically modify the mild rough eye phenotype exhibited by hntpeb at 29°. Each box above the chromosomes represents the region that is missing in a particular fly line (deficiency). The boxes below the chromosomes represent the region that is duplicated in a certain fly line (duplication). The results from the genetic interactions are grayscale coded: white, no interaction; dark gray, moderate suppressor; black, suppressor; and light gray, enhancer. A list of the interacting regions can be found in Table 1.
TABLE 1.
Regions of the second and third chromosomes that genetically interact withhntpeb
| Stock | Deficiency (Df) or duplication (Dp) |
Cytology |
|---|---|---|
| Suppressor | ||
| Dp(2;2)Cam6 (4518) | Dp | 35B;36C |
| Df(3L)kto2 (3617) | Df | 76B1–2;76D5 |
| Df(3L)XS533 (5126) | Df | 76B4;77B |
| Moderate suppressor | ||
| Df(2L)E110 (490) | Df | 25F3–26A1;26D3–11 |
| Df(2R)H3E1 (201) | Df | 44D1–4;44F12 |
| Df(2R)stan2 (596) | Df | 46F1–2;47D1–2 |
| Df(2R)vg135 (1642) | Df | 48C–48D;49D |
| Df(2R)vg-C (754) | Df | 49A4–13;49E7–F1 |
| Dp(2;2)Cam16 (2622) | Dp | 57C4–6;60E4 |
| In(2LR)lt616-L BR27-R | Dp | 60C;60E |
| Df(2R)Px2 (2604) | Df | 60C5–6;60D9–10 |
| Df(3L)HR119 (3649) | Df | 63C2;63F7 |
| Df(3L)vin2 (2547) | Df | 67F2–3;68D6 |
| Df(3L)vin5 (2611) | Df | 68A2–3;69A1–3 |
| Df(3L)W10 (2608) | Df | 75A6–7;75C1–2 |
| Df(3L)rdgC-co2 (2052) | Df | 77A1;77D1 |
| Df(3R)D605 (823) | Df | 97E3;98A5 |
| Enhancer | ||
| Df(3R)Dl-BX12 (3012) | Df | 91F1–2;92D3–6 |
| Moderate enhancer | ||
| Dp(2;2)Cam2 (3394) | Dp | 23D1–2;26C1–2 |
| Df(2L)Dwee-delta5 (3571) | Df | 27A;28A |
| Df(2L)r10 (1491) | Df | 35D;36A6–7 |
| Df(2R)knSA3 (1150) | Df | 51B5–11;51D7–E2 |
| Df(3L)GN24 (3686) | Df | 63F6–7;64C13–15 |
| Df(3L)ZN47 (3096) | Df | 64C;65C |
| Df(3L)Delta1AK (4370) | Df | 79E5–F1;79F2–6 |
| Df(3R)Antp17 (1842) | Df | 84B1–2;84D11–12 |
| Df(3R)p712 (1968) | Df | 84D4–6;85B6 |
| Df(3R)by10 (1931) | Df | 85D8–12;85E7–F1 |
| Df(3R)DG2 (4431) | Df | 89E1–F4;91B1–B2 |
Dose-dependent modifiers of the mild rough eye phenotype observed in hntpeb adult fly eyes. The stock name is followed by the Bloomington stock number in parentheses. Each line represents either a deficiency (Df) or a duplication (Dp) that enhances or suppresses the hntpeb rough eye phenotype. The region of the chromosome that is either duplicated or absent is listed in the cytology column.
Mutations in 63 autosomal loci dominantly interact with hntpeb:
To identify interacting genes in the autosomal regions defined by the deficiencies and duplications, 438 individual P-element lethal lines mapping to the 19 identified regions were tested for their ability to dominantly modify the hntpeb rough eye phenotype (for details, see materials and methods; Figure 3). Whenever possible, the interactions were confirmed with additional alleles of each putative modifier gene (Table 2; Figure 1D). In addition, we tested mutations in 12 candidate genes, including members of the JNK pathway (anterior open and jun-related antigen) and the small GTPase, RhoA (see Table 2).
In total, 470 crosses were performed and 89 interacting mutant lines were identified (Figure 3; Table 2): 77 dominantly suppress and 12 dominantly enhance the hntpeb rough eye phenotype. These represent 63 different loci: 45 with genetically and/or molecularly characterized gene products and 18 with novel or uncharacterized products. The interacting genes can be grouped into several different functional classes on the basis of the cellular and molecular functions of their encoded proteins (Table 2): components of the cytoskeleton (e.g., profilin and βHeavy-spectrin), the extracellular matrix (e.g., collagen type IV, α1 and α2 chains), signal transduction pathways (e.g., Delta and Puckered), nucleic acid binding proteins (e.g., Slow border cells and Apontic), proteins localized to cell membranes (e.g., lamin), and miscellaneous and novel genes (Table 2).
Most of the identified loci are general rather than stage- or tissue-specific dominant modifiers of hnt:
HNT is expressed in several different tissues during development, including the extra-embryonic amnioserosa, the tracheal system, and the larval eye imaginal disc (Yip et al. 1997). It has specific roles in each of these tissues as well as general roles in all tissues in which it is expressed (Lamka and Lipshitz 1999; Wilk et al. 2000; Reed et al. 2001; Pickup et al. 2002). Since our primary screen was performed utilizing the severity of the eye phenotype as readout, we wanted to distinguish between eye-specific and more general genetic interactions.
hnt308 is a P-element insertion in the 5′ regulatory region of the hnt gene and causes embryonic lethality with some larval, pupal, and adult escapers (Reed et al. 2001). We therefore retested hntpeb-interacting mutations in 11 loci for modification of hnt308 by assaying for dominant enhancement or suppression of embryonic lethality (see materials and methods). The genes retested encode transcription factors (apt and slbo), cytoskeletal regulatory proteins (RhoA, kst, and chic), members of signal transduction pathways (tkv, Dl, and puc), membrane-associated proteins (turtle and heixuedian), and components of the extracellular matrix (vkg and Cg25C). Since both laminin and collagen IV are major components of the extracellular matrix, we also tested mutations in a candidate gene, LanA, not identified in the initial screen; LanA encodes Drosophila laminin-α. As a control for genetic interactions with different hnt alleles we also tested a deficiency, Df(3L)kto2, which had been identified in our screen as a strong suppressor of the hntpeb rough eye phenotype (Figure 1C; Table 1).
For most genes tested (7 of 10) at least 1 allele exhibits a significant dominant genetic interaction with both hntpeb and hnt308 (Figure 4; Table 3). Considering all alleles tested, 70% show an interaction with both hnt alleles (12 of 17; Table 3). Of these, half of the interactions (6 of 12) are in the same direction (i.e., suppressor or enhancer of both hntpeb and hnt308). Df(3L)kto2 dominantly suppresses both hntpeb and hnt308 (Figure 4; Table 3), suggesting that an unknown hnt-interacting gene maps within this deficiency (reptin, which maps distal to the deficiency breakpoint, weakly suppresses and therefore cannot explain the strong interaction seen with the deficiency). In addition, two different alleles of LanA interact with hnt308 (Figure 4; Table 3). Of the genes tested only three—heix (1 allele), puc (2 alleles), and slbo (1 allele)—failed to show significant interactions with hnt308. We conclude that the majority of genes tested in both the adult and the embryo define general rather than stage- or tissue-specific dominant modifiers of hnt. Detailed results for a subset of the hnt-interacting genes are presented below.
TABLE 3.
Genetic interactions withhnt308
| Gene | Allele | Genetic interactions with hntpeb |
Genetic interactions with hnt308 |
P | N | AS | TR | Eye |
|---|---|---|---|---|---|---|---|---|
| chickadee | 01320 | Su | NS (1.23) | 0.1–0.5 | 454 | + | + | + |
| 221 | Su | E (1.38) | 0.01–0.025** | 1347 | + | + | + | |
| RhoA | J3.8 | Su | Su (ND) | —a | 1195 | + | + | + |
| k07236 | ND | NS (ND) | —a | 810 | + | + | + | |
| karst | 01318 | Su | E (1.37) | ≪0.005*** | 1121 | ND | + (apical) | + |
| 1 | ND | NS (1.65) | 0.1-0.5 | 787 | ND | + (apical) | + | |
| 2 | ND | NS (1.05) | 0.5-0.9 | 682 | ND | + (apical) | + | |
| viking | 01209 | Su | Su (0.46) | ≪0.005*** | 1478 | − | + (basal lamina)b | + (peripodial epithelium)b |
| 177 | Su | Su (0.75) | 0.05–0.1(*) | 538 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| k16721 | Su | E (1.28) | 0.05–0.1(*) | 406 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| k00236 | Su | E (1.36) | 0.01–0.025 ** | 411 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| k07138 | Su | E (1.33) | 0.01–0.025 ** | 446 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| k16502 | Su | NS (1.01) | 0.5–0.9 | 396 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| Cg25C | k00405 | Su | Su (0.75) | 0.05–0.1(*) | 343 | − | + (basal lamina)b | + (peripodial epithelium)b |
| 234-9 | Su | Su (0.54) | <0.005*** | 447 | − | + (basal lamina)b | + (peripodial epithelium)b | |
| LanA | 3A1 | ND | Su (0.72) | 0.025–0.05* | 613 | ND | + (basal lamina)b | + |
| 4A8 | ND | Su (0.72) | 0.01–0.025** | 997 | ND | + (basal lamina)b | + | |
| thickveins | k16713 | Su | Su (0.72) | 0.05–0.1(*) | 408 | ND | + | + |
| puckered | A251.1f3 | Su | NS (0.92) | 0.5–0.9 | 446 | + | ND | + |
| 1 | ND | NS (ND) | —a | 610 | + | ND | + | |
| Delta | 05151 | E | Su (0.48) | ≪0.005*** | 401 | +b | + | + |
| slow border cells | ry8 | Su | NS (0.85) | 0.1–0.5 | 335 | ND | + | ND |
| turtle | k14703 | Su | Su (0.69) | 0.01–0.025** | 546 | ND | ND | ND |
| heixuedian | k11403 | Su | NS (0.8) | 0.1–0.5 | 310 | ND | ND | ND |
| Df(3L)kto2 | NA | Su | Su (0.26) | ≪0.005*** | 471 | NA | NA | NA |
Comparisons are shown between genetic interactions with hnt308 vs. the ones observed with hntpeb. E denotes enhancement and Su denotes suppression of hnt308 embryonic lethality (EL; see materials and methods). The normalized EL is shown in parentheses. The last three columns show whether (+) or not (−) the gene is expressed in the amnioserosa (AS), embryonic tracheal system (TR), or the developing eye (Eye). NS, no significant interaction; ND, not determined; NA, not applicable; P, the P-value for the χ2 test; N, number of embryos assayed. Asterisks are as in Figure 4.
Method of analysis was as in Reed et al. (2001) and differed slightly from that used in this study; thus, P-values were not calculated.
Expression was determined in this study.
Mutations in genes encoding proteins with a role in F actin cytoskeletal organization dominantly interact with hnt:
Three of the hnt-interacting loci encode proteins that have a role in the assembly or function of the F actin-based cytoskeleton: chic, kst, and RhoA. These were of particular interest in light of the previously reported defects in the actin-based cytoskeleton in hnt mutants (Reed et al. 2001; Pickup et al. 2002). kst encodes Drosophila βHeavy-spectrin, which has actin crosslinking activity and associates with the plasma membrane (Thomas and Kiehart 1994). chic encodes profilin, a central player in the regulation of actin polymerization (Cooley et al. 1992; Verheyen and Cooley 1994). Small GTPases such as RhoA (also called Rho1) function in organization of the actin cytoskeleton as well as adherens junction formation, intracellular targeting of proteins, phosphorylation of catenins, and regulation of cell signaling pathways (reviewed by Tepass et al. 2001; Van Aelst and Symons 2002; Wilk et al. 2004).
Four alleles at the chic locus—chic11, chic01320, chic221, and chick13321—interact genetically with hntpeb; three suppress and one enhances the rough eye phenotype (Table 2). Two alleles were tested for interaction with hnt308: one, chic01320, enhances the embryonic lethality but not at a statistically significant level, while the other, chic221, significantly enhances the lethality (Figure 4; Table 3). The direction of the chic01320 interaction differs in the eye (suppressor) vs. the embryo (enhancer). One allele of kst, kst01318, suppresses the rough eye phenotype of hntpeb (Table 2) while it enhances the embryonic lethality of hnt308 (Table 3; Figure 4); two additional alleles, kst1 and kst2, show slight—but not statistically significant—enhancement of the embryonic lethality (Table 3; Figure 4). The recessive lethal allele RhoAJ3.8 suppresses the hntpeb and the hnt308 phenotypes, while the milder, nonlethal allele RhoAK07236 does not interact with hnt308 (Tables 2 and 3).
We conclude that kst, chic, and RhoA are general rather than tissue- or stage-specific dominant modifiers of hnt. The opposite direction of the genetic interaction in the eye vs. the embryo seen for kst and chic may reflect either differences in the role of hnt in regulating these proteins in distinct tissues or the different character of each of the hnt alleles (see discussion).
Genes that encode components of several signal transduction pathways genetically interact with hnt:
We have previously shown that basket (which encodes JNK) and dpp (which encodes a TGFβ/BMP homolog and is a potential transcriptional target of JNK signaling) act as dominant suppressors of hnt308 (Reed et al. 2001). Furthermore, by assaying the intracellular localization of JUN and FOS, as well as dpp and puc transcription (puc encodes a JNK phosphatase and is also a transcriptional target of JNK signaling), we showed that HNT downregulates the JNK signaling pathway (Reed et al. 2001).
Here we detected dominant genetic interactions between hntpeb and members of several signal transduction pathways (Table 2), including those mediated by JNK, TGFβ/BMP, Notch/Delta, and epidermal growth factor receptor (EGFR). Of particular interest in light of our previous results on JNK signaling, tkv mutations act as dominant suppressors of both the hntpeb rough eye phenotype and hnt308 embryonic lethality (tkv encodes a DPP receptor; Figure 4; Tables 2 and 3) while pucA251 acts as a mild dominant suppressor of the hntpeb eye phenotype and also shows mild, albeit not statistically significant, dominant suppression of hnt308 (Figure 4; Table 3). Three Dl alleles dominantly enhance the hntpeb rough eye phenotype (Figure 1D; Table 2); one of these, Dl05151, was tested in the embryo and significantly suppresses hnt308 embryonic lethality (Figure 4; Table 3).
There are several possible interpretations—not mutually exclusive—of the hnt genetic interactions with multiple signaling pathways. First, HNT may primarily regulate JNK signaling, with only indirect effects on, for example, the DPP/BMP pathway since this pathway is transcriptionally regulated in response to JNK. Second, HNT may independently regulate the production of components of the JNK, DPP/BMP, and Notch/Delta signaling pathways. Third, HNT may directly regulate production of proteins that are required for more than one cell-cell signaling pathway (e.g., components of the extracellular matrix that regulate ligand binding).
Genes that encode components of the ECM genetically interact with hnt:
The basal lamina, a specialized ECM, is composed mainly of collagen type IV and laminin. hntpeb is dominantly suppressed by six different alleles of viking (collagen IV α2 chain) and two alleles of Cg25C (collagen IV α1 chain; Table 2). Five of the six tested alleles of viking and both of the Cg25C alleles also genetically interact with hnt308 (Table 3; Figure 4). Moreover, two LanA alleles (LanA3A1 and LanA4A8; LanA encodes laminin-α chain) suppress hnt308 embryonic lethality (Table 3; Figure 4). The direction of the genetic interactions among alleles of viking and hnt308 varies (Table 3; Figure 4): two alleles suppress embryonic lethality (vkg01209 and vkg177), three are enhancers (vkgk16721, vkgk00236, and vkgk07138), and one shows no significant genetic interaction (vkgk16502). Some of the differences in the direction of the genetic interaction between specific vkg and hnt alleles may derive from the genetic complexity of the vkg locus (see Table 4 and discussion).
TABLE 4.
Complementation ofviking alleles
| 01209 S | 177-27 S | k00236 E | k07138 E | k16502 NI | |
|---|---|---|---|---|---|
| 01209 S | − | ||||
| 177-27 S | −a | − | |||
| k00236 E | +b | −a | − | ||
| k07138 E | −b | −a | −a | − | |
| k16502 NI | +a,b | −a | −b | −a | − |
The first column and the first row show the genetic results obtained with hnt308 with different vkg alleles. S, suppressor; E, enhancer; NI, no interaction; −, fails to complement; +, complements. vkg177-27 fails to complement the semilethal allele vkgk16721 (not shown).
Lethal complementation tests done by us.
Complementation tests reported by FlyBase (http://flybase.bio.indiana.edu).
HNT controls the expression of genetically interacting genes both tissue autonomously and tissue nonautonomously:
To establish which interacting genes might be directly regulated by HNT, we analyzed their expression in hnt mutants. Tests were carried out on a subset of interacting genes selected because they are expressed in at least two of three HNT-expressing tissues (amnioserosa, tracheal system, and/or the larval eye disc). To carry out the tests we used antibodies or enhancer trap lines where the P element is inserted in the gene of interest and there is detectable β-galactosidase reporter gene expression in the amnioserosa, the tracheal system, and/or the larval eye disc: Dl, RhoA, puc, apontic, and dpp could be examined in the embryo and Dl, dpp, apontic, and kst in the eye disc. dpp and puc served as a controls since they had already been shown to be downregulated by HNT in the amnioserosa and eye (Reed et al. 2001; Pickup et al. 2002). In the case of the developing eye disc, since the mutations assayed are embryonic lethals, patches of hntXE81 or hntEH704a mutant tissue were generated using the FLP/FRT system (see materials and methods). The results of our analyses are presented in Figures 5 and 6 and are categorized by cellular process below.
Figure 5.—
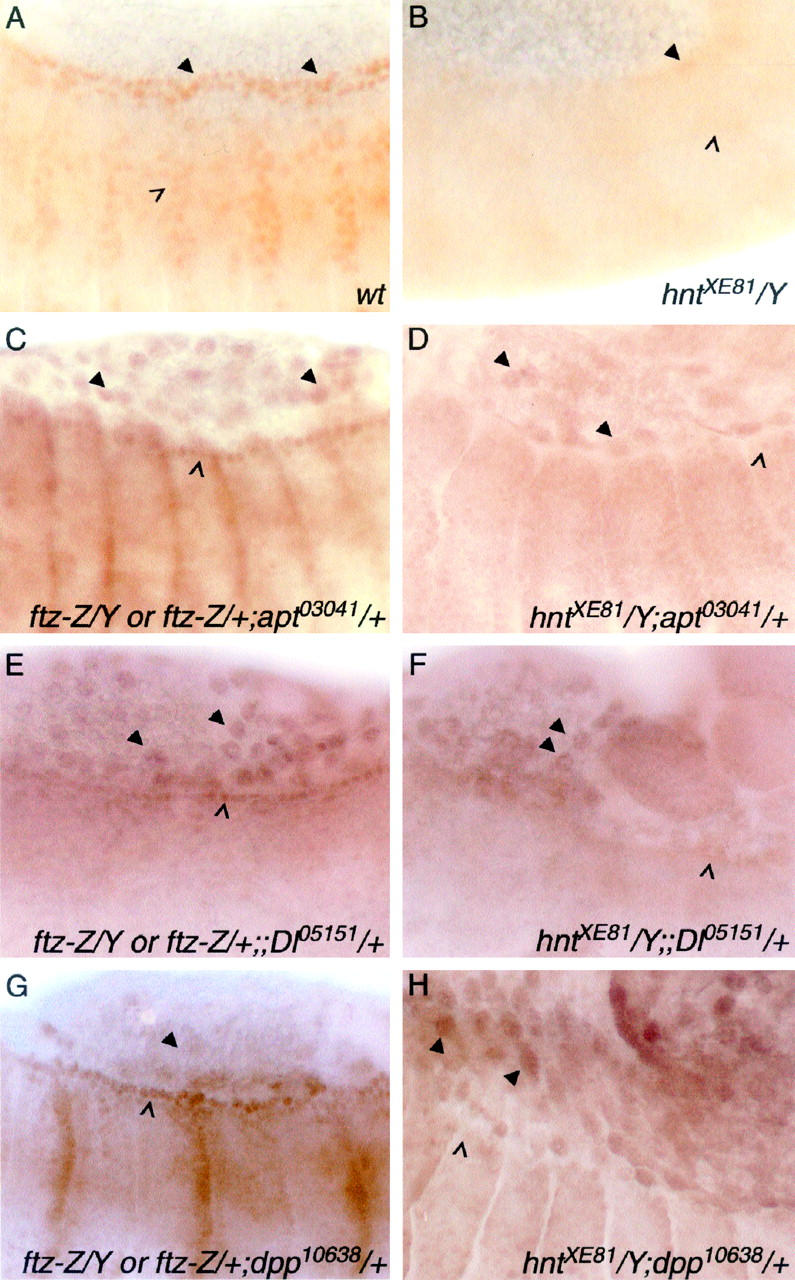
HNT regulates candidate interacting genes tissue autonomously and nonautonomously in the embryo. (A and B) Wild-type (A) and hntXE81 (B) stage 14 embryos showing APT protein in the dorsal vessel (arrowheads) and the tracheal system (open arrowhead). APT expression can be seen to be very reduced in the hnt mutant. (C–H) Expression reported by lacZ enhancer trap lines detected with anti-β-galactosidase antibody. The left column shows embryos with one copy each of the lacZ insertion and the FM7, ftz-lacZ balancer chromosome. The latter distinguishes these embryos from their hntXE81 male siblings (right column). apontic (apt) expression in the leading edge (open arrowhead in C) is almost absent in hnt mutant embryos (open arrowhead in D), whereas the amnioserosal expression is not altered (arrowheads in C and D). A similar result is seen with Delta (Dl) expression (E vs. F; leading edge, open arrowheads; amnioserosa, arrowheads). dpp expression is upregulated in the amnioserosa of hnt mutant embryos (arrowheads in G vs. H) while epidermal leading edge expression is nonautonomously reduced (open arrowheads in G and H).
Figure 6.—
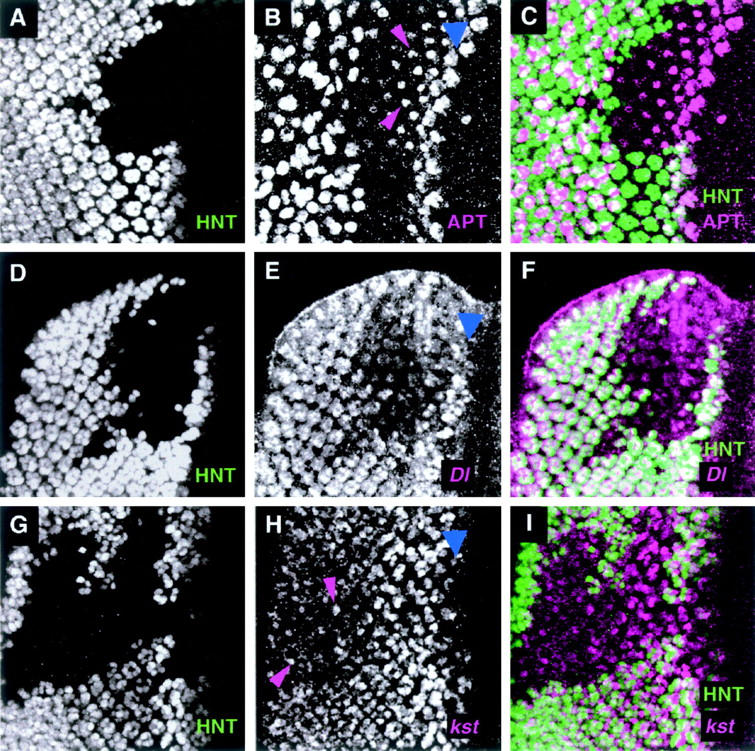
HNT regulates candidate genes in the larval eye disc. (A–C) Confocal images of a third instar larval eye disc, which contains an hntEH704a mutant patch. The discs were double immunostained with anti-HNT (A) to visualize the patch and an anti-APT antibody (B). (C) The two single channels are merged. APT expression persists or is elevated in a single R cell for several rows just posterior to the furrow (magenta arrowheads) compared to the adjacent wild-type tissue. (D–F) Confocal images of an eye disc containing a clone of hntXE81 tissue and marked with the Dl05151 enhancer trap line. The disc is double stained with anti-HNT antibody (D) and a chicken anti-β-galactosidase antibody (E), which reports Dl-lacZ expression. The two single channels are merged in F. Within the hnt patch, Dl-lacZ expression levels are reduced in all of the R cell precursors posterior to the furrow. (G–I) Confocal images of a disc with an hntXE81 clone and marked with the kst03041 enhancer trap line. The disc is double stained with anti-HNT antibody (G) and a rabbit anti-β-galactosidase antibody (H), which reports the kst-lacZ expression. (I) The two single channels are merged. The initial kst expression looks unaffected in the hnt mutant tissue but by rows 10 and 11 the kst expression level is somewhat reduced and/or more diffuse in the R precursor cells than in the neighboring wild-type tissue. Occasionally a few apical cells are seen in this posterior region of the mutant tissue that have elevated kst expression (for examples see magenta arrowheads). Blue arrowheads mark the morphogenetic furrow in B, E, and H.
Transcription (apt):
During embryonic development apontic is expressed in the dorsal vessel, the tracheae, the amnioserosa, and the epidermal leading edge (Figure 5, A and C). In hnt mutant embryos the expression of apontic in the amnioserosa is not significantly altered (compare arrowheads in Figure 5, C vs. D) but leading edge expression is greatly reduced (compare open arrowhead in Figure 5, C vs. D). To visualize dorsal vessel and tracheal expression of apontic, which is not detectable using a single copy of the apontic enhancer trap, we used an APT-specific antibody. Wild-type embryos show APT expression in the dorsal vessel (arrowheads in Figure 5A), the embryonic tracheal system (open arrowhead in Figure 5A, brown staining), and the head (data not shown). In hnt mutant embryos all of these tissues show very reduced APT levels (Figure 5B). When HNT expression is removed specifically from tracheal cells using Df(1)rb1 (Wilk et al. 2000), APT levels are reduced only in the tracheal system (data not shown). We conclude that HNT regulates apontic in both a tissue autonomous (tracheal cells) and a tissue nonautonomous (dorsal vessel, leading edge, and head) manner.
In the developing eye disc, APT protein is expressed in all peripodial membrane cells, as well as in the disc epithelium where APT is found in clusters of cells in the morphogenetic furrow, in the emerging R8 cell precursors, and then, more posteriorly, in basal undifferentiated disc cells (see wild-type tissue in Figure 6, B and C). In hnt mutant patches (n = 10) the peripodial membrane and basal epithelial staining is unaffected (data not shown), but APT expression in the early R8 precursor cell persists or is elevated for two to three additional, more posterior, rows compared to that in wild-type tissue (magenta arrowheads in Figure 6B). This effect is subtle but reproducible and suggests that HNT may be necessary tissue or cell autonomously for downregulation of apontic expression in the R8 precursor cell.
Signal transduction (Dl, dpp, and puc):
In embryos, Dl-lacZ enhancer trap expression is found in the amnioserosa (arrowheads in Figure 5E), the leading edge (open arrowhead in Figure 5E), and the tracheal system (not detectable with a single copy of this Dl enhancer trap line). In hnt mutant embryos, leading edge expression is greatly reduced (compare open arrowhead in Figure 5, E vs. F), while amnioserosal expression remains unchanged (compare arrowheads in Figure 5, E vs. F). Since HNT itself is not expressed in the leading edge, HNT must regulate Dl expression in the leading edge cells in a cell and tissue nonautonomous manner. In the third instar eye disc, Dl-lacZ enhancer trap expression is found in all of the R cell precursor cells posterior to the furrow (refer to wild-type tissue in Figure 6E). In hntEH704a mutant tissue (n = 8) Dl-lacZ expression is reduced in all of the R cells (Figure 6, E and F). This effect is seen specifically with the chicken anti-β-galactosidase antibody (ab-cam) and has been confirmed with X-GAL staining (data not shown). The same effect is not obvious with the rabbit anti-β-galactosidase antibody (Cappel) used in other experiments, suggesting that the reduction in Dl-lacZ expression is moderate and can be detected only at a certain threshold of staining.
HNT downregulates dpp and puc in the amnioserosa of hnt308 mutant embryos (Reed et al. 2001). Here we analyzed an amorphic hnt allele (hntXE81). In wild type, dpp and puc expression in the amnioserosa is very weak (for dpp, see Figure 5G, arrowhead; data not shown for puc) whereas, in hntXE81 mutant embryos, dpp and puc expression in the amnioserosa is significantly elevated (for dpp, compare arrowheads in Figure 5, G vs. H), consistent with tissue autonomous downregulation of dpp and puc expression by HNT. Downregulation of dpp by HNT has been shown previously in hnt mutant eye tissue where the expression of a dpp-lacZ reporter is elevated in photoreceptor precursor cells posterior to the furrow (Pickup et al. 2002). In the embryo, HNT may have an additional, tissue nonautonomous, effect on dpp expression levels: dpp leading edge expression is clearly reduced in hnt mutant embryos (compare open arrowhead in Figure 5, G vs. H), suggesting that upregulation of dpp in the leading edge cells requires HNT function in the amnioserosa.
Cytoskeleton (RhoA and kst):
Amnioserosal expression of RhoA is unchanged in hnt mutant embryos (data not shown) and was not assayed in the eye disc. In the eye disc, kst-lacZ expression is found in clusters of cells in the furrow and then in all of the emerging R cell precursors (see wild-type area of Figure 6H). In clones of hntXE81 mutant tissue (n = 7) the early expression of kst-lacZ looks normal, but in more posterior regions of the clones (rows 10 and 11 and more posteriorly) the kst-lacZ staining declines or is absent in most cells when compared to the adjacent nonmutant tissue (Figure 6H). When we examined mosaic clusters along the borders of hnt mutant clones, we found examples of clusters with only a single hnt+ cell. In 50% of these cases, this hnt+ cell exhibited the same reduced level of kst-lacZ expression as its neighboring, mutant precursor cells, suggesting that the regulation of kst by HNT may have a cell nonautonomous component to it. This result is consistent with previous observations in which we showed that some of the mutant phenotypes in hnt mutant eye tissue are partially cell nonautonomous (Pickup et al. 2002). At this stage there are also a few dispersed R precursor cells (defined as such because the nuclei are apical and stain with anti-ELAV antibody; data not shown) that have higher than wild-type levels of kst-lacZ staining (Figure 6H, magenta arrowheads).
In summary, we have identified five genes whose expression levels are regulated tissue autonomously by HNT (apt, Dl, dpp, kst, and puc). Three of these (apt, Dl, and dpp) are also regulated tissue nonautonomously by HNT.
Collagen IV and laminin deposition and/or maintenance in the basal lamina of the developing tracheal system are affected in hnt mutant embryos:
In Drosophila, procollagen IV (Lunstrum et al. 1988) and laminin-α (Kusche-Gullberg et al. 1992) are synthesized in the circulating blood cells, which are known as hemocytes. Subsequently, collagen IV and laminin are deposited in the basement membranes of major organs (Fessler and Fessler 1989; Montell and Goodman 1989; Yarnitzky and Volk 1995; Martin et al. 1999). Since HNT is not expressed in any mesodermal derivatives, including the hemocytes, any effects on collagen IV and laminin distribution in HNT-expressing tissues must derive from HNT-dependent defects in processing, deposition, or maintenance of these molecules in the basal lamina of that tissue. The basal lamina plays a pivotal role in maintenance of tissue integrity (reviewed by Yurchenco and O'Rear 1994; Ashkenas et al. 1996; Wilk et al. 2004). Our previous studies of the role of HNT during tracheal development clearly showed that HNT regulates tracheal tissue integrity (Wilk et al. 2000). Because the embryonic tracheal system has a defined basal lamina (Tepass and Hartenstein 1994) and mutations in collagen IV and laminin exhibit particularly strong dominant genetic interactions with hnt, we chose to analyze collagen IV and laminin deposition during embryonic tracheal development in wild-type and hnt mutant embryos utilizing specific antibodies and tracheal markers (see materials and methods).
Collagen IV is present at high levels in hemocytes (asterisks in Figure 7A), the fat body (not shown), and basement membranes (arrowheads in Figure 7, A and A′; see also Fessler and Fessler 1989). As the tracheae develop, collagen IV (stage 14 onward) and laminin (stage 13 onward) can be detected on the basal side of the tracheal cells (Figure 7, A, A′, and D; arrowheads, stage 15 embryos). To determine whether basal collagen IV and laminin deposition are affected in the tracheae of hnt mutant embryos, we analyzed the tracheal tissue of two amorphic hnt alleles (hntXE81 and hnt1142; identical results were obtained for both alleles). As expected, hnt mutant embryos show normal levels of collagen IV in hemocytes (Figure 7, C and C′; asterisks). Basal localization of collagen IV in the developing tracheal epithelia occurs in hnt mutants (Figure 7, B′ and C′). However, hnt mutant tracheae show a patchy and discontinuous collagen IV distribution when compared to wild-type tracheae. By late embryonic stage 14, this phenotype is more pronounced: each embryo has areas with marked reductions or complete absences of collagen IV (Figure 7, B and B′; open arrowhead and arrows, respectively) as well as patches of overaccumulation (Figure 7, C and C′; solid arrowheads). Tracheal laminin distribution at stage 13 is identical in hnt and wild-type embryos (data not shown). As for collagen, by stage 14, hnt embryos have patchy or discontinuous laminin staining in the basal lamina of their tracheae compared to wild type (arrowhead in Figure 7D vs. arrowhead in Figure 7E).
Figure 7.—
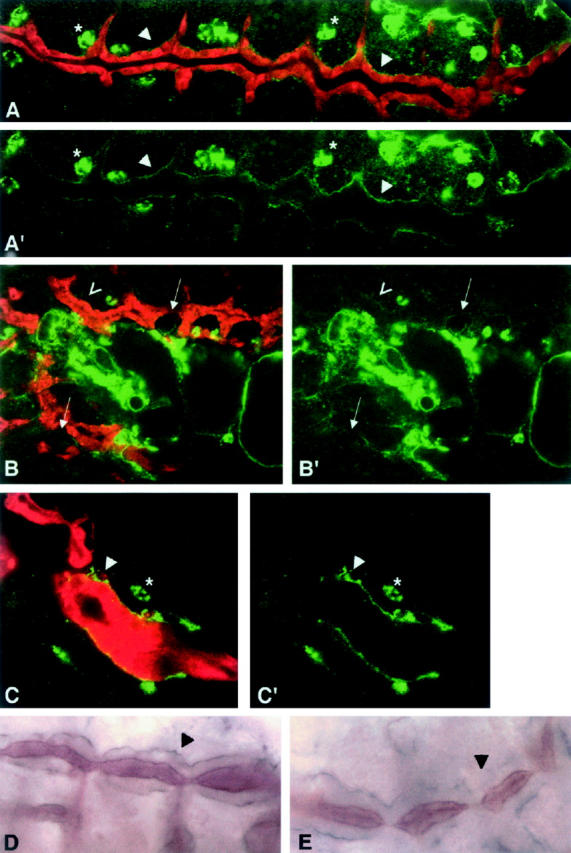
Collagen type IV and laminin are abnormally distributed in the basal lamina of hnt mutant tracheae. Confocal images from either +/+;1-eve-1 (A) or hnt;1-eve-1 mutant embryos (B and C) that were double stained for a tracheal cell marker, shown in red (see materials and methods), and with mouse monoclonal anti-collagen IV antibody, shown in green. Primes show the green channel only (only the collagen IV staining). Collagen IV expression can be seen in the hemocytes (asterisks) and in the basal lamina of the tracheal system (arrowheads). The distribution of collagen IV is less uniform in hnt mutant tracheae than in wild type: absent (arrows in B), weaker (open arrowhead in B), or stronger and patchy (arrowhead in C). Light microscope images from wild-type (D) or hnt (E) embryos immunostained for laminin (purple) and tracheal lumen antibody 55 (brown). Arrowheads show laminin staining in the basal lamina of the developing tracheae, evenly distributed in wild type (D) but uneven and patchy in hnt mutant embryos (E).
We conclude that, while hnt does not directly regulate the expression of either collagen IV or laminin, it is required in HNT-expressing tissues, such as the trachea, for collagen IV and laminin deposition, distribution, or maintenance in the basal lamina.
DISCUSSION
Here we have identified >60 loci that exhibit dose-sensitive genetic interactions with hntpeb, a viable rough-eyed hnt allele, and have shown that the majority of a subset of these that were tested for interaction with hnt308 also modify the embryonic lethality associated with that allele. The direction of the dominant genetic interactions is not always the same in the eye and embryo. We do not believe that this difference is due to a difference in the nature of the two hnt alleles since both behave as hypomorphs (Yip et al. 1997; Reed et al. 2001; Pickup et al. 2002): hnt308 is caused by a P-element insertion 509 nucleotides upstream of the transcription start site and results in reduced accumulation of HNT protein, particularly in the amnioserosa (Reed et al. 2001). hntpeb shows defects only in eye development, and HNT expression levels are normal in hntpeb mutant eyes. We therefore presume that hntpeb is caused by a point mutation that alters the function rather than the expression level or pattern of HNT protein (we have not yet been able to detect any sequence alterations in the open reading frame; A. T. Pickup and H. D. Lipshitz, unpublished observations).
An alternative hypothesis is that the role of hnt in regulating the cellular process affected by the interactor may differ in the eye and the embryo. Differences in direction of interaction occur for Dl (enhancer of eye phenotype, suppressor of embryonic lethality), chic (suppressor of eye phenotype, enhancer of embryonic lethality), and kst (suppressor of eye phenotype, enhancer of embryonic lethality). The fact that the direction of interaction changes in the same way for both cytoskeletal regulatory proteins (chic and kst), which function to regulate F actin assembly, is consistent with this alternative hypothesis. However, since the exact cause of embryonic lethality in hnt308 is unknown (Reed et al. 2001) and the hntpeb eye phenotype is complex (Pickup et al. 2002), understanding the reason for the particular direction of any specific genetic interaction is likely to come only with a more detailed understanding of the molecular pathways regulated by HNT and the particular role of HNT in transcriptional control. In regard to the latter, for example, it will be important to assess whether different HNT cofactors might be present in different tissues.
In the case of vkg (which encodes collagen IV), the direction of the genetic interaction with hnt differs for different vkg alleles. For example, all six vkg alleles are suppressors of hntpeb; however, two of the five alleles that interact with hnt308 are suppressors and three are enhancers. Thus, the direction of interaction differs not only for the two hnt alleles, but also for different vkg alleles. It is likely that this additional layer of complexity derives from the fact that vkg alleles themselves show complex interallelic complementation (Table 4; see also Gellon et al. 1997), possibly because collagen forms multimers (of two α1 chains and one α2 chain) in the extracellular matrix.
We have previously shown that the HNT zinc-finger protein is expressed in specific tissues in each of which it regulates cell differentiation, epithelial integrity, and cell survival (Yip et al. 1997; Lamka and Lipshitz 1999; Wilk et al. 2000; Reed et al. 2001; Pickup et al. 2002). During at least two morphogenetic processes—embryonic dorsal closure and retinal differentiation—we have reported defects in the F actin-based cytoskeleton in hnt mutants. In the embryo, these defects are tissue nonautonomous, since they occur in the leading edge cells that are not themselves expressing HNT but are closely apposed to HNT-expressing amnioserosal cells (Reed et al. 2001). In the eye, the defects may be cell autonomous, occurring in the photoreceptor cells, each of which expresses HNT (Pickup et al. 2002). In addition to these cytoskeletal defects, we have presented evidence that HNT downregulates JNK signaling in both the amnioserosa and, possibly, the eye (Reed et al. 2001; Pickup et al. 2002). Finally, HNT is required in the amnioserosa, tracheal system, and eye disc to maintain epithelial integrity; in hnt mutants, these tissues fall apart and the cells subsequently undergo apoptosis (Frank and Rushlow 1996; Lamka and Lipshitz 1999; Wilk et al. 2000; Pickup et al. 2002; Reed et al. 2004).
Since HNT is a nuclear Zn-finger protein with all of the hallmarks of a transcription factor, the cellular and tissue phenotypes seen in hnt mutants are likely to be an indirect consequence of defects in transcriptional control. However, the particular molecular pathway(s) affected in hnt mutants are unknown. Our screen for dominant genetic interactors with hnt mutants was thus motivated in large part by the desire to identify potential genetic pathways that are regulated, directly or indirectly, by HNT. Because the screen initially focused on modification of the hntpeb rough eye phenotype and then retested the interactors for modification of the hnt308 embryonic lethal phenotype, both eye-specific and general modifier genes could be identified. Our initial focus was primarily on loci that act as dominant modifiers of both phenotypes and are expressed in the same tissues as HNT and thus are candidates for HNT regulation—directly or indirectly—in all tissues in which it is expressed. In this regard, it is clear that mutations in several types of genes modify the hnt phenotype: these include genes that encode other transcription factors (e.g., apt), signal transduction molecules (e.g., Dl, and dpp), and regulators of the actin-based cytoskeleton (e.g., chic, RhoA, and kst).
Our assays of effects on expression of a subset of these interactors allowed us to identify which might be direct targets of HNT transcriptional control and which are unlikely to fall into this category. Candidates for direct regulation by HNT include apt, Dl, dpp, kst, and puc since their expression is altered in a tissue autonomous way in hnt mutants. In the cases of dpp and puc, the role of HNT appears to be to downregulate their expression, while HNT's role is to upregulate Dl and kst expression. For apt, HNT functions either to downregulate (in the eye) or to upregulate (in the tracheae) expression. It has recently been reported that the mammalian homolog of HNT, RREB-1/Finb, can function as a DNA-binding protein that acts to “potentiate” transcriptional activation by the BETA2/NeuroD basic helix-loop-helix protein (Ray et al. 2003). Whether HNT acts as a transcriptional potentiator and, additionally, as a DNA-binding “antipotentiator,” remains to be determined. Of interest in this regard is the fact that the effects on candidate target gene expression that we have seen in the eye disc appear to involve a reduction or increase in levels, not an absolute on/off control.
Two of the candidate HNT target genes, dpp and puc, are transcriptional targets of JNK signaling, presumably of the AP-1 transcription factor (Glise and Noselli 1997; Hou et al. 1997; Riesgo-Escovar and Hafen 1997a,b; Sluss and Davis 1997; Zeitlinger et al. 1997). This raises the interesting possibility that one of HNT's roles may be to regulate AP-1 activity. For example, HNT might prevent AP-1 from activating some or all of its target genes by competing for AP-1 binding sites, by binding to AP-1 components and preventing them from binding to DNA, or by binding to the same target genes but functioning as a repressor.
Another hnt-interactor, chic (which encodes profilin, Cooley et al. 1992), plays a role in embryonic dorsal closure and larval eye morphogenesis (Jasper et al. 2001; Benlali et al. 2002), two processes in which HNT functions. Furthermore, the chic gene has been identified as a JNK pathway target gene in a screen that used serial analysis of gene expression (Jasper et al. 2001). It is therefore possible that defects in AP-1 target gene regulation in hnt mutants may underlie the genetic interaction between hnt and chic reported here. An alternative is that the genetic interaction results from nonautonomous effects of hnt mutants on the leading edge of the epidermis (Reed et al. 2001). chic mutants show defects in leading edge filopodia during dorsal closure (Jasper et al. 2001). It is thus possible that chic mutants enhance the embryonic lethality of hnt308 by further increasing the disruption of actin-rich structures at the leading edge that is caused nonautonomously by hnt mutant amnioserosal tissue.
Several interactors are regulated both tissue autonomously and tissue nonautonomously by hnt. For example, HNT tissue autonomously regulates apt, dpp, kst, and Dl in the developing retina; apt in the tracheae; and dpp in the amnioserosa. However, in hnt mutants but not in wild type, apt, Dl, and dpp expression is absent from the epidermal leading edge cells. Similarly, apt is expressed in the dorsal vessel in wild type but not in hnt mutants. Since HNT is not expressed in leading edge cells or the dorsal vessel, these effects must be tissue nonautonomous. We have previously presented extensive evidence that HNT-dependent downregulation of JNK signaling in the amnioserosa is required for assembly of the F actin-based purse string in the epidermal leading edge and have hypothesized that this occurs only at a high-low JNK signaling boundary (Reed et al. 2001). It is therefore possible that HNT-dependent upregulation of genes such as apt, Dl, and dpp in the leading edge also requires such a boundary. A recent screen for cardiogenic genes has reported a requirement for hnt in assembly of the heart tube and for heart patterning (Kim et al. 2004). Since dorsal closure is required for assembly of the heart but fails in hnt mutants, some of the cardiogenic defects in hnt mutants may derive indirectly from the dorsal closure defect. However, the absence of apt expression in the dorsal vessel of hnt mutants that we have observed here may underlie specific heart pattern defects observed in that study.
In light of the previously reported tissue integrity defects in hnt mutants (Lamka and Lipshitz 1999; Wilk et al. 2000; Pickup et al. 2002), we were particularly interested in the strong genetic interactions between hnt and components of the extracellular matrix (collagen IV subunits and laminin). The most detailed analyses of these defects had been carried out in the embryonic tracheal system, where we previously showed that hnt mutant tracheal cells have normal crumbs and DE-cadherin distribution, adherens junctions, and apical-basal polarity (Wilk et al. 2000); however, in that study, the basal lamina was not investigated. Given the strong genetic interactions between hnt and genes encoding components of the basal lamina, together with the known role of the basal lamina in maintenance of epithelial integrity, we focused here on the distribution of collagen IV and laminin in the basal lamina of hnt mutant tracheae. We have shown that deposition or maintenance of collagen IV and laminin is abnormal in the basal lamina of hnt mutant tracheae, although we cannot at present definitively determine whether this is the cause of the loss of integrity. Interestingly, when collagen IV levels are reduced during Drosophila embryogenesis, transgenic embryos show defects in germ band retraction and dorsal closure (Borchiellini et al. 1996), two processes for which HNT is essential (Yip et al. 1997; Lamka and Lipshitz 1999; Reed et al. 2001). Moreover, LanA embryos have defects in the tracheal dorsal trunk that are similar those described for hnt mutant embryos (Stark et al. 1997; Wilk et al. 2000). The less severe defect in the LanA mutants than in hnt may reflect the fact that, even without laminin, collagen IV can still assemble a meshwork in basal epithelia (both alleles used here are amorphic, see Henchcliffe et al. 1993; Yarnitzky and Volk 1995).
Since both collagen and laminin are synthesized primarily in the circulating hemocytes, which deposit these molecules in the basal lamina during its construction, and HNT is not expressed in the hemocytes, the defects in deposition of these basal lamina components in hnt mutants must be indirect. We have recently demonstrated that integrin-dependent membrane interactions between the yolk sac membrane and the amnioserosa are essential for amnioserosal epithelial integrity and to prevent anoikis (Reed et al. 2004). We hypothesized that membrane apposition may be abnormal in hnt mutants, leading to premature anoikis. Since it is known that laminin-integrin physical interaction plays a key role in assembly of the basal lamina (reviewed in Wilk et al. 2004), it will be of interest to determine whether integrin expression is normal in HNT-expressing epithelia. Defects in integrin expression in hnt mutant epithelia may underlie abnormalities in the basal lamina and genetic interactions with extracellular membrane proteins, as well as many of the other genetic interactions we have reported here (e.g., with signal transduction pathway and cytsokeletal components). Alternatively, since it is known that the ECM plays a key role in signal transduction (reviewed by Gumbiner 1996; Lauffenburger and Horwitz 1996; Wilk et al. 2004), the phenotypic effects of HNT may be indirect via regulation of ECM deposition and maintenance. In βPS and αPS3 integrin mutants, germ band retraction and dorsal closure fail and there are defects in tracheal development (Wieschaus and Noell 1986; Leptin et al. 1989; Bunch et al. 1992; Stark et al. 1997). The similarity of the integrin and hnt mutant phenotypes is thus consistent with the possibility that integrin expression or function is regulated by HNT.
Acknowledgments
We thank the Bloomington Drosophila Stock Center for hundreds of the stocks provided for this study. Thanks also go to J. Duffy, S. L. Zipursky, N. McGinnis, B. Shilo, M. Krasnow, L. I. Fessler, U. Tepass, and R. Schuh for providing fly stocks and reagents. R.W. was supported in part by an Ontario Graduate Scholarship and a studentship from the Ontario Student Opportunity Trust Fund-Hospital for Sick Children Foundation Student Scholarship Program; A.T.P. was supported in part by a postdoctoral fellowship from the Hospital for Sick Children Research Training Centre; J.K.H. was supported in part by an Eli Lilly Canada-Medical Research Council/Pharmaceutical Manufacturer's Association of Canada Health Program Fellowship. H.D.L. is Canada Research Chair (CRC, Tier 1) in Developmental Biology at the University of Toronto. This research was supported by funds from the CRC Program and an operating grant to H.D.L. from the National Cancer Institute of Canada with funds from the Canadian Cancer Society.
References
- Ashkenas, J., J. Muschler and M. J. Bissell, 1996. The extracellular matrix in epithelial biology: shared molecules and common themes in distant phyla. Dev. Biol. 180: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlali, A., I. Draskovic, D. Hazelett and J. Treisman, 2002. act up controls actin polymerization to alter cell shape and restrict Hedgehog signaling in the Drosophila eye disc. Cell 101: 271–281. [DOI] [PubMed] [Google Scholar]
- Borchiellini, C., J. Coulon and Y. Le Parco, 1996. The function of type IV collagen during Drosophila muscle development. Mech. Dev. 58: 179–191. [DOI] [PubMed] [Google Scholar]
- Bunch, T. A., R. Salatino, M. C. Engelsgjerd, L. Mukai, R. F. West et al., 1992. Characterization of mutant alleles of myospheroid, the gene encoding the beta subunit of the Drosophila PS integrins. Genetics 132: 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley, L., E. Verheyen and K. Ayers, 1992. chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell 69: 173–184. [DOI] [PubMed] [Google Scholar]
- Dixon, W. J., and F. J. Massey, Jr., 1957 Introduction to Statistical Analysis. McGraw-Hill, New York.
- Eulenberg, K. G., and R. Schuh, 1997. The tracheae defective gene encodes a bZIP protein that controls tracheal cell movement during Drosophila embryogenesis. EMBO J. 16: 7156–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler, J. H., and L. I. Fessler, 1989. Drosophila extracellular matrix. Annu. Rev. Cell Biol. 5: 309–339. [DOI] [PubMed] [Google Scholar]
- Fessler, L. I., A. G. Campbell, K. G. Duncan and J. H. Fessler, 1987. Drosophila laminin: characterization and localization. J. Cell Biol. 105: 2383–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, L. H., and C. Rushlow, 1996. A group of genes required for maintenance of the amnioserosa tissue in Drosophila. Development 122: 1343–1352. [DOI] [PubMed] [Google Scholar]
- Geiger, B., A. Bershadsky, R. Pankov and K. M. Yamada, 2001. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell. Biol. 2: 793–805. [DOI] [PubMed] [Google Scholar]
- Gellon, G., K. W. Harding, N. McGinnis, M. M. Martin and W. McGinnis, 1997. A genetic screen for modifiers of Deformed homeotic function identifies novel genes required for head development. Dev. Suppl. 124: 3321–3331. [DOI] [PubMed] [Google Scholar]
- Glise, B., and S. Noselli, 1997. Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes Dev. 11: 1738–1747. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B. M., 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84: 345–357. [DOI] [PubMed] [Google Scholar]
- Henchcliffe, C., L. Garcia-Alonso, J. Tang and C. S. Goodman, 1993. Genetic analysis of laminin A reveals diverse functions during morphogenesis in Drosophila. Development 118: 325–337. [DOI] [PubMed] [Google Scholar]
- Hou, X. S., E. S. Goldstein and N. Perrimon, 1997. Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev. 11: 1728–1737. [DOI] [PubMed] [Google Scholar]
- Jasper, H., V. Benes, C. Schwager, S. Sauer, S. Clauder-Munster et al., 2001. The genomic response of the Drosophila embryo to JNK singnaling. Dev. Cell 1: 579–586. [DOI] [PubMed] [Google Scholar]
- Kim, Y.-O., S.-J. Park, R. S. Balaban, M. Nirenberg and Y. Kim, 2004. A functional genomic screen for cardiogenic genes using RNA interference in developing Drosophila embryos. Proc. Natl. Acad. Sci. USA 101: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusche-Gullberg, M., K. Garrison, A. J. MacKrell, L. I. Fessler and J. H. Fessler, 1992. Laminin A chain: expression during Drosophila development and genomic sequence. EMBO J. 11: 4519–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamka, M. L., and H. D. Lipshitz, 1999. Role of the amnioserosa in germ band retraction of the Drosophila melanogaster embryo. Dev. Biol. 214: 102–112. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D. A., and A. F. Horwitz, 1996. Cell migration: a physically integrated molecular process. Cell 84: 359–369. [DOI] [PubMed] [Google Scholar]
- Leptin, M., T. Bogaert, R. Lehmann and M. Wilcox, 1989. The function of PS integrins during Drosophila embryogenesis. Cell 56: 401–408. [DOI] [PubMed] [Google Scholar]
- Lunstrum, G. P., H.-P. Barchinger, L. I. Fessler, K. G. Duncan, R. E. Nelson et al., 1988. Drosophila basement membrane procollagen IV. J. Biol. Chem. 263: 18318–18327. [PubMed] [Google Scholar]
- Martin, D., S. Zusman, X. Li, E. L. Williams, N. Khare et al., 1999. wing blister, a new Drosophila laminin α chain required for cell adhesion and migration during embryonic and imaginal development. J. Cell Biol. 145: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell, D. J., and C. S. Goodman, 1989. Drosophila Laminin: sequence of B2 subunit and expression of all three subunits during embryogenesis. J. Cell Biol. 109: 2441–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup, T. A., M. L. Lamka, Q. Sun, M. L. R. Yip and H. D. Lipshitz, 2002. Control of photoreceptor cell morphology, planar polarity and epithelial integrity during Drosophila development. Development 129: 2247–2258. [DOI] [PubMed] [Google Scholar]
- Ray, S. K., J. Nishitani, M. W. Petry, M. Y. Fessing and A. B. Leiter, 2003. Novel transcriptional potentiation of BETA2/NeuroD on the secretin gene promoter by the DNA-binding protein Finb/RREB-1. Mol. Cell. Biol. 23: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, B. H., 1992 The genetic analysis of endoreduplication in Drosophila melanogaster, Ph.D. Thesis, University of Cambridge, Cambridge, UK.
- Reed, B. H., R. Wilk and H. D. Lipshitz, 2001. Downregulation of Jun kinase signaling in the amnioserosa is essential for dorsal closure of the Drosophila embryo. Curr. Biol. 11: 1098–1108. [DOI] [PubMed] [Google Scholar]
- Reed, B. H., R. Wilk, F. Schock and H. D. Lipshitz, 2004. Integrin-dependent apposition of Drosophila extraembryonic membranes promotes morphogenesis and prevents anoikis. Curr. Biol. 14: 372–380. [DOI] [PubMed] [Google Scholar]
- Reichman-Fried, M., B. Dickson, E. Hafen and B.-Z. Shilo, 1994. Elucidation of the role of breathless, a Drosophila FGF receptor homolog, in tracheal cell migration. Genes Dev. 8: 428–439. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar, J. R., and E. Hafen, 1997. a Common and distinct roles of DFos and DJun during Drosophila development. Science 278: 669–672. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar, J. R., and E. Hafen, 1997. b Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and the AP-1 transcription factor DJun during dorsal closure. Genes Dev. 11: 1717–1727. [DOI] [PubMed] [Google Scholar]
- Sluss, H. K., and R. J. Davis, 1997. Embryonic morphogenesis signaling pathway mediated by JNK targets the transcription factor JUN and the TGF-beta homologue decapentaplegic. J. Cell. Biochem. 67: 1–12. [PubMed] [Google Scholar]
- Stark, K. A., G. H. Yee, C. E. Roote, E. L. Williams, S. Zusman et al., 1997. A novel α integrin subunit associates with βPS and functions in tissue morphogenesis and movement during Drosophila development. Dev. Suppl. 124: 4583–4594. [DOI] [PubMed] [Google Scholar]
- Tepass, U., and V. Hartenstein, 1994. The development of cellular junctions in the Drosophila embryo. Dev. Biol. 161: 563–596. [DOI] [PubMed] [Google Scholar]
- Tepass, U., G. Tanentzapf, R. Ward and R. Fehon, 2001. Epithelial cell polarity and cell junctions in Drosophila. Annu. Rev. Genet. 35: 747–784. [DOI] [PubMed] [Google Scholar]
- Thomas, G. H., and D. P. Kiehart, 1994. βHeavy-spectrin has a restricted tissue and subcellular distribution during Drosophila embryogenesis. Development 120: 2039–2050. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L., and M. Symons, 2002. Role of Rho family GTPases in epithelial morphogenesis. Genes Dev. 16: 1032–1054. [DOI] [PubMed] [Google Scholar]
- Verheyen, E. M., and L. Cooley, 1994. Profilin mutations disrupt multiple actin-dependent processes during Drosophila development. Dev. Suppl. 120: 717–728. [DOI] [PubMed] [Google Scholar]
- Wieschaus, E., and E. Noell, 1986. Specificity of embryonic lethal mutations in Drosophila analyzed in germ line clones. Roux's Arch. Dev. Biol. 195: 63–73. [DOI] [PubMed] [Google Scholar]
- Wilk, R., I. Weizman and B.-Z. Shilo, 1996. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 10: 93–102. [DOI] [PubMed] [Google Scholar]
- Wilk, R., B. H. Reed, U. Tepass and H. D. Lipshitz, 2000. The hindsight gene is required for epithelial maintenance and differentiation of the tracheal system in Drosophila. Dev. Biol. 219: 183–196. [DOI] [PubMed] [Google Scholar]
- Wilk, R., A. T. Pickup and H. D. Lipshitz, 2004 Epithelial morphogenesis, pp. 277–304 in Encyclopedia of Molecular Cell Biology and Molecular Medicine, edited by R. A. Meyers. Wiley-VCH, Weinheim, Germany.
- Xu, T., and G. M. Rubin, 1993. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Yarnitzky, T., and T. Volk, 1995. Laminin is required for heart, somatic muscles, and gut development in the Drosophila embryo. Dev. Biol. 169: 609–618. [DOI] [PubMed] [Google Scholar]
- Yip, M. L. R., M. L. Lamka and H. D. Lipshitz, 1997. Control of germ-band retraction in Drosophila by the zinc-finger protein HINDSIGHT. Development 124: 2129–2141. [DOI] [PubMed] [Google Scholar]
- Yurchenco, P. D., and J. J. O'Rear, 1994. Basal lamina assembly. Curr. Opin. Cell Biol. 6: 674–681. [DOI] [PubMed] [Google Scholar]
- Zeitlinger, J., L. Kockel, F. A. Peverali, D. B. Jackson, M. Mlodzik et al., 1997. Defective dorsal closure and loss of epidermal decapentaplegic expression in Drosophila fos mutants. EMBO J. 16: 7393–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]