

Abstract
Cyclin E together with its kinase partner Cdk2 is a critical regulator of entry into S phase. To identify novel genes that regulate the G1- to S-phase transition within a whole animal we made use of a hypomorphic cyclin E mutation, DmcycEJP, which results in a rough eye phenotype. We screened the X and third chromosome deficiencies, tested candidate genes, and carried out a genetic screen of 55,000 EMS or X-ray-mutagenized flies for second or third chromosome mutations that dominantly modified the DmcycEJP rough eye phenotype. We have focused on the DmcycEJP suppressors, S(DmcycEJP), to identify novel negative regulators of S-phase entry. There are 18 suppressor gene groups with more than one allele and several genes that are represented by only a single allele. All S(DmcycEJP) tested suppress the DmcycEJP rough eye phenotype by increasing the number of S phases in the postmorphogenetic furrow S-phase band. By testing candidates we have identified several modifier genes from the mutagenic screen as well as from the deficiency screen. DmcycEJP suppressor genes fall into the classes of: (1) chromatin remodeling or transcription factors; (2) signaling pathways; and (3) cytoskeletal, (4) cell adhesion, and (5) cytoarchitectural tumor suppressors. The cytoarchitectural tumor suppressors include scribble, lethal-2-giant-larvae (lgl), and discs-large (dlg), loss of function of which leads to neoplastic tumors and disruption of apical-basal cell polarity. We further explored the genetic interactions of scribble with S(DmcycEJP) genes and show that hypomorphic scribble mutants exhibit genetic interactions with lgl, scab (αPS3-integrin—cell adhesion), phyllopod (signaling), dEB1 (microtubule-binding protein—cytoskeletal), and moira (chromatin remodeling). These interactions of the cytoarchitectural suppressor gene, scribble, with cell adhesion, signaling, cytoskeletal, and chromatin remodeling genes, suggest that these genes may act in a common pathway to negatively regulate cyclin E or S-phase entry.
REGULATION of the G1- to S-phase transition by external signals is critical to the decision to proliferate or to differentiate. Progression through G1 phase is controlled by the activity of the Cyclin-dependent ser/thr protein kinases (Cdks) associated with their regulatory Cyclin partners (Ekholm and Reed 2000). In mammalian cells, the G1 cyclins, Cyclin D (D1, D2, and D3) in association with Cdk4(6), and Cyclin E (E1 and E2) in association with Cdk2, play distinct roles in the G1- to S-phase transition. Cyclin D/Cdk4 functions early in G1, while cyclin E/Cdk2 functions at the G1- to S-phase transition, triggering DNA replication initiation and centrosome duplication. In mammalian cells, Cyclin D/Cdk4 and Cyclin E/Cdk2 act to phosphorylate and inactivate the tumor suppressor protein, Retinoblastoma (Rb; Lundberg and Weinberg 1998), which functions by binding to and inactivating the E2F/Dp transcription factor required for the transcription of S-phase genes (Dyson 1998). Binding and phosphorylation of Rb by Cyclin D/Cdk4 and Cyclin E/Cdk2 inactivate Rb, allowing the E2F/Dp transcription factor to function. G1 Cyclin-Cdks are also regulated by the binding of Cdk inhibitory proteins (Sherr and Roberts 1999), such as the p21CIP1 class of inhibitors, which bind to Cyclin E/Cdk2, inhibiting its activity and leading to G1 arrest.
The key players in the regulation of the G1- to S-phase transition are highly conserved between mammals and Drosophila (Edgar and Lehner 1996). Genetic analysis in Drosophila has shown that both Cyclin E and Cyclin D act to regulate Drosophila Rb (Rbf; Xin et al. 2002). Drosophila Cyclin E is essential for the G1- to S-phase transition during embryogenesis and is downregulated in G1-arrested cells (Richardson et al. 1993, 1995; Knoblich et al. 1994). In contrast, Drosophila Cyclin D primarily acts to regulate cell growth (increase in cell mass) and through the coupling of cell growth to G1- to S-phase progression, stimulates cell proliferation (Datar et al. 2000; Meyer et al. 2000). As in mammalian cells, Drosophila Cyclin E/Cdk2 activity is regulated via a homolog of p21CIP1, Dacapo, which is required during exit into a terminal G1 arrest prior to differentiation (de Nooij et al. 1996; Lane et al. 1996). Degradation of Cyclin E protein also plays an important role in limiting cell proliferation, and mutations in the ago gene (encoding a homolog of Cdc4, an F-box-containing component of the G1 phase ubiquitin ligase) result in increased Cyclin E protein stability and excessive cell proliferation during eye development (Moberg et al. 2001). However, relatively little is known about the upstream signals that regulate Drosophila cyclin E transcription or the downstream targets of Drosophila Cyclin E/Cdk2 that lead to the initiation of DNA replication within a whole-animal context.
The developing Drosophila eye presents an ideal system to study the relationship between cell proliferation and differentiation. The eye develops from a single cell layer epithelium at the third larval instar stage, where a wave of morphogenesis moves from the posterior to the anterior of the eye imaginal disc (Thomas and Wassarman 1999). Associated with this wave of morphogenesis is the morphogenetic furrow (MF), where the cell cycle becomes coordinated with differentiation. Within and anterior to the MF cells are arrested in G1, while posterior to the MF a subset of cells begins to differentiate into the photoreceptor cell preclusters and the surrounding cells enter a synchronous S phase, after which a subset of these cells undergoes mitosis. Hedgehog signaling has been shown to be important for Cyclin D and Cyclin E expression in this post-MF cell division (Duman-Scheel et al. 2002). Perturbations to the organized arrangement of cell division in the developing eye by, for example, ectopic expression of S-phase inducers, Cyclin E or E2F/Dp, or the negative cell cycle regulators, human p21 or Drosophila Rbf, result in defects in eye development leading to disorganized or rough adult eyes (de Nooij and Hariharan 1995; Richardson et al. 1995; Asano et al. 1996; Du et al. 1996; Xin et al. 2002). The eye phenotypes resulting from overexpression of Cyclin E, E2F/Dp, or Rbf in the posterior differentiating cells of the eye disc have been used as the basis of genetic screens of EMS-mutagenized flies to identify dominant modifiers, revealing novel regulators of the cell cycle (Staehling-Hampton et al. 1999; Boulton et al. 2000; Lane et al. 2000; Duman-Scheel et al. 2002).
A hypomorphic mutation in Drosophila cyclin E, DmcycEJP, which results in a rough eye phenotype, has provided an opportunity to carry out genetic screens to identify novel genes involved in the regulation of cyclin E expression and function. We have previously shown that DmcycEJP exhibits a rough eye phenotype due to a reduction in Cyclin E levels and S phases in the developing eye and that this phenotype is sensitive to the dosage of G1- to S-phase genes known to interact with Cyclin E (Secombe et al. 1998). This article reports the results of mutagenesis and deficiency screens to identify genes that dominantly modify the DmcycEJP rough eye phenotype and presents initial characterization of DmcycE suppressor genes, predicted to act as negative regulators of Cyclin E and/or the G1- to S-phase transition.
MATERIALS AND METHODS
Mutagenesis screen:
For X-ray mutagenesis, 3- to 5-day-old Drosophila males were placed into empty vials (∼100 in each) and treated with 4000 rad of X rays in a CIS Biointernational X-ray machine using a 137Cs radiation source (activity 3400 Ci). Mutagenized flies were then allowed to recover for 4 hr with food before being added to 3-day-old virgin females. The flies were turned into new bottles after 2 days and removed after 4 days. EMS mutagenesis was carried out as previously described (Grigliatti 1998). For both EMS and X-ray mutageneses, DmcycEJP males isogenic on the second and third chromosomes were mutagenized and crossed en masse to b DmcycEJP females. The progeny from this cross were scored for dominant modification of the DmcycEJP rough eye phenotype. In addition, F1 progeny were scored for black-bodied flies to estimate the mutation frequency. From the number of black mutant flies obtained, we calculated that the X-ray mutagenesis frequency was 2.3 × 10−3 and the EMS mutagenesis frequency was ∼3 × 10−4, which are within the ranges described by previous studies (Grigliatti 1998).
Flies selected as having enhanced or suppressed eyes were crossed to a DmcycEJP strain to ensure that the modification of the DmcycEJP rough eye phenotype observed initially was heritable and reproducible and then crossed to second or third chromosome balancers to generate stocks. To simplify the screen and stock generation, only interactors that mapped to the second or third chromosome were kept. Once a stock was generated, flies were crossed to a DmcycEJP strain to ensure that the enhancer or suppressor mutation segregated away from the balancer chromosome. Any mutations that resulted in a dominant eye roughening in the absence of DmcycEJP were discarded.
For complementation analysis, inter se crosses were carried out between all lethal alleles on each chromosome and allele combinations that resulted in trans-heterozygous lethality to >98% were considered to be within the same gene group.
Genetic mapping of second chromosome genes was carried out using either the b1, cn1, bw1 or al1, dpov1, b1, pr1, c1, px1, sp1 multiply marked chromosomes, while third chromosome genes were mapped using the ru1, h1, th1, st1, cu1, sr1, es, ca1 multiply marked chromosomes. The deficiency kit (Bloomington Stock Center) was used for deficiency mapping. Chromosome cytology of third chromosome suppressors was analyzed after Giemsa staining of polytene chromosomes prepared from non-Tubby larvae from a cross of the suppressor (over TM6B) to Canton-S.
DmcycE interactions:
To test X and third chromosome deficiencies for interaction with DmcycEJP, stocks were generated using balancers that contained the deficiency chromosome and DmcycEJP and the stock was crossed to homozygous DmcycEJP flies and progeny containing the deficiency and DmcycEJP were examined. To examine second chromosome candidate genes for interaction with DmcycEJP, the candidate gene mutant was recombined onto a marked DmcycEJP chromosome (using a dp, b, DmcycEJP, cn, bw chromosome) balanced over CyO and then crossed to a homozygous DmcycEJP stock and non-Curly flies were examined. X or third chromosome candidate genes were tested for interaction with DmcycEJP after generating stocks containing the candidate gene mutant and DmcycEJP, by crossing to b, DmcycEJP, bw flies and examining non-FM7 or non-TM6B flies. The eyes of at least 50 flies of the appropriate genotype were examined and compared with b, DmcycEJP, bw/DmcycEJP flies.
Phenotypic analysis of cyclin E suppressors:
To determine whether a suppressor was acting at the level of S-phase regulation, second chromosome modifiers were crossed to the Curly-Tubby (Cy-Tb) second chromosome balancer, which carries the Tubby dominant larval marker, and were crossed to homozygous DmcycEJP flies, and non-Tubby larvae were selected for examination of S phases by BrdU labeling. Third chromosome modifiers were balanced over TM6B (marked by Tubby) and crossed to homozygous DmcycEJP flies and non-Tubby larvae were picked for BrdU labeling. BrdU labeling was carried out as described previously (Secombe et al. 1998). Cyclin E antibody staining was carried out using a polyclonal Cyclin E antibody raised in rats, as previously described (Crack et al. 2002). To determine whether the stage of lethality was before or after the third instar larval stage, each suppressor stock balanced over Cy-Tb or TM6B was examined for the presence of any homozygous modifier (non-Tubby) larvae that survived to or beyond the third larval instar stage. Scanning electron microscopy of adult eyes was carried out as previously described (Secombe et al. 1998).
RESULTS
Identification of X and third chromosome deficiencies that dominantly modify DmcycEJP:
We have previously demonstrated that the DmcycEJP rough eye phenotype is sensitive to the gene dose of known cyclin E-interacting genes (Secombe et al. 1998). To obtain an estimate of how many interactors were expected from a random mutagenesis, available X and third chromosome deficiencies were tested to determine how many of these were able to modify the DmcycEJP phenotype.
A total of 20 suppressor regions and 16 enhancer regions on the X and third chromosomes were identified by generating homozygous DmcycEJP flies that were also heterozygous for the deficiency chromosome (Table 1). Consistent with results described previously (Secombe et al. 1998), deficiencies removing genes already known to interact with DmcycEJP such as RBF, roughex, E2F1, and string behaved as expected (Table 1), with the exception of Df(3R)vin2 that removes cyclin A. We have previously shown that cyclin A mutants dominantly enhance DmcycEJP phenotype (Secombe et al. 1998), while Df(3L)vin2 and the overlapping deficiency Df(3L)vin5 suppressed the DmcycEJP rough eye phenotype. The most likely explanation for this is that these deficiencies also delete a dose-sensitive suppressor of DmcycEJP (Table 1). Suppression of DmcycEJP was also observed with a deficiency (of the region 63F4–64C15) removing the Drosophila cdc4(ago) gene, which encodes an F-box protein of the Skp1-cullin-F-box (SCF) ubiquitin ligase complex involved in Cyclin E protein degradation and can dominantly suppress the DmcycEJP rough eye phenotype (Moberg et al. 2001).
TABLE 1.
X and third chromosome regions that modify theDmcycEJP phenotype
| Deficiency | Region removed by deficiency | Effect on DmcycEJP | Candidate genes in the region |
|---|---|---|---|
| X chromosome | |||
| Suppressors | |||
| Df(1)tBA1 | 1A1 to 2A | Suppression | Rbfa |
| Df(1)JC19 | 2F6 to 3C5 | Suppression | NC |
| Df(1)N73 and Df(1)sqh | 5D1–2 to 5D5–6 | Suppression | roughex, air4 (−) |
| Df(1)KA14 | 7F1–2 to 8C6 | Mild suppression | lawc (−), air11 |
| Df(1)vN48 | 9F to 10C3–5 | Suppression | dlg |
| Df(1)C246 | 11D–E to 12A1–2 | Suppression | BAP60b |
| Df(1)RK4 | 12F5–6 to 13A9–B10 | Suppression | NC |
| Df(1)sc72b | 13F1 to 14B1 | Suppression | NC |
| Df(1)ma13 | 19A1–2 to 20E–F | Suppression | RpS6 |
| Enhancers | |||
| Df(1)4b18 | 14B8 to 14C1 | Enhancement | NC |
| Df(1)N19 | 17A1 to 18A2 | Enhancement | fused |
| Third chromosome | |||
| Suppressors | |||
| Df(3L)HR232 and Df(3L)HR119 | 63C6 to 63D3 | Suppression | sprouty |
| Df(3L)GN24 | 63F4–7 to 64C13–15 | Suppression | cdc4 (ago)c |
| Df(3L)vin2 and Df(3L)vin5 | 68A2–3 to 68D6 | Suppression | NC 3.2 region (See Table 5) |
| Df(3L)brm11 | 71F1–4 to 72D1–10 | Suppression |
Brahma3.5 region (see Table 5) |
| Df(3L)81K19 | 73A3 to 74F | Suppression |
argos, Abl, Dab, sina, sinahd3.1 region (Table 5) |
| Df(3R)p712 | 84D4–6 to 85B6 | Suppression | pyd(Z01) |
| Df(3R)by10 | 85D8–12 to 85F1 | Suppression | hyd (−) |
| Df(3R)Cha7 | 90F1–4 to 91F5 | Suppression | NC |
| Df(3R)XS | 96A1–7 to 96A21–25 | Mild suppression | NC |
| Df(3R)T1P | 97A to 98A1–2 | Mild suppression |
l(3)mbt (−), scribble63S15 region (Table 5) |
| Df(3R)awdKRB | 100C6–7 to 100D3–4 | Suppression | tramtrack (−) |
| Enhancers | |||
| Df(3L)emc5 | 61C3–4 to 62A8 | Enhancement | emc (−), trio (S) Rac1b |
| Df(3L)RG7 | 62B8–9 to 62F2–5 | Enhancement | Roughened (Rap1) (−) |
| Df(3L)HR370 | 63A1 to 63D10 | Enhancement | NC |
| Df(3L)GN50 | 63E1–2 to 64B17 | Enhancement | cdc2-63E (−), Ras64Bb, RfC40 |
| Df(3L)66CG28 | 66B8–9 to 66C10 | Enhancement | DNApolα50b |
| Df(3L)hi22 | 66D10–11 to 66E1–2 | Enhancement | h (−), dally (−), mcm7b |
| Df(3L)AC1 | 67A2 to 67D7–13 | Enhancement | shc, eif-4E, cdk8b |
| Df(3L)Cat | 75B8 to 75F1 | Enhancement | Replication-deficient regionb |
| Df(3L)rdgC | 77A1 to 77D1 | Enhancement | DNA primase |
| Df(3L)PcMK | 78A3 to 79E1 | Enhancement | cyclin H DNApol-etab |
| Df(3R)Tp110 | 83C1–2 to 84B1–2 | Enhancement | plx |
| Df(3R)eN19 | 93B to 94A | Enhancement | E2F1 |
| Df(3R)crbS874 | 95E8–F1 to 95F15 | Enhancement | crb |
| Df(3R)3450 | 98E3 to 99A6–8 | Enhancement | string |
Where deficiencies that overlap have the same effect on DmcycEJP, the region common to both deficiencies is given as the cytological region. Candidate genes that are underlined have the same effect on the DmcycEJP phenotype as the corresponding deficiency. Those indicated by (−) have been tested and shown to have no effect on DmcycEJP, while those indicated by (S) have been shown to suppress rather than enhance. NC, there was no candidate satisfying our criteria in the interval. Gene descriptions are as follows: Rbf, Retinoblastoma; roughex (inhibitor of Cyclin A/Cdk1 in the MF); air4, aberrant immune response 4 (blood cell tumor suppressor); lawc (enhancer of TxG mutants); air11, aberrant immune response 11 (blood cell tumor suppressor); dlg, discs-large (cytoarchetectual protein, neoplastic tumor suppressor); BAP60, Brahma-associated protein 60 (Brahma complex protein, chromatin remodeling); RpS6, Ribosomal protein S6 (translation factor, tumor suppressor); fused (protein kinase required for Hh signaling); sprouty (acts antagonistically to the Egfr); brahma (SWI2-related ATPase, chromatin remodeling, negative growth regulator); argos (Egfr ligand, anatogonist of Egfr signaling); Abl (nonreceptor tyrosine protein kinase); dab, disabled (acts synergistically with Abl); sina and sinah (ring finger E3 ubiquitin ligases, protein degradation); pyd, ZO1, and tamou (membrane-associated guanylate kinase); hyd, hyperplastic discs (HECT domain E3 ubiquitin ligase, protein degradation); l(3)mbt, lethal (3) malignant brain tumor (translation factor, tumor suppressor); scribble (cytoarchitectual protein, neoplastic tumor suppressor); tramtrack (neural differentiation inhibitor); emc, extra-macrochaetae (Id-related HLH repressor protein required for cell proliferation in the wing and with hairy for MF progression in the eye); trio (Rac-GEF, required for Rac activation); Rac1 (Rac family GTPase); Roughened (Rap1; Ras-like GTPase); cdc2-63E (cdc2-related protein kinase); Ras64B (Ras-related); RfC40, Replication factor-C40 (DNA replication initiation); DNApolα50, DNA polymerase-α 50-kD subunit (DNA replication); h, hairy (see emc); dally (glypican, cooperates with Wg and other growth factor receptors); mcm7, minichromosome maintenance 7 (DNA replication initiation); shc (adaptor protein required for Egfr signaling); eif-4E (translational initiation factor); cdk8 (cdc2-related protein kinase); DNA primase (DNA replication); cyclin H (Cyclin required for activation of cdk8 protein kinase); DNApol-eta (DNA replication); plx, pollux (a cell adhesion protein related to the human oncogene TRE17; Zhang et al. 1996); E2F1 (S-phase transcription factor); crb, crumbs (apical-lateral membrane protein involved in cell polarity); and string (Cdc25 phosphatase, activator of Cdc2). An unidentified gene essential for DNA replication is located within the 75B8–75F1 region (Smith et al. 1993). For more details see text.
Specific alleles of RBF have been shown to suppress the DmcycEJP phenotype and overexpression of RBF enhances the DmcycEJP rough eye phenotype (Secombe et al. 1998; our unpublished data).
No specific mutation is available to test the interaction.
cdc4 (ago) alleles have been shown to suppress DmcycEJP (Moberg et al. 2001).
sina alleles showed only weak dominant suppression of the DmcycEJP rough eye phenotype; however, a sina-related gene is located next to sina (sinah), and removal of both may account for the suppression observed by the deficiency (M. Coombe, L. Quinn, R. Dickins, J. Secombe and H. Richardson, unpublished results).
For the remaining DmcycEJP interacting deficiencies, the regions were searched for possible candidate modifying genes using the cytosearch function at Flybase. Candidate cyclin E-interactors were genes expected to either promote S-phase entry for enhancers or inhibit S-phase entry for suppressors. These include homologs of tumor suppressors or oncogenes, genes involved in the initiation of DNA replication, in ubiquitin-mediated degradation pathways, or in chromatin remodeling (Table 1). Genes involved in chromatin remodeling were considered candidates, on the basis of the observation in mammalian cells that components of the SWI/SNF-Brahma chromatin remodeling complex negatively regulate cell proliferation (Harbour and Dean 2001). In a number of cases, specific mutations in these candidate genes were tested for modification of the DmcycEJP phenotype. This approach enabled the identification of a number of novel cyclin E-interacting genes (Table 1, and see below). For the most part, however, identification of candidates within the modifying deficiency, based on the expected classes of interactors, was not successful. Of the 36 regions that modify DmcycEJP, candidate genes for only 12 of these were shown to modify DmcycEJP in a way that would account for the modification by the deficiency. In addition to Rbf1, roughex, ago (cdc4), E2F1, and string discussed above, discs-large (dlg), RpS6, brahma, sina, Abl, scribble, and crumbs were identified in this way and are discussed in detail below. Many interactors did not have an obvious candidate gene within the deficiency breakpoints, or possible candidates were tested but did not interact with cyclin E, or specific mutations were not available in the candidate genes.
Tumor suppressors and oncogenes:
From the DmcycEJP deficiency screen, a number of regions that showed suppression removed Drosophila tumor suppressor genes, while many that enhanced removed potential oncogenes (Table 1). These candidate genes, as well as other potential oncogenes or tumor suppressors, were specifically tested where possible (Table 2).
TABLE 2.
Interaction of tumor suppressor and signaling pathway mutants withDmcycEJP
| Gene: allele | Function | Effect on DmcycEJP |
|---|---|---|
| Tumor suppressors | ||
| dlg: dlg6 | Cell polarity | Suppression |
| l(3)mbt: l(3)mbtE2 | No effect | |
| brat: l(2)37Cf1 | Translation | No effect |
| air2 | No effect | |
| air4 | No effect | |
| air6 | Mild suppression | |
| air7 | Suppression | |
| air8 (RpS6) | Translation | Suppression |
| air10 | Suppression | |
| air13 | No effect | |
| hop-air (activated allele of Jak) | Mitogenic signaling | Suppression |
| air16 | Mild suppression | |
| lats/warts: latsP1,91 | Protein kinase | No effect |
| hyd: hyd15 | HECT domain Ubiquitin ligase | No effect |
| fat: fat4,G-rv | Atypical Cadherin | Suppression |
| expanded: ex01270 | FERM domain 4.1 superfamily | Suppression |
| shotgun: shgk03401,2 | E-cadherin | Mild suppression |
| Negative regulators of signaling pathways | ||
| patched: ptcIIIa,G12 | Inhibitor of Hh signaling | Suppression |
| Gap1: Gap11 | Ras-GAP—inactivates Ras | Suppressed |
| yan/aop: aop1 | Inhibitor of Ras pathway—Pnt transcription upregulation | No effect |
| axin: axnE77 | Inhibitor of Wg signaling | Mild enhancement |
| Positive regulators of signaling pathways | ||
| Egfr-topQY1—hypomorphic | Receptor tyrosine protein kinase | LOF—enhancement |
| ElpB1—ligand independent activated allele | (RTK) | GOF—suppression |
| Ras85D: Ras85e1b | Effector of Egfr signaling | Enhancement |
| drk: drk10626 | Effector of Egfr signaling | Slight enhancement |
| pointed: pnt7825 | Transcription factor—downstream of Ras signaling | Slight suppression |
| rhoA: rhoA72R,720 | Actin cytoskeleton reorganization | Enhancement |
| Roughened (Rap1): Rap1CD3 | Actin cytoskeleton reorganization | LOF—no effect |
| Notch: N264-39 | Signaling protein | No effect |
| hedgehog: hh18,21,AC | Signaling protein | Mild enhancement |
| wingless: wgIL1-8,1-17 | Signaling protein | Mild suppression |
| armadillo: armYD35 | Transcriptional factor mediator of Wg signaling | Suppression |
| disheveled: dsh3,6 | Mediator of Wg signaling | Mild suppression |
| spen (poc): poc261-18,361-6 | RNA-binding protein | Suppression |
LOF, loss of function; GOF, gain of function.
Deficiencies removing potential oncogenes that enhanced DmcycEJP include those removing a Ras-like GTPase Rac1 (61C3–4; 62A8), a Rap-related GTPase Roughened/Rap1/dRas3 (62B8–9; 62F2–5), and a Ras-like GTPase Ras64B (63E1–2; 64B17). Loss-of-function mutations in Rap1 did not affect DmcycEJP (not shown) and we have not yet tested Rac1. However, we have shown that trio, which encodes a Rac activator, dominantly suppresses DmcycEJP (see below). Moveover, Rac2, which plays a redundant role with Rac1, was isolated in a screen for genes that when overexpressed inhibit cell proliferation in the Drosophila eye, which was rescuable by ectopic expression of Cyclin E (Tseng and Hariharan 2002). Taken together these data suggest that Rac is a negative regulator of G1-S progression in Drosophila and thus it is unlikely that halving the dosage of Rac1 accounts for the dominant enhancement of the 61C3–4 to 62A8 region. We were also unable to test Ras64B since there were no available mutants in this gene. However, we tested whether mutants in other oncogenic GTPases, Ras85D and Rho1, could enhance DmcycEJP (Table 2). Ras has a well-established role in oncogenesis in mammalian cells (Malumbres and Barbacid 2003) and overexpression of an activated form of Ras85D in Drosophila results in a hyperplastic phenotype (Karim and Rubin 1998). Ras85D has also been shown to increase Cyclin E protein levels post-transcriptionally in the wing and eye discs (Prober and Edgar 2000; Brumby and Richardson 2003). Consistent with its expected role as positive regulator of G1-S progression, mutations in Ras85D dominantly enhanced the DmcycEJP rough eye phenotype (Table 2; data not shown). In mammalian cells, Rho promotes cell proliferation and is required for Ras-induced transformation (Sahai and Marshall 2002). Indeed, overexpression of wild-type and dominant active forms of mammalian Rho have been shown to upregulate Cyclin E/Cdk2 activity and induce progression from G1 into S phase. Although, no role for Rho1 has been revealed in G1-S progression in Drosophila, we observed that mutants in rho1 dominantly enhanced the DmcycEJP rough eye phenotype (Table 2; data not shown), revealing a novel role for Drosophila Rho1 that warrants further investigation.
In addition, a deficiency removing fused (17A1–18A2), an effector of the Hh pathway, showed enhancement of DmcycEJP. Although we have not specifically tested fused to determine whether it represents the interacting gene, this interaction is consistent with the recent observation that the Hedgehog (Hh) pathway acts to upregulate cyclin E transcription in the eye (Duman-Scheel et al. 2002) and that upregulation of the Hh pathway is oncogenic in mammals (Wetmore 2003). To explore this further, we analyzed the effect of halving the dose of Hh or patched (a negative regulator of the Hh receptor, Smoothened) on the DmcycEJP rough eye phenotype (Table 2; data not shown). As expected, Hh alleles dominantly enhanced while patched alleles dominantly suppressed DmcycEJP, consistent with a role for the Hh pathway in positively regulating cyclin E and inducing S-phase entry. We also examined other signaling pathways for dominant interactions with DmcycEJP (Table 2; data not shown). In mammalian cells, the EGF receptor, the Wnt/Wingless, and Notch signaling pathways have a growth and/or cell cycle stimulatory role in many cells and can be oncogenic when upregulated (Allenspach et al. 2002; Chang et al. 2003; Giles et al. 2003). Consistent with the interaction of Ras85D with DmcycEJP, loss-of-function mutations in the EGF receptor (Egfr) enhanced DmcycEJP while gain-of-function mutations (Ellipse) suppressed. Other downstream components of the Egfr-Ras pathway also interacted with DmcycEJP in a manner consistent with the Egfr having a positive role in regulating Cyclin E and entry into S phase (Table 2). Reducing the dose of Notch, however, showed no effect on the DmcycEJP phenotype. Interestingly, halving the dosage of wingless (wg), disheveled (encoding a Wg-signaling mediator), and armadillo (arm; encoding a β-catenin homolog, the Wg signaling transcriptional effector) resulted in suppression of DmcycEJP. In contrast, halving the dosage of axin (encoding an inhibitor of Wg signaling) enhanced DmcycEJP. While contrary to the expected role of the Wg pathway, an inhibitory proliferative function for Wg has been observed in the zone of nonproliferation in the third instar wing pouch (Johnston and Edgar 1998; Johnston et al. 1999; Johnston and Sanders 2003). Similarly, we have previously shown that the Dpp (TGFβ homolog), although growth stimulatory earlier in development, acts to negatively regulate cell cycle progression in the third instar eye imaginal disc and mutants that disable the Dpp signaling pathway dominantly suppress DmcycEJP (Horsfield et al. 1998).
Several DmcycEJP suppressor regions on the X chromosome and on the third chromosome remove known Drosophila tumor suppressor genes. Specific mutations were available for some of the candidate genes encoding tumor suppressors and were therefore tested for a genetic interaction with cyclin E. Specific mutations in Ribosomal protein S6 (RpS6 air8), the best candidate for the cyclin E suppressor in the 19A–20F region, were tested and shown to suppress the DmcycEJP rough eye phenotype (Table 2; not shown). Mutations in RpS6 were identified as loss-of-function mutations that result in overproliferation of larval hematopoietic tissues and give rise to variable melanotic tumor phenotypes (Gateff et al. 1996). RpS6 is phosphorylated in response to mitogen stimulation and phosphorylated RpS6 is preferentially incorporated into polysomes, resulting in an increased rate of translation of a subset of transcripts (Amaldi and Pierandrei-Amaldi 1997; Martin and Blenis 2002). However, disruption of Drosophila S6 kinase leads to reduced growth and smaller flies and mutation of the upstream kinase Tor causes cell cycle arrest that can be rescued by cyclin E expression (Zhang et al. 2000). Furthermore, conditional knockout of RpS6 in mice results in a specific block in cyclin E expression (Volarevic et al. 2000). Given this role for RpS6 in mammalian cells, it is unknown how halving the dosage of RpS6 leads to the suppression of DmcycEJP; however, it is consistent with the tumor suppressor function of Drosophila RpS6.
Other Drosophila tumor suppressors were tested for interaction with DmcycEJP (Table 2), and those that showed suppression were hop-air (an activating mutation in JAK kinase), consistent with a role for Drosophila Jak in cell proliferation and that Cyclin D-Cdk4 and Cyclin E-Cdk2 bind and regulate STAT92E protein stability (Chen et al. 2003); fat (encoding an atypical Cadherin involved in planar polarity); expanded (encoding a FERM domain protein involved in actin remodeling); and the unidentified air7, air10, and air16 (Gateff et al. 1996; de Lorenzo et al. 1999). The Drosophila E-cadherin gene, shotgun (shg; Tepass et al. 1996; Uemura et al. 1996), when halved in dosage, was also shown to slightly suppress DmcycEJP. In contrast, lethal (3) malignant brain tumor [l(3)mbt] and hyperplastic discs (hyd; Gateff et al. 1996; de Lorenzo et al. 1999), which were considered candidates for the regions 97A–98A2 and 85D8–85E13, respectively (Table 1), did not modify the DmcycEJP phenotype when specific alleles were tested (Table 2; data not shown). Taken together these data suggest that there are specific pathways that show rate-limiting effects on Cyclin E and thereby entry into S phase, in the eye imaginal disc.
Identification of cyclin E interactors using a mutagenic dominant modifier screen:
As described above, screening for dominant genetic modifiers of DmcycEJP using deficiencies and candidate gene approaches has revealed some interesting interactors. However, this approach is limited in that the deficiencies may remove more than one modifier, confounding the identification of interacting genes. For these reasons, an unbiased genetic screen for DmcycEJP modifiers using mutagenized flies was carried out, to generate specific modifier mutations that could be further characterized. To randomly generate mutations that could then be examined for their effect on the DmcycEJP phenotype, we utilized X-ray mutagenesis, which causes deletions and chromosomal rearrangements (Sankaranarayanan and Sobels 1976) that are expected to aid in the identification of the modifier, and EMS mutagenesis, which causes nucleotide substitutions resulting in missense or nonsense mutations (Lifschytz and Falk 1968). For the X-ray mutagenesis, 39,234 F1 flies were screened for modification of the DmcycEJP rough eye phenotype and stocks of 104 suppressors and 59 enhancers that consistently modified the DmcycEJP phenotype on the second or third chromosomes were generated (summarized in Table 3). For the EMS mutagenesis a total of 15,049 F1 flies were screened and 29 suppressors and 54 enhancer mutations on the second or third chromosomes were isolated (Table 3).
TABLE 3.
Summary of the 246 modifiers identified in the screen
| Second chromosome
|
Third chromosome
|
|||
|---|---|---|---|---|
| Homozygous lethal |
Homozygous viable |
Homozygous lethal |
Homozygous viable |
|
| Suppressors | ||||
| EMS | 19 | 5 | 5 | 0 |
| X ray | 60 | 12 | 26 | 3 |
| Enhancers | ||||
| EMS | 13 | 1 | 38 | 2 |
| X ray | 13 | 8 | 29 | 9 |
Summary of the number of homozygous viable and homozygous lethal second and third chromosome modifiers obtained from the EMS and X-ray mutageneses. Not included are three X-ray-generated suppressor mutations likely to be translocations to the Y, for which it was not possible to know whether they were homozygous viable or lethal.
DmcycEJP suppressor complementation groups:
For the second chromosome homozygous lethal suppressors, complementation analysis revealed that there were 10 complementation groups containing more than one allele, as well as many with single alleles (Tables 4 and 5; and data not shown). In addition, these stocks were crossed to a number of alleles on the second chromosome identified in the screens for enhancers of the eye phenotypes generated by overexpression of cyclin E or E2F1/Dp (Staehling-Hampton et al. 1999; Lane et al. 2000). This analysis revealed that 62S9 was allelic to E(sev-cycE)e93 (and was termed group 2.11). Further analysis revealed that some members of group 2.6 contained a second lethal allele that was distinct from the lethal common to group 2.6 members, forming two new groups, 2.12 (containing the 2.6 allele, 42S13, and a single allele 22S9) and 2.13 (containing the 2.6 alleles 42S14 and 66S4 and the 2.7 allele 55S2). Thus there were a total of 13 second chromosome suppressor groups with multiple members. For the third chromosome suppressors, complementation analysis revealed that there were 5 groups containing >1 allele, and there were many single alleles (Tables 4 and 5). Groups 3.3 and 3.4, however, cannot truly be considered as groups with more than one allele as there were only two members in each and they both contained a common member, 65S55, which appears to contain a large deletion. The suppression of the DmcycEJP adult eye phenotype by representatives of the identified suppressor groups is shown in Figure 1.
TABLE 4.
Summary of identified suppressors
| Group | Alleles | Cytological location | Stage lethal | Identified genes |
|---|---|---|---|---|
| Second chromosome | ||||
| 2.1 | 23S9, 27S3, E2S31, E6S2 | Genetically linked to aristaless (21C) | Third larval instar | lethal-2-giant larvae (lgl) |
| FTC: Df(2L)net-PMF | ||||
| C: Df(2L)al | ||||
| 21A1–B8 | ||||
| 2.2 | 28S2, 38S4, 39S2 | Genetically between cinnabar (43E) and brown (59E) |
Embryonic | phyllopod |
| FTC: Df(2R)trix | ||||
| C: Df(2R)3072r and Df(2R)CX1 | ||||
| 51A1–5 | ||||
| 2.5 | 42S11, 58S12 | FTC: Df(2R)nap9 and Df(2R)ST1 | Pupal | l(2)04524 dEB1 |
| C: Df(2R)nap1 and In(2R)pk78s | ||||
| 42B3–42C7 | ||||
| 2.11 | 62S9, E(sev-cycE)e93 | FTC: Df(2R)Jp1 and Df(2R)XTE18 | Embryonic |
l(2)01288 scab (α-integrin) |
| C: Df(2R)03072 and Df(2R)Jp4 | ||||
| 51D3–51F13 | ||||
| 2.12 | 42S13, 22S9 | FTC: Df(2L)TW137, Df(2L)H20, and Df(2L)M36F-55 |
Larval/pupal | cadNa |
| C: Df(2L)TW50 and Df(2L)TW3 | ||||
| 36D1–E4 | ||||
| Third chromosome | ||||
| 3.5 | 25S14, E6S8 | FTC: Df(3R)brm11b | Embryo | brahma |
| C: Df(3R)BK10 and Df(3R)st-f13 | ||||
| 71F1–72D1 | ||||
| 2S1 | FTC: Df(3L)emc5c | Before third larval instar |
trio (Rac GEF) | |
| C: Df(3L)emc-E12, Df(3L)Ar11, and Df(3L)RG5 | ||||
| 61E–62A8 | ||||
| 35S1 | Genetically between stripe (86D1) and curled (90F7) |
Embryo | moira | |
| FTC: Df(3R)sbd-105d | ||||
| C: Df(3R)ea and Df(3R)PO4 | ||||
| 89A11–89B10 | ||||
| 43S2 | Genetically left of scarlet (73A4) | Pupal | l(3)72Dk zn72D | |
| FTC: Df(3L)st-f13,d Df(3L)st-g24,d and Df(3L)th102d | ||||
| C: Df(3L)st-b11 and Df(3L)brm11 | ||||
| 72D1–72D10 | ||||
| 63S15 | FTC: Df(3R)T1P,b Df(3R)Tl-X, and Df(3R)Tl-I | Third larval instar | scribble | |
| C: Df(3R)XTAI and Df(3R)3450 | ||||
| 97B–97D2 | ||||
| 65S19 | Genetically between veinlet (62A) and thread (72D) |
Semilethal | trithorax-like | |
| FTC: Df(3L)fz-M21d | ||||
| C: Df(3L)fz-GR3b and Df(3L)BK10 | ||||
| 70D4–71C3 | ||||
Underlined alleles are members of more than one group. FTC, failed to complement; C, complemented.
Not confirmed by testing specific allele for suppression of DmcycEJP.
Deficiency also suppressed DmcycEJP (Table 1 and data not shown).
Deficiency did not suppress and in fact enhanced DmcycEJP (see Table 1).
Deficiency did not suppress DmcycEJP (data not shown).
TABLE 5.
Summary of unidentified suppressors
| Group | Alleles | Cytological location | Effect on DmcycEJP eye disc S phases |
Stage lethal |
|---|---|---|---|---|
| Second chromosome | ||||
| 2.3 | 59S16, 65S12 | Genetically between black (34D) and cinnabar (43E) |
Increased | Pupal |
| FTC: Df(2L)TW137, Df(2L)TW50, Df(2L)E71, Df(2L)TW3 and Df(2L)OD15 | ||||
| C: Df(2L)H20 and Df(2L)PR-A16 | ||||
| 36F7–37B8 | ||||
| 2.4 | 26S8, 57S6, 59S3 | Genetically between black (34D) and cinnabar (43E) |
Increased | Postembryonic |
| FTC: In(2R)bwVDe2L and Df(2R)nap1 | Before third larval instar |
|||
| C: Df(2R)nap9 | ||||
| 41D2–42A2 | ||||
| 2.6 |
41S1, 42S7, 42S13, 42S14, 65S4, 66S4, 67S7, E3S17, E3S18, E3S31 |
Unknown | Increased | Larval/pupal |
| 2.7 |
55S2, 64S19, 65S39 (14S3, 57S1, 65S23, E10S15) |
FTC: Df(2R)M60E | Increased | Before third instar larvae |
| C: Df(2R)ES1 | ||||
| 60E2–8 | ||||
| 2.8 | E6S4,a E6S19 | FTC: Df(2L)Dwee1Δ5 and Df(2L)spdj2 | Increased | Larval lethal |
| C: Df(2L)Dwee1w05, Df(2L)J-H, and Df(2L)E110 | ||||
| 27B2–C3 | ||||
| 2.9 | 25S11, E1S4 | FTC: Df(2R)X58-12 and Df(2R)X58-8 | Increased | Postembryonic |
| C: Df(2R)X58-7, Df(2R)59AD, and Df(2R)pu-D17 |
Before third larval instar |
|||
| 58E4–59A | ||||
| 2.10 | 65S5, 65S13, E10S34b | FTC: In(2R)bwVDe2lCyR | Increased | Embryonic |
| C: Df(2R)M41A4 and Df(2R)nap9 | ||||
| 41A–41E1 | ||||
| 2.13 | 42S14, 66S4, 55S2 | FTC: Df(2R)M60E | Increased | Larval/pupal |
| C: Df(2R)ES1 | ||||
| 60E2–8 | ||||
| Third chromosome | ||||
| 3.1 | 19S5, 24SX, 58S5, 62S2 | Genetically left of thread (72D) | Increased | Larval |
| FTC: Df(3L)81k19d | ||||
| C: Df(3L)st-b11 and Df(3L)W10 | ||||
| 73D–74F | ||||
| (male recombination mapping to 74B1–B4) | ||||
| 3.2 |
41S13, 44S18, 59S26, 65S1, E6S25, E8S13 |
FTC: Df(3L)lxd6, Df(3L)vin5,d and Df(3L)vin2d |
Increased | Pupal |
| C: Df(3L)vin7 and Df(3L)AC1 | ||||
| 68A9–68B3 | ||||
| 3.3 | 34S3, 65S55 | Genetically between veinlet (62A) and hairy (66D10) |
Increased | Larval |
| 62A–66D10e | ||||
| 3.4c | 1S2 | Genetically left of scarlet (73A3) | Increased | Before third instar larval |
| 65S55 | FTC: Df(3L)29A6f and Df(3L)Rd1-2d | |||
| C: Df(3L)AC1 | ||||
| 66F5 | ||||
| Breakpoint at 66F | ||||
| Third chromosome, single alleles | ||||
| 1S3 | Genetically right of ebony (93D2) | Increased | Before third instar | |
| FTC: Tp(3;Y)J55 | larval/pupal | |||
| C: Df(3R)T1-P, Df(3R)D605, Df(3R)3459, Df(3R)Dr-rv1, Df(3R)L127, and Df(3R)B81 | ||||
| 98A–100B | ||||
| (98A5–98E3) | ||||
| Breakpoint at 98C | ||||
| 13S1 | Genetically right of curled (86D1) | ND | Before third instar | |
| FTC: Df(3R)Dl-BX12f | larval | |||
| C: Df(3R)Cha7, Df(3R)KX18, and Df(3R)FX3 | ||||
| 91F5–91F11 | ||||
| 20S1 | Genetically to the left of hairy (66D10) |
Increased | Before third instar larval |
|
| 61A–66D10e | ||||
| Breakpoint at 63E | ||||
| 42S12 | Genetically to the left of hairy 66D10) |
Increased | Before third instar larval |
|
| 61A–66D10e | ||||
| Breakpoint at 62E/F | ||||
| 42S33 | Genetically to the left of thread (72D) |
Increased | Second instar larval | |
| 61A–72De | ||||
| 43S1 | Unknown | Increased | Before third instar larval |
|
| 47S8 | Unknown | Increased | Before third instar larval |
|
| 59S9 | Genetically between veinlet (62A) and thread (72D) |
Increased | Semilethal | |
| FTC: Df(3L)RG7g | ||||
| C: Df(3L)Aprt-1 and Df(3L)M21 | ||||
| 62D2–62F5 | ||||
| Breakpoint at 62B | ||||
| 59S18 | FTC: Df(3L)RG5f | Increased | Before third instar larval |
|
| C: Df(3L)Aprt-1, Df(3L)Aprt-32, and Df(3L)RG7 | ||||
| 62A10–62B1 | ||||
| 68S10 | FTC: Df(3R)3-4f | Increased | Before third instar | |
| C: Df(3R)110 and Df(3R)e1025-14 | ||||
| 82F3–82F10 | ||||
| E9S1 | Genetically between veinlet (62A) and thread (72D) |
Increased | Pupal | |
| 62A–72De | ||||
Underlined alleles are members of more than one group. 55S2 is a member of 2.7 and 2.13, but other 2.7 alleles complement the 2.13 alleles, 42S14 and 66S4. Weak alleles are in parentheses. These gave escapers that showed the Rehow phenotype with other 2.7 alleles, although they failed to complement Df(2R)M60E. FTC, failed to complement; C, complemented; ND, not determined.
E6S4 also contains another lethal at 29D1–2 to 30C4–D1.
All three alleles failed to complement each other, but 65S13 and E10S34 were not completely lethal over the deficiency and gave rise to escapers with rough eyes and wing defects.
Mapping data are for 1S2.
Deficiency also suppressed DmcycEJP (Table 1 and data not shown).
The given cytological interval was determined only by genetic mapping of the lethal. No deficiencies uncovering this mutant were identified by deficiency mapping.
Deficiency did not suppress DmcycEJP (data not shown).
Deficiency did not suppress but rather enhanced DmcycEJP (see Table 1).
Figure 1.—
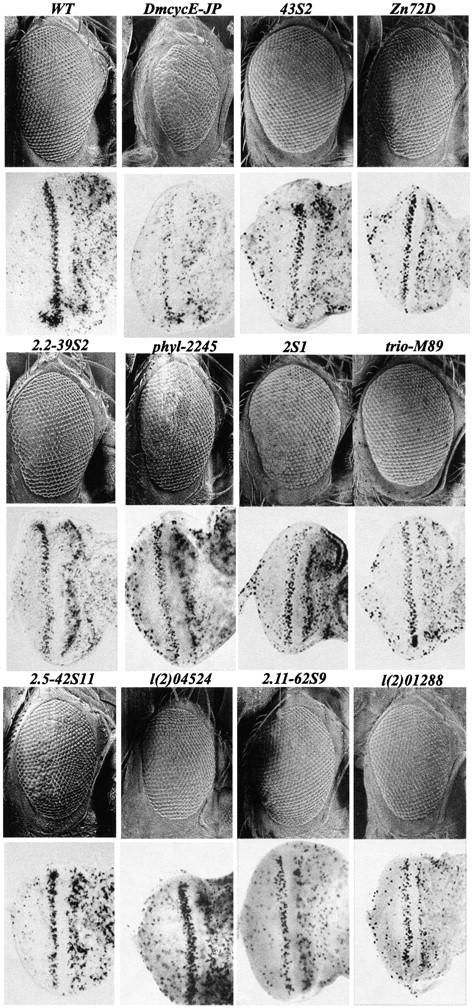
The identified dominant suppressors of DmcycEJP: scanning electron-micrographs of adult eyes and BrdU labeling of eye imaginal discs from DmcycEJP individuals heterozygous for the identified suppressor alleles. Genotypes are as indicated: wild type (WT); DmcycEJP; DmcycEJP; 43S2/+; DmcycEJP; zn72D/+; DmcycEJP, 2.2-39S2/+; DmcycEJP, phyl2245/+; DmcycEJP; 2S1/+; DmcycEJP; trioM89/+; DmcycEJP, 2.5-42S11/+; DmcycEJP, dEB1-l(2)04524/+; DmcycEJP, 2.11-62S9/+; and DmcycEJP, 2.11-l(2)01288/+.
Complementation crosses revealed genetic interactions between many DmcycEJP modifiers. Some alleles when trans-heterozygous showed reduced numbers and/or a striking phenotype characterized by rough eyes, held-out wings, and poor viability [the rough eyes and held-out wings (Rehow) phenotype] and in some cases the trans-heterozygous females were sterile. The Rehow phenotype occurred between the severe group 2.7 alleles (55S2, 64S19, and 65S39) and the weak 2.7 alleles (65S23, E10S15) or the single alleles 19S3, 40S5, 42S3, 64S10, 61S10, or 62S9 (group 2.11). These single alleles also showed the Rehow phenotype when crossed with each other. The trans-heterozygous Rehow phenotype of 61S10 and the severe 2.7 alleles was dependent on the presence of the DmcycEJP mutant, since it was observed only when DmcycEJP was homozygous. For 55S2, 64S19, 65S39, 19S3, 40S5, 42S3, 62S9, 65S23, E10S15, or 64S10, the Rehow phenotype occurred independent of DmcycEJP homozygosity (in the background of DmcycEJP/+). Crosses between the third chromosome single-allele suppressors; 47S8 and 59S9, 20S1, 63S15, or 65S19; also gave rise to the Rehow phenotype. In these cases, genetic and deficiency mapping data suggest that none of these alleles are weak alleles of the same complementation group.
Basic characterization was then carried out on second chromosome multimember complementation groups and most of the third chromosome suppressors (summarized in Tables 4 and 5). We determined whether the suppression of DmcycEJP was occurring at the level of S phases during eye development (shown for representatives of the identified suppressors in Figure 1). In all cases examined, there was a significant increase in the size of the eye disc as well as in the number of S phases in the anterior and the post-MF S-phase band. Thus all suppressors tested act to suppress DmcycEJP by increasing S phases in the normal pattern. The stage of lethality of the homozygous modifier mutation was also determined by counting the number of hatched embryos and examining whether homozygous third instar larvae were present (Tables 4 and 5). This analysis led to the observation that group 2.1 and 63S15 homozygotes died as overgrown larvae, a phenotype that occurs with Drosophila neoplastic tumor suppressors (Gateff et al. 1996; de Lorenzo et al. 1999; see below).
Mapping and identification of DmcycEJP suppressors:
The cytological location of the lethal mutation for the complementation groups and some of the single alleles was determined by crossing suppressors to the deficiency collection (Bloomington Stock Center). In addition, a crude map position was determined for most of the third chromosome interactors and some of the second chromosome interactors by genetic mapping of the DmcycEJP suppressor mutation. In all cases tested, the map location of the suppressor by genetic mapping was consistent with the map location of the lethal by deficiency mapping. In some cases, chromosome cytology was examined to map aberrations (Tables 4 and 5). Knowledge of the location of the modifier gene then enabled likely candidate genes to be investigated by testing mutant alleles, where available, for failure to complement the modifier mutant.
This strategy enabled the identification of 5 of the 13 second chromosome (2.1, 2.2, 2.5, 2.11, and 2.12) and 6 of the 20 third chromosome suppressors (3.5, 2S1, 35S1, 43S2, 63S15, and 65S19; summarized in Table 4). Of the remaining groups, although map positions were well defined for 7 of 13 of the second (2.3, 2.4, 2.7, 2.8, 2.9, 2.10, 2.13) and 8 of 20 third chromosome suppressor groups (3.1, 3.2, 3.4, 1S3, 13S1, 59S9, 59S18, 68S10), and available candidate gene alleles were tested for each of the suppressor genes, the identity of the suppressors is not yet known (Tables 5 and 7). In these cases, it is likely that these suppressor mutations define novel genes. For two of the suppressor groups (2.6 and 3.3) and five of the single alleles (20S1, 42S12, 42S33, 43S1, 47S8) a precise location for the suppressor was not determined, since none of the available deficiencies failed to complement the suppressor. In these cases the lethal mutation must map to a region not covered by the deficiency collection. A brief description of the identification of the more precisely localized suppressors is detailed below and summarized in Tables 4 and 5.
The identified suppressors:
By complementation tests to known gene alleles the identities of five second chromosome suppressors (2.1, 2.2, 2.5, 2.11, 2.12) and six third chromosome suppressors (3.5, 2S1, 35S1, 43S2, 63S15, 65S19) were revealed. These suppressor genes fall into the functional groups of chromatin remodeling and transcription factors (four genes), signaling (two genes), cytoskeletal (one gene), cell adhesion (two genes), and neoplastic tumor suppressors (two genes). Specific details on the verification and characterization of these suppressors are discussed under these functional groupings (summarized in Table 4).
Chromatin remodeling and transcription factor genes:
3.5. (Brahma):
3.5 was mapped to 71F1–72D1 (Table 4) and alleles of brahma, a SWI2 homolog, encoding a component of the SWI/SNF chromatin-remodeling complex (Papoulas et al. 1998), failed to complement both 3.5 alleles. Furthermore, Df(3R)brm11 (71F1–4; 72D1–10) was identified as a dominant suppressor of DmcycEJP in the screen of third chromosome deficiencies (Table 1). Consistent with suppressor 3.5 being brahma, we showed that previously isolated alleles of brahma also dominantly suppressed DmcycEJP (Brumby et al. 2002).
35S1 (Moira):
35S1 was mapped to 89A11–89B10 (Table 4). Candidate mutants in this region were tested for allelism with 35S1, and alleles in moira, a SWI3 (BAP155) homolog, failed to complement 35S1. Consistent with the suppressor 35S1 being moira, we demonstrated that previously isolated alleles of moira also dominantly suppressed DmcycEJP (Brumby et al. 2002).
Brahma and Moira are components of the Drosophila Brahma (SWI/SNF-related) chromatin remodeling complex (Papoulas et al. 1998), which has been shown to play a role in negatively regulating S phase (Staehling-Hampton et al. 1999; Harbour and Dean 2000). Consistent with this notion, alleles of other Brahma complex genes, snr1 and osa, as well as a deficiency that removes the brahma-associated protein 60 (BAP60) or BAP111, dominantly suppress the DmcycEJP phenotype (Brumby et al. 2002; Table 1).
65S19 (Trithorax-like):
65S19 was only semilethal; however, genetic and deficiency mapping was still possible, and 65S19 was located to 70D4–71C3 (Table 4). Complementation tests of candidate genes in the region revealed that Trithorax-like (Trl) was allelic to 65S19. Consistent with this, previously characterized alleles of Trl also dominantly suppressed DmcycEJP (Brumby et al. 2002).
43S2[l(3)72Dk (zn72D)]:
43S2 was localized to 72D1–72D10 (Table 4) and complementation tests of mutations in the 72D1–10 region revealed that In(3)Taf4XS-2884, an inversion affecting expression of Taf4 (Taf110) and Zn72D (Sauer et al. 1996), failed to complement 43S2. A specific EMS allele of Taf4, l(3)72Dj, however, complemented 43S2, suggesting that 43S2 is most likely allelic to zn72D (CG5215). Indeed, another EMS allele in the region, l(3)72Dk, which failed to complement In(3)Taf4XS-2884, also failed to complement 43S2, suggesting that l(3)72Dk is an allele of zn72D. The zn72D gene encodes a zinc finger protein, but has not been characterized. In an attempt to verify the identity of 43S2 suppression as being due to a mutation of zn72D, l(3)72Dk was crossed into the DmcycEJP background. However, l(3)72Dk did not suppress the DmcycEJP adult eye phenotype or the S-phase defect of DmcycEJP eye discs as effectively as 43S2 did (Figure 1), which may be due to l(3)72Dk being a weaker allele than 43S2. Molecular characterization of the 43S2 and l(3)72Dk lesion will be required to confirm this. Interestingly, Zn72D was identified in a differential expression screen as a gene expressed specifically in the differentiating region of the eye disc (Jasper et al. 2002), consistent with a role for Zn72D in cell cycle arrest or differentiation.
Signaling pathway genes:
2.2. (phyllopod):
2.2 was localized to 51A1–51A5 (Table 4). Consistent with this, Df(2R)trix (51A1–2; 51B6) dominantly suppressed the DmcycEJP rough eye phenotype (data not shown). Mutations and P alleles within the 51A region were tested for allelism with 2.2 alleles, revealing that a null allele of phyllopod, phyl2245, failed to complement all three S(DmcycEJP) 2.2 alleles. To verify that 2.2 was indeed phyl, previously identified phyl alleles (2245 and 2366) were tested and shown to dominantly suppress the rough eye phenotype and the S-phase defects of DmcycEJP (Figure 1; M. Coombe, L. Quinn, R. Dickins, J. Secombe and H. Richardson, unpublished results). These data are consistent with the mutation of phyl being responsible for the observed suppression of DmcycEJP by the 2.2 alleles.
Phyl expression is induced by the Sevenless receptor tyrosine kinase signaling pathway and is a rate-limiting component in R7 photoreceptor cell differentiation in the eye imaginal disc, but also has other roles in neural differentiation during development (Dickson 1998). Phyl is a pioneer protein (containing no homology to other known proteins) that functions with the Ring finger protein Seven in absentia (Sina) and the F-box protein Ebi, to bind to and target the two isoforms of the neural differentiation inhibitor, Tramtrack (Ttk69 and Ttk88) and probably other proteins for destruction by the ubiquitin/proteosome pathway, allowing neural cell differentiation (Li et al. 1997; Tang et al. 1997; Boulton et al. 2000). Consistent with this, homozygous viable mutants in sina (sina1) strongly suppressed the DmcycEJP adult rough eye and S-phase defects, while a stronger sina allele (sina2) showed weak dominant suppression (M. Coombe, L. Quinn, R. Dickins, J. Secombe and H. Richardson, unpublished results). However, a deficiency removing sina showed strong dominant suppression of DmcycEJP (Table 1). This deficiency removes a sina-related gene (sina-h), located adjacent to sina, as well as Abl, which has been shown to dominantly suppress DmcycEJP (see below). Consistent with the involvement of the Sina complex in negative regulation of G1-S, ebi alleles have been shown to dominantly suppress DmcycEJP (Boulton et al. 2000). The mechanism by which the Sina complex acts to regulate G1-S does not involve targeting Cyclin E or E2F for ubiquitin-dependent degradation (Boulton et al. 2000) and remains to be determined.
2S1 (trio):
2S1 was mapped to 61E–62A8 (Table 4) and by crosses to mutations within the region it was revealed that trio [encoding a Rac guanine nucleotide exchange factor (Rac-GEF; Bateman et al. 2000)] failed to complement 2S1. To confirm this interaction, a previously isolated allele of trio (trioM89) was crossed into the DmcycEJP background. trioM89 was shown to dominantly suppress the DmcycEJP rough eye phenotype and S-phase defect (Figure 1). Rac-GEFs are involved in the activation of Rac family GTPases, which have roles in actin cytoskeletal remodeling (Blanchard 2000). In mammalian cells, Rac can lead to repression of Rho activity (Sander et al. 1999), and therefore mutation of trio may lead to higher levels of Rho activity. Rho activation in mammalian cells has been shown to promote cell cycle progression by leading to downregulation of the Cyclin/Cdk inhibitors p21 and p27 (Aznar and Lacal 2001; Pruitt and Der 2001; Sahai and Marshall 2002). trio has been shown to genetically interact with Abl, encoding a nonreceptor tyrosine kinase also involved in actin cytoskeleton remodeling (Luo 2000). Consistent with this, the deficiency removing Abl (73A3; 74F) dominantly suppressed the DmcycEJP rough eye phenotype; however, this deficiency also removes sina, sina-h (see above), and the Abl pathway gene, Disabled (Dab). The Abl alleles Abl04674 and Abl1 were then tested and shown to also suppress the DmcycEJP rough eye phenotype (not shown). The precise mechanism by which reducing the dosage of trio and Abl leads DmcycEJP suppression remains to be determined.
Cytoskeletal genes:
2.5. (dEB1):
2.5 was localized to 42B3–42C7 (Table 4). 2.558S12 was also lethal over the adjacent deficiency, Df(2R)nap1 (41D2–E1; 42B1–3), indicating that this allele is a deficiency or rearrangement that affects a larger region than 2.542S11. S(DmcycEJP)2.542S11 was crossed to P-element alleles available in the region and l(2)04524, was found to be semilethal in combination with 2.542S11. The few escaper flies, trans-heterozygous for 2.542S11 and l(2)04524, did not have any gross abnormalities, but generally died within a few days of eclosing, and the females were sterile. l(2)04524 is inserted within the 5′-UTR of the Drosophila homolog of the EB1 gene (BDGP). dEB1 encodes a cytoskeleta1 protein that binds to microtubules and plays an important role in adherens junction integrity and cell polarity (Lu et al. 2001; Rogers et al. 2002). EB1 was identified in mammalian cells as a binding partner of the adenomatous polyposis coli (APC) colon cancer tumor suppressor (Su et al. 1995); however, Drosophila APC1 and APC2 both lack the EB1-binding domain. Consistent with the identity of 2.5 being dEB1, l(2)04524 and the EMS dEB1 alleles, dEB15 (1DL) and dEB16 (GJ63/9) (obtained from J. Roote), dominantly suppressed DmcycEJP rough eye and S-phase defects (Figure 1 and data not shown). Moreover 2.542S11 and l(2)04524 disrupt dEB1 transcription (D. Coates, L. Quinn, R. Dickins, J. Secombe, A. Brumby and H. Richardson, unpublished results). How the EB1 microtubule protein is involved in G1-S regulation remains to be determined.
Cell adhesion genes:
2.11. (scab) (α-Integrin):
Group 2.11 was defined by S(DmcycEJP)62S9 from this screen and E(sev-cycE)e93 was from the Lane et al. (2000) genetic screen (see above). 2.11 was mapped to the region 51D3–51F13 (Table 4), and by testing mutations within this region, it was revealed that the P allele, l(2)01288, failed to complement both 2.11 alleles. The insertion point of l(2)01288 has been defined (BDGP) and disrupts the scab gene, encoding an α-integrin, αPS3, thought to play a role in tissue morphogenesis (Stark et al. 1997). To further confirm that 2.11 is allelic to scab, previously identified EMS-derived alleles of scab (scb1 and scb2) were tested and shown to also fail to complement 2.11 alleles. Consistent with the suppressing gene being scab, l(2)01288, scb1, and scb2 were recombined onto the DmcycEJP and were shown to also suppress the rough eye phenotype and the S-phase defect of DmcycEJP (Figure 1 and data not shown). In mammalian cells, integrins in association with the extracellular matrix have a well-established role in promoting anchorage-dependent cell proliferation (Danen and Yamada 2001). However, recent studies have shown that integrins can also inhibit G1-S progression (Hazlehurst et al. 2000; Mettouchi et al. 2001). Our identification of scab in the DmcycEJP genetic screen suggests that in Drosophila integrins also act as negative regulators of G1-S.
2.12. (CadN):
2.12 alleles 42S13 (also an allele of group 2-6) and 22S9 (Figure 1 and data not shown) were mapped to 36D1–36E4 (Table 4). Mutations and P alleles in the region were tested by complementation analysis, revealing that an allele of CadN (CadNM12) failed to complement both 2.12 alleles. CadN encodes a cadherin-like transmembrane protein (Lee et al. 2001; Iwai et al. 2002) that can bind to α-catenin and β-catenin (Armadillo), components of the adherens junction (Perez-Moreno et al. 2003). In mammalian cells, downregulation of N-Cadherin leads to upregulation of G1 Cyclin activity (Charrasse et al. 2002). Due to the close location of CadN and DmcycE, it was not possible to obtain a recombinant of the CadN allele with DmcycEJP to confirm that CadN exhibits the same modifier effect as S(DmcycEJP)2.12.
Cytoarchitectural tumor suppressor genes:
2.1. [lethal-(2)-giant larvae]:
2.1 was localized to 21A1–21B7–8 by deficiency mapping (Table 4). The mapping of 2.1 was initially confounded by the fact that two deficiencies in the deficiency kit, Df(2L)Prl (32F1–3; 33F1–2) and Df(2L)J39 (31D1–11; 32D1–E5), also contained lesions in the 21A region and therefore failed to complement 2.1. The localization of 2.1 was confirmed by genetic mapping of 2.1 alleles, which indicated that the lethal mapped to the left of UbcD1 (32A4–5) and close to al (21C2–4). Since 2.1 homozygous mutants die as giant larvae, an allele of the lethal-(2)-giant-larvae (lgl) gene, which also gives giant larvae and is localized at 21A, was tested for complementation of 2.1 alleles and failed to complement, whereas mutations in other genes in this region that have been identified as negative cell cycle regulators in previous screens, spen (poc; Staehling-Hampton et al. 1999; Lane et al. 2000) and net (I. Harriharan, personal communication), both complemented 2.1 alleles. Taken together these data suggest that lgl corresponds to 2.1. To confirm that a lesion in lgl suppresses the DmcycEJP phenotype, a null allele of lgl (lgl4) was tested for suppression of DmcycEJP. However, lgl4 did not suppress the S-phase defect or the rough eye phenotype of DmcycEJP to the same extent as 2.1 alleles did (Figure 2; and data not shown). However, halving the dosage of 2.1 alleles resulted in a greater increase in Cyclin E protein levels in DmcycEJP eye discs than halving the dosage of lgl4/+ (Figure 3). It is possible that additional mutations in the lgl4 background may account for its poorer ability to dominantly suppress DmcycEJP compared with 2.1 alleles. Consistent with lgl mutations being responsible for the suppression of DmcycEJP, lgl-2.1 and other lgl mutant clones in the eye imaginal disc showed ectopic expression of Cyclin E, which could be suppressed by expression of lgl using a UAS-lgl transgene (N. Amin, A. Brumby, J. Secombe and H. Richardson, unpublished results).
Figure 2.—
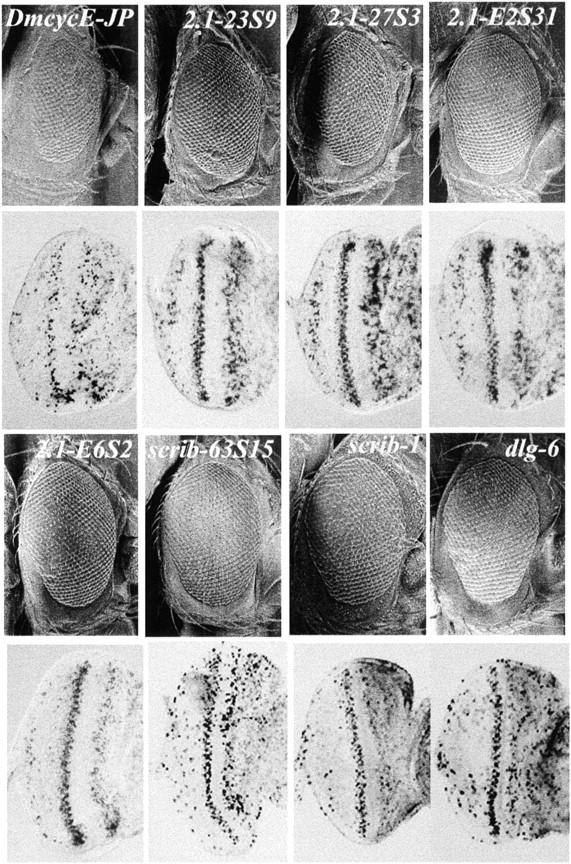
Scanning-electron micrographs of adult eyes and BrdU labeling of eye imaginal discs from lgl, scrib, or dlg heterozygotes in a DmcycEJP background. Genotypes are as indicated: DmcycEJP; 2.1-23S9/+, DmcycEJP; 2.1-27S3/+, DmcycEJP; 2.1-E2S31/+, DmcycEJP; 2.1-E6S2/+, DmcycEJP; DmcycEJP; scrib-63S15/+; DmcycEJP; scrib1/+; and dlg6/+; DmcycEJP.
Figure 3.—
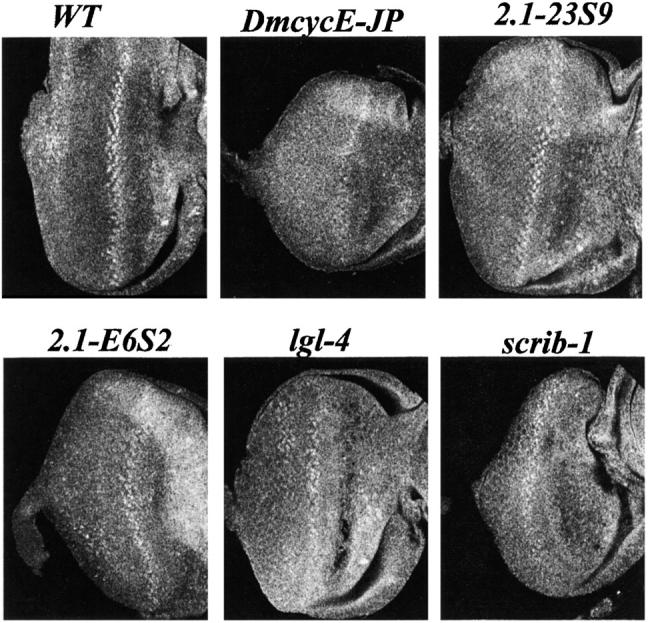
Cyclin E protein levels in eye imaginal discs from third instar larvae. Genotypes are as indicated: wild type (WT); DmcycEJP; 2.1-23S9/+, DmcycEJP; 2.1-E6S2/+, DmcycEJP; lgl4/+, DmcycEJP; and DmcycEJP; scrib1/+.
63S15 (scribble):
63S15 was localized to 97B–97D2 (Table 4), and consistent with a suppressor mapping in this region, Df(3R)T1P, which failed to complement 63S15, was identified as a suppressor of DmcycEJP in the screen of third chromosome deficiencies (Table 1). Cytological analysis of 63S15 showed that there was a lesion in the 97D region involving a translocation to the second chromosome (data not shown). By crosses to P alleles in the region, 63S15 was found to be allelic to l(3)j7b3, which is located in the first intron of a gene now known as scribble (Bilder and Perrimon 2000).
Scribble is a four-PDZ95-Dlg-ZO1 and multi-leucine-rich repeat containing protein localized to septate junctions and required for apical-basal polarity (Bilder and Perrimon 2000; Humbert et al. 2003). When homozygous, 63S15, like scribble null alleles, arrest as giant overgrown larvae due to amorphous overgrowth of imaginal discs and brain lobes, which is characteristic of neoplastic tumor suppressor mutants (Gateff et al. 1996; de Lorenzo et al. 1999; Bilder 2001). To confirm that lesions in scribble suppress DmcycEJP, the l(3)j7b3 allele and stronger EMS alleles of scrib, scrib1 and scrib2 (Bilder and Perrimon 2000), were crossed into a DmcycEJP background. The weak P allele, l(3)j7b3, did not suppress DmcycEJP, although mild suppression was observed with scrib1 and scrib2 alleles, but not as well as with 63S15 (Figure 2 and data not shown). This suggests that 63S15 may be a stronger scribble allele than scrib1 or scrib2. In confirmation that scribble alleles suppress the DmcycEJP phenotype, halving the dosage of scribble in DmcycEJP eye discs leads to higher levels of Cyclin E (Figure 3 and data not shown) and scrib1 and scrib2 eye imaginal disc clones show ectopic expression of Cyclin E (Brumby and Richardson 2003).
lgl and scribble are neoplastic tumor suppressor genes that together with discs-large (dlg) act in the same pathway to regulate apical-basal cell polarity (Bilder et al. 2000; Humbert et al. 2003). Because of this function, we have termed these proteins cytoarchitectural tumor suppressors to highlight their role in cell structure. Consistent with this pathway being important in regulation of G1- to S-phase progression, a deficiency removing dlg, Df(1)vN48, as well as a specific dlg allele (dlg6) showed suppression of DmcycEJP (Tables 1 and 2; Figure 2). Scribble, Dlg, and Lgl have been recently shown to act antagonistically to the Crumbs cell polarity complex (Bilder et al. 2003; Tanentzapf and Tepass 2003), and consistent with this, a deficiency removing crumbs and a crumbs allele (crb2) dominantly enhanced DmcycEJP (Table 1).
Scribble-interacting genes:
To determine whether a common pathway is involved in the mechanism by which the DmcycEJP suppressors lead to deregulation of cell proliferation, we analyzed weak scribble mutant combinations for a dominant genetic interaction with other genes identified in the DmcycE genetic screen (Table 6). The trans-heterozygous combination of scrib5/scribl(3)jB709 or scrib1/scrib5 results in adults with eye, bristle, and thorax-closure defects (not shown). Reducing the dose of the lgl (27S3, E2S31, and lgl4) showed a strong genetic interaction with the weak scrib allele phenotype, resulting in no scrib mutant progeny heterozygous for lgl. This is consistent with the previous observations that scribble mutations exhibit strong genetic interactions with lgl and dlg in the embryo (Bilder et al. 2000). Strikingly, halving the dosage of several other suppressor genes identified in the screen also resulted in very low numbers of scrib mutant progeny, most notably with dEB1 (2.5), phyl (2.2), the αPS3 integrin gene scab (2.11), the Brahma complex gene moira, and to a lesser extent brahma, as well as the unidentified 2.3, 2.4, and 2.9 genes. The mechanism of these interactions requires further analysis and relies on identifying the 2.3, 2.4, and 2.9 genes.
TABLE 6.
Genetic interactions withscribble—effect of halving the dosage of otherS(DmcycEJP) genes on the viability of hypomorphicscribble allele combinations
| Total no. of progeny
|
||||
|---|---|---|---|---|
| Mutant allele |
x−/+ or CyO/+; scrib1or5/TM6B |
x−/+; scrib1/scrib5 | % of expected no. of x−/+ scrib1/scrib5 progenya | |
| 2.1 (lgl) | 27S3 | 304 | 0 | 0 |
| E2S31 | 145 | 0 | 0 | |
| lgl4 | 248 | 0 | 0 | |
| 2.2 (phyl) | 28S2 | 231 | 0 | 0 |
| 2.3 | 65S12 | 227 | 0 | 0 |
| 36Fd | 343 | 1 | 2 | |
| 2.4 | 57S6 | 235 | 0 | 0 |
| 59S3 | 210 | 8 | 25 | |
| 2.5 (dEB1) | 42S11 | 147 | 3 | 13 |
| 2.9 | E1S4 | 256 | 0 | 0 |
| 25S11 | 242 | 0 | 0 | |
| 2.11 (scab) | E(sev-cycE)e93 | 86 | 1 | 8 |
| Total no. of progeny
| ||||
| Mutant allele | x− scribJ7b3or5 /TM6B | x− scribJ7b3/scrib5 | % of expected no. of x−/+ scrib1/scrib5 progenya | |
| brm | 2 | 401 | 47 | 24 |
| mor | 1 | 323 | 13 | 8 |
| 3.1 | 19S5 | 276 | 91 | 66 |
| 24SX | 279 | 58 | 42 | |
The expected number of scrib1/scrib5 progeny = total TM6B progeny/4 × 0.61 was derived as follows: The control cross of scrib1/TM6B × scrib5/TM6B gave 127 scrib1/scrib5 out of 627 total progeny, i.e., 61% of the expected number. To control for this, the percentage of the expected number of x−/+ scrib1/scrib5 progeny has been adjusted by this factor. The weaker scrib allele combination, −/+ scribJ7b3/scrib5, gave expected Mendelian numbers of progeny. The expected number of scribJ7b3/scrib5 progeny = total number of TM6B progeny/2.
The unidentified suppressors:
The map positions for suppressor groups 2.3, 2.4, 2.7, 2.8, 2.9, 2.10, 2.13, 3.1, 3.2, and 3.4 (1S2) and the single alleles 1S3, 13S1, 59S9, 59S18, and 68S10 were defined by genetic and deficiency mapping (Table 5). For the third chromosome suppressors, 3.1, 3.2, and 3.4 (1S2), the location of a suppressor within the defined region could be confirmed since the corresponding deficiencies dominantly suppressed DmcycEJP (Table 1; and data not shown). However, for 13S1, 59S9, 59S18, and 68S10, the deficiencies that failed to complement these suppressors did not suppress DmcycEJP (Table 1; and data not shown). For most of the unidentified suppressors complementation tests of all likely mutations and P alleles within the respective regions and Southern analysis of candidates have so far failed to identify the affected gene (Table 7); therefore, these suppressor mutations affect novel genes, which will require further analysis to identify. The exception is 2.3, where there are two candidates (Table 7 and see below). Potential candidates, with links to identified DmcycEJP suppressors and thereby G1-S regulation, were found for many of the unidentified suppressors (see Table 7). Some of these candidates have been tested by complementation tests or Southern analysis and have been ruled out as being affected by the suppressor mutation (Table 7). Details on mapping and potential candidates for 2.3, 3.1, 1S3, and 59S9 are described below. For the details on other unidentified suppressors, see Tables 5 and 7.
TABLE 7.
Candidates for the unidentified suppressors
| Suppressor group |
Candidate gene | Gene function | Possible links to cell cycle regulation |
|---|---|---|---|
| 2.3 | l(2)36Fd? | Unknown | Unknown |
| hamlet? | Transcription factor in Dendrite morphogenesis (Mooreet al. 2002) |
May be involved in regulating cytoskeletal, cell adhesion or cytoarchitectural tumor suppressor genes |
|
| 2.4 | Act42A | Actin 42A | Brahma complex component (Papoulas et al. 1998) |
| CG12792 | WD40 domain | May be involved in proteolysis as is Cdc4/Ago (Moberg et al. 2001) |
|
| CG10412 | Dbl-related (RhoGEF) | Activator of Rho family proteins and may regulate Rac (Blanchard 2000) |
|
| 2.7 | CG2727 (Emp)a | CD36-like | CD36-like proteins encode cell surface signaling |
| CG2736a | CD36-like | proteins that may have a role in adhesion and | |
| CG3829a | CD36-like | signaling pathways regulating cell proliferation | |
| (Greenwalt et al. 1992) | |||
| CG3770 | Claudin-like | A tight junction protein involved in cell-cell adhesion and may have a role in inhibiting cell proliferation (Michl et al. 2003; Tepass et al. 2001) |
|
| 2.8 | wee1b | Cdc2 inhibitor (Campbell et al. 1995) |
In combination with Cyclin A can drive entry into S phases (Dong et al. 1997; Thomas et al. 1997) |
| neuroligin | Cell adhesion protein | The mammalian homolog binds to Dlg4 (Bolliger et al. 2001) |
|
| 2.9 | Jitterbug (Filamin)a | Actin-binding protein | Possibly acts to regulate Rho family members (Sokol and Cooley 2003; Stossel et al. 2001) |
| moa | Cell adhesion | Possible role in Integrin signalling (Prout et al. 1997; Walsh and Brown 1998) |
|
| 2.10 | p120-catenina | Adherens junction component | Binds to E-cadherin and regulates Rho in mammalian cells (Aznar and Lacal 2001; Blanchard 2000; Jaffe and Hall 2002) |
| Gprk-1 | G-protein-coupled receptor protein kinase with a RGS domain |
Negative regulator of heterotrimeric G proteins, responsible for the rapid turnoff of G-protein-coupled receptor signaling pathways (De Vries and Gist Farquhar 1999) |
|
| 2.13 | CG2727 (Emp) | CD36-like | (See 2.7) |
| CG2736 | CD36-like | ||
| CG3829 | CD36-like | ||
| CG3770 | Claudin-like | ||
| 3.1 | CG6445a | Cadherin-like | Possible role at adherens junctions |
| CG3885 | Sec3-like exocyst component | Involved in docking at the plasma membrane, which is a function that Lgl has also been implicated in Lehman et al. (1999) and Musch et al. (2002) |
|
| 3.2 | CG6190 | Ubiquitin ligase - HECT domain protein |
A HECT domain ubiquitin ligase gene related to hyd, a tumor suppressor (de Lorenzo et al. 1999; Gateff et al. 1996) |
| 3.4 | CG5263 (smg) | Translational repressor | Role in neural cells (Clark et al. 2002); possible role in G1-S regulation, given the identification of RpS6 in the screen |
| 1S3 | APC1b | Adenomatous polyposis coli tumor suppressor |
Possible role at adherens junction and may function with EB1 (Ahmed et al. 1998; Lu et al. 2001) |
| pins (rapsinoid) | Asymmetric division of neuroblasts | Directly interacts with Dlg (Bellaiche et al. 2001; Parmentier et al. 2000) |
|
| 13S1 | CG5555 | Ring finger domain | Homologous to a protein shown to interact with the BRCA1 tumor suppressor protein in mammals (Sharan and Bradley 1998) |
| 59S9 | Spinophilin (neurabin) | Actin-binding scaffold protein | In mammalian cells is involved in binding to and upregulating Rac and p70-S6K activity (Buchsbaum et al. 2003) |
| 59S18 | draper | Protein with multiple extracellular EGF repeats, similar to laminin γ3 and Notch |
Predicted to be involved in cell adhesion and signaling and involved in differentiation of neural cells (Egger et al. 2002) |
| 68S10 | canoeb | Component of the Adherens junction | Acts antagonistically to the Ras signaling pathway (Matsuo et al. 1997) |
| CG12591 | Ig C2-domain—cell adhesion | Possible role at adherens junction or in signaling (Tepass et al. 2001) |
?, failed to complement 2.3 alleles, but testing did not confirm whether the mutations dominantly suppress DmcycEJP.
Tested by Southern analysis and no obvious disruptions were observed.
Mutants were tested and shown to complement the DmcycEJP suppressor.
2.3. (59S16, 65S12) location (36F7–37B8):
While 2.359S16 carries a deletion removing at least six complementation groups within the 36F7–37B8 region, including l(2)36Fd and l(2)37Ac, 2.365S12 was found to be lethal over the unidentified lethal gene l(2)36Fd, but gave ∼5% escapers over l(2)37Ac. 2.365S12 is therefore likely to be a smaller lesion affecting both of these uncharacterized genes. A recently characterized gene in the 36F region, hamlet, which is a transcription factor involved in dendrite morphogenesis (Moore et al. 2002), was also tested for allelism with 2.3 and failed to complement 2.359S16 and 2.365S12 but not l(2)36Fd. Further analysis is required to determine whether hamlet or l(2)36Fd corresponds to the 2.3 suppressor.
3.1. (19S5, 24SX, 58S5, 62S2) location [73D–74F (74B1–74C1)]:
Consistent with the map position defined by deficiency mapping, chromosome cytology revealed that 58S5 contained a deletion in the 74A–F region, and it failed to complement several lethal alleles in the region. The cadherin-like gene, CG6445 (Cad74A), was considered a candidate, since the cadherin-like protein, Fat, is a tumor suppressor in Drosophila (Gateff et al. 1996; de Lorenzo et al. 1999). Southern analysis failed to reveal any alterations in this gene in 3.1 alleles (data not shown). The method of male recombination (Svoboda et al. 1995) was then used to further define the map position of the 3.1 alleles, 19S5 and 24S10 relative to several P alleles, revealing that the lethal associated with 3.1 mapped to the right of blot (74B1–2) and to the left of l(3)S070006 (allelic to l(3)L6750 = frc at 74B4), l(3)00073 (74C1–2), and EIP74EF (74D2–5). Taken together these data suggest that 3.1 maps between 74B1 and 74B4. A candidate gene within this region, CG3885, encodes a Sec3-like protein, a component of the exocyst complex involved in docking at the plasma membrane, which is a function that Lgl has also been implicated in (Lehman et al. 1999; Musch et al. 2002).
1S3 location [98A–100B (98A5–98E3)]:
Chromosome cytology showed that 1S3 contained a translocation breakpoint at 98C (data not shown). Since there is a hole in the deficiency collection between 98A5 and 98E3, it is likely that 1S3 maps within this region. A candidate in the 98A5–98E3 region was APC1 (encoding the Adenomatous polyposis tumor suppressor; Ahmed et al. 1998); however, mutations in APC1 (APCQ8 and APCX1) complemented 1S3. Another candidate is raps (pins), which encodes a protein involved in asymmetric division of neuroblasts and directly interacts with Dlg (Parmentier et al. 2000; Bellaiche et al. 2001). Further analysis is required to test whether raps mutations are allelic to 1S3.
59S9 location (62D2–62F5):
Consistent with this location for 59S9, cytological analysis revealed a breakpoint at 62B. A possible candidate in this region is spinophilin (neurabin), encoding an actin-binding scaffold protein, which in mammalian cells is involved in binding to and upregulating Rac and p70-S6K activity (Buchsbaum et al. 2003). Since another gene involved in Rac activation, trio, was identified as a suppressor of DmcycEJP it is possible that spinophilin is also a suppressor. Furthermore, Drosophila mutations in spinophilin are semilethal (Keegan et al. 2001), as is 59S9.
Further analysis is needed to investigate whether the potential candidates for these suppressors, listed above and in Table 7, are disrupted by the suppressor mutations and for the identification of the suppressors.
DISCUSSION
In this study, we have identified genetic interactors of cyclin E by screening deficiencies, by testing candidate genes, and through EMS and X-ray mutagenesis screens. This work has led to the identification of many genes that when mutated have the ability to dominantly modify the DmcycEJP adult rough eye phenotype and S-phase defect in third instar larval eye imaginal discs. In addition to genes already known to be regulators of Drosophila cyclin E or G1-S progression, such as E2F1; retinoblastoma (Rbf); ago (cdc4) encoding a protein involved in Cyclin E degradation (Moberg et al. 2001); the EGF receptor pathway genes Egfr and Ras85D, which act to promote Cyclin E protein accumulation (Prober and Edgar 2000; Brumby and Richardson 2003); and Hh signaling pathway genes, which act to promote cyclin E transcription (Duman-Scheel et al. 2002); this screen led to the identification of many novel cyclin E interactors. This study has mainly concentrated on the suppressors of DmcycEJP, although from the deficiency screen and specifically testing candidates, we identified axin (an inhibitor of Wg signaling), rho1, and crumbs as enhancers of DmcycEJP, which therefore may act as novel positive regulators of G1-S progression. The suppressors of DmcycEJP identified include the following classes: (1) chromatin remodeling genes brm, mor, Trl, or the transcription factor Zn72D; (2) signaling pathway genes phyl, sina, trio, Abl, RpS6, wg and Wg pathway effectors dsh and arm; (3) genes encoding cytoskeletal proteins dEB1 (encoding a microtubule-binding protein) and expanded (encoding a FERM domain cytoskeletal protein and hyperplastic tumor suppressor); (4) genes encoding cell adhesion proteins scab (encoding an α-integrin), cadN (N-Cadherin), shg (E-Cadherin), and fat (encoding an atypical-cadherin and hyperplastic tumor suppressor); and (5) cytoarchitectural tumor suppressor genes scribble, lgl, and dlg, required for apical-basal cell polarity and cell proliferation inhibition. While some of these genes (brm, mor, expanded, fat, scribble, and lgl) have been previously shown or implicated to play a role in negatively regulating G1-S (Gateff et al. 1996; de Lorenzo et al. 1999; Staehling-Hampton et al. 1999; Bilder et al. 2000), a potential role for Trl, Znf72D, phyl, sina, trio, Abl, RpS6, wg, dsh, arm, dEB1, scab, cadN, and shg in inhibiting G1-S progression in Drosophila is novel. Further studies are required to determine whether Abl, RpS6, wg, dsh, arm, and shg do indeed suppress DmcycEJP by acting at the S-phase level and to understand the mechanism by which these genes act in G1-S regulation. The identification of novel classes of presumptive negative regulators of cyclin E or G1-S progression highlights the power of Drosophila whole-animal genetics as a tool for revealing new cell proliferation pathways.
It is unclear at present how many of the DmcycEJP modifiers identified in our screen bear upon the role of Cyclin E in DNA replication or centrosome duplication (see Introduction). Brahma and Moira are likely to be downstream targets of Cyclin E/cdk2 that may impact upon transcriptional regulation or DNA replication (Brumby et al. 2002), but whether other interactors act upstream or downstream of Cyclin E remains to be determined. The only cyclin E interactor we identified that has been shown to be associated with the centrosome is EB1 (Rehberg and Graf 2002); however, whether this reflects upon the role for Cyclin E in centrosome duplication in Drosophila is unclear. A recent study has shown that the Drosophila SkpA, a component of SCF ubiquitin ligases, regulates centrosome duplication independently of Cyclin E accumulation (Murphy 2003).
Similar genetic screens carried out using phenotypes generated by overexpression of cyclin E (Lane et al. 2000) or the G1-S regulators E2F1/Dp (Staehling-Hampton et al. 1999), Rbf (Duman-Scheel et al. 2002), and human p21 (Cdk2 inhibitor; I. Hariharan, personal communication) have revealed a more restricted set of interacting genes than that obtained in our cyclin E hypomorphic allele genetic screen. The GMR-E2F1/Dp screen (Staehling-Hampton et al. 1999) revealed alleles of the chromatin remodeling genes brm, mor, and osa and of the transcription factor pointed, an effector of the Egfr-Ras signaling pathway, as enhancers. This is consistent with our identification of brm and mor as suppressors of the hypomorphic cyclin E phenotype in our mutagenesis screen. In addition, we tested alleles of osa and showed that they suppressed the hypomorphic cyclin E phenotype (Brumby et al. 2002). The sevenless-cyclin E screen (Lane et al. 2000) revealed alleles in identified cell cycle genes cdk2 (as a suppressor), dacapo (as an enhancer), and E2F1 (a suspected gain-of-function allele as an enhancer) and identified as an enhancer the novel gene spen (poc), also identified in the GMR-E2F1/Dp screen (Staehling-Hampton et al. 1999). Spen (Poc) is a RNP-type RNA-binding protein that has recently been shown to be required for Wg signaling in imaginal discs (Lin et al. 2003). We have not identified spen (poc) as a suppressor in our genetic screen, but alleles of spen (poc) were tested and shown to suppress DmcycEJP (Table 2), consistent with the Wg signaling pathway acting to negatively regulate G1-S progression in the eye disc. As detailed above, we have shown that one of the single alleles identified as an enhancer in the sevenless-cyclin E screen is allelic to our DmcycEJP suppressor 2.11, which we have identified as scab. In the GMR-Rbf screen, alleles of patched, encoding an inhibitor of Hedgehog (Hh) signaling, were identified as dominant suppressors (Duman-Scheel et al. 2002). Although our mutagenesis screen did not reveal alleles of patched, patched alleles strongly suppressed DmcycEJP (Table 2), consistent with the notion that Hh signaling leads to increased transcription of cyclin E (Duman-Scheel et al. 2002). The greater number of interactors that we obtained in our screen may be due to the fact that our screen was of a cyclin E hypomorphic phenotype that affected cell proliferation in early eye development as well as the post-MF S phases and may therefore have been more sensitive to gene dosage than the overexpression screens. Furthermore, unlike the overexpression screens, the cyclin E hypomorphic screen is more likely to reveal genes that are upstream of cyclin E expression.
The DmcycEJP suppressor genes we have identified from our mutagenic screen are mostly distinct from Drosophila tumor suppressors previously described (Torok et al. 1993; Gateff et al. 1996; de Lorenzo et al. 1999). Recently, clonal screens have revealed a novel pathway involved in inhibiting G1-S progression and cell death in the Drosophila eye (Hay and Guo 2003). This pathway includes lats (warts), salvador, and hippo, and although this pathway has been recently shown to regulate cyclin E at possibly both a transcriptional and protein stability level, we did not identify alleles of these genes in our genetic screen. Alleles of hippo, at least, have been shown to suppress the DmcycEJP phenotype (Wu et al. 2003). The fact that we did not identify hippo in our mutagenesis screen may have been because the screen was not saturating. However, lats (warts) alleles did not show appreciable suppression of DmcycEJP (Table 2); therefore it is possible that only certain mutations of this pathway are capable of dominant suppression.
Also pertinent to our study is the recent Drosophila protein interaction map determined by yeast two-hybrid analyses (Giot et al. 2003). None of our identified cyclin E genetic interactors were identical to the 15 interactors identified by the protein interaction study (Giot et al. 2003), but many proteins identified in our screen were not analyzed in their screen (e.g., Brahma, Moira, Scab, CadN, Dsh, Scribble, Crumbs, Expanded, and Abl). Most of the 15 yeast two-hybrid interactors with Cyclin E are uncharacterized, but of the characterized proteins, Combgap, a transcription factor, has been implicated in cell proliferation via its effect on Ci expression (Campbell and Tomlinson 2000). Of the other characterized interactors, Gliolectin is involved in cell adhesion in axon pathfinding (Sharrow and Tiemeyer 2001) and Traf2 is involved in Dorsal activation (Shen et al. 2001), but no cell proliferation role has been described for these proteins. Some of the Cyclin E yeast two-hybrid interacting genes map to regions where cyclin E genetic interactors have been mapped (not shown) and are candidates for future analysis.
Whether the genetic suppressors of cyclin E identified in our screen can all be connected in a common pathway or represent several converging pathways acting upon G1-S progression in the eye imaginal disc remains to be determined. As a first step to explore this we examined interactions between a weak scrib mutant and S(DmcycEJP) alleles, which revealed genetic interactions with lgl, phyl, dEB1 scab, mor, the unidentified suppressors 2.3, 2.4, and 2.9, and to a lesser extent brm. This analysis provides a connection between chromatin remodeling, signaling, cytoskeletal, cell-cell adhesion, and cytoarchitectural suppressor genes. How exactly these pathways may be connected and whether other genes identified in the DmcycEJP screen are also functionally connected now warrant further investigation.
Interestingly, many of the genes identified in the screen have roles in cell polarity; for example, scrib, dlg, lgl, and crumbs are involved in apical-basal cell polarity, while dlg, fat, expanded, and the Wg pathway, via Rho and Jnk, have roles in planar polarity (Blaumueller and Mlodzik 2000; Bellaiche et al. 2001; Yang et al. 2002; Eaton 2003; Fanto et al. 2003). Moreover, E-cadherin (shg) and β-catenin (arm) function at the adherens junction, which is important in both apical-basal cell polarity and cell-cell adhesion (Tepass et al. 2001). Whether other cell polarity genes, such as bazooka, par3, apkc, patj, and stardust (Humbert et al. 2003), are also DmcycEJP modifiers and the molecular mechanism by which this occurs require further analysis. Pertinent to this, a recent study has shown that apkc clones have reduced cell division and that apkc mutants can suppress the overgrowth of lgl mutants, suggesting that upregulation of apkc contributes to the overgrowth phenotype of lgl, and perhaps also scrib and dlg, mutants (Rolls et al. 2003).
How are junctional components connected to signaling pathways or to the cell cycle machinery? In mammalian cells, the Frizzled receptors, Fz1, Fz2, Fz4, and Fz7, have been shown to bind to mammalian Dlg1 (Hering and Sheng 2002), which may therefore provide a connection between apical-basal and the Frizzled-Rho-Jnk planar polarity pathway (Adler and Lee 2001), as well as to the canonical Wg-Arm (β-catenin) pathway to effect S-phase entry (Figure 4). Furthermore, mammalian scrib genetically and physically interacts with the planar polarity gene, vang (strabismus) (Kallay et al. 2003; Montcouquiol et al. 2003; Murdoch et al. 2003). Mammalian Vang is a potential tumor suppressor that can act to regulate the Wg-Arm pathway (Katoh 2002). If Vang acts similarly in Drosophila, it would provide another connection between planar polarity, apical-basal polarity, and Wg signaling pathways. Connections between polarity proteins and the Egfr signaling pathway have also been observed in Caenorhabditis elegans and mammalian cells (Simske et al. 1996; Huang et al. 2003). Consistent with this, antagonistic interactions between E-cadherin in adherens junction function and the Egfr signaling pathway have been observed in the Drosophila nervous system (Dumstrei et al. 2002), and if this also occurs in the eye imaginal disc then decreasing E-cadherin levels would be expected to cause an increase in Egfr-Ras signaling that would lead to increased Cyclin E protein (Brumby and Richardson 2003). Furthermore, there is evidence that the FERM domain protein Expanded, which functions together with another FERM domain protein, Merlin, a homolog of the NF2 tumor suppressor, modulates the Dpp signaling pathway (McCartney et al. 2000) and in mammalian cells NF2 can inhibit Ras signaling (Lim et al. 2003). There is also a precedent for a connection between Integrin signaling and cell polarity pathways, since the transmembrane Laminin receptor Dystroglycan has been shown to have a role in epithelial cell apical-basal polarity (Deng et al. 2003). This now raises the question of whether Scab (αPS3-Integrin) plays a role in apical-basal cell polarity. In mammalian cells, integrins act via focal adhesion kinase (Fak) to activate Rho-family GTPases (Schoenwaelder and Burridge 1999) and recently it has been reported that integrins are important for the localization of aPKC (Datta et al. 2003). In Drosophila, a role for scab in modulation of Dpp signaling has been described in wing vein formation (Araujo et al. 2003), suggesting a mechanism by which scab may also affect cell proliferation. Furthermore, there is a connection between the Trio-Rac-Abl pathway and polarity, since Trio interacts with the Lar receptor-like tyrosine phosphatase, which has recently been shown to have a role in epithelial planar polarity (Frydman and Spradling 2001).
Figure 4.—

Possible pathways connecting cyclin E-interacting genes. Interactors identified in our cyclin E screen are shaded. Direct protein interactions between cyclin E interactors or other relevant proteins are indicated by the double-headed arrows. Arrows indicate positive interactions while barred lines indicate negative interactions. Not shown are interactions between Scab and the Dpp pathway, between E-cadherin and the Egfr pathway, between Fat and Atrophin (a nuclear corepressor), and between Expanded/Merlin and the Dpp and Egfr pathways. *, genes that genetically interact with scribble. See the text for details.
The Drosophila microtubule-binding protein dEB1 has also been implicated in playing a role in adherens junction function and cell polarity by RNA ablation studies (Lu et al. 2001; Rogers et al. 2002). Interestingly, the recently published study on Drosophila protein interactions using yeast two-hybrid analysis has revealed that dEB1 binds to the Sina homolog CG13030, providing a connection to the Sina-Phyl pathway (Giot et al. 2003). Sina and the Sina homolog also bind to Rasputin (Rin), a homolog of the RasGAP-binding protein G3BP, which has a role in planar polarity via effects on the Rho signaling pathway (Pazman et al. 2000). Thus the Sina-Phyl complex may act via Rasputin to negatively regulate Ras and Rho signaling and thereby G1-S progression (Figure 4). The protein interaction study (Giot et al. 2003) has also revealed that RpS6, identified as a suppressor in our screen, binds to the planar polarity protein Vang/Strabismus, which was not tested in our screen. Interestingly in mammalian cells, Cdc42, a Rho-family GTPase component of the apical Par6 complex, functions via p70-S6 kinase to upregulate cyclin E transcription (Chou et al. 2003) and disruption of RpS6 in mice results in a specific block in cyclin E expression (Volarevic et al. 2000). The yeast two-hybrid analysis study (Giot et al. 2003) also revealed protein interactions between Zn72D and Actin 5C, a component of the Brahma complex (Papoulas et al. 1998), between the Brahma-associated protein Bap60 and the apical zone polarity protein aPKC (Humbert et al. 2003), and between Dlg or Lgl and zinc finger transcription factors. There are precedents for functional interactions between cell polarity proteins and nuclear corepressors, for example, between Drosophila Fat (atypical cadherin involved in planar polarity) and Atrophin (Fanto et al. 2003), suggesting that yeast two-hybrid interactions between the Brahma complex or the zinc finger transcription factors and cell polarity proteins may be functionally relevant, although further investigation is required. Although there may be many pathways that connect the Cyclin E interactors identified in this screen to G1-S progression, the examples above suggest ways in which cell polarity proteins may link to signaling pathways or directly to chromatin remodeling, corepressors, or transcription factors to regulate cyclin E or the transcription of other G1- to S-phase genes (Figure 4).
In summary, the identification in our cyclin E screen of genes that were not necessarily predicted to play roles in G1-S progression highlights the importance of using whole-animal genetics to investigate G1-S regulation. The identified cyclin E genetic suppressors are conserved in mammals and given their demonstrated or presumptive roles as inhibitors of G1-S progression in Drosophila are candidates for tumor suppressors in mammalian cancers.
Acknowledgments
We thank Dr. Leonie Quinn for critical comments on this article. We are grateful to Dr. C. Lehner, N. Dyson, and I. Hariharan for supplying fly stocks from their screens and exchanging unpublished information. Also we thank D. Bilder, P. Bryant, S. Campbell, R. Hynes, Y.N. Jan, J. Kennison, E. Liebl, B. Mechler, J. Roote, K. Watson, E. Wieshaus, and the Bloomington and Sveged Stock Centers for supplying fly stocks. We acknowledge the ARC and the National Health and Medical Research Council of Australia (NHMRC) for supporting this project and the Australian Research Council, Wellcome Foundation, and NHMRC for fellowship support for H.R. H.R. is currently a NHMRC senior research fellow.
References
- Adler, P. N., and H. Lee, 2001. Frizzled signaling and cell-cell interactions in planar polarity. Curr. Opin. Cell Biol. 13: 635–640. [DOI] [PubMed] [Google Scholar]
- Ahmed, Y., S. Hayashi, A. Levine and E. Wieschaus, 1998. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell 93: 1171–1182. [DOI] [PubMed] [Google Scholar]
- Allenspach, E. J., I. Maillard, J. C. Aster and W. S. Pear, 2002. Notch signaling in cancer. Cancer Biol. Ther. 1: 466–476. [DOI] [PubMed] [Google Scholar]
- Amaldi, F., and P. Pierandrei-Amaldi, 1997. TOP genes: a translationally controlled class of genes including those coding for ribosomal proteins. Prog. Mol. Subcell. Biol. 18: 1–17. [DOI] [PubMed] [Google Scholar]
- Araujo, H., E. Negreiros and E. Bier, 2003. Integrins modulate Sog activity in the Drosophila wing. Development 130: 3851–3864. [DOI] [PubMed] [Google Scholar]
- Asano, M., J. R. Nevins and R. P. Wharton, 1996. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev. 10: 1422–1432. [DOI] [PubMed] [Google Scholar]
- Aznar, S., and J. C. Lacal, 2001. Rho signals to cell growth and apoptosis. Cancer Lett. 165: 1–10. [DOI] [PubMed] [Google Scholar]
- Bateman, J., H. Shu and D. Van Vactor, 2000. The guanine nucleotide exchange factor trio mediates axonal development in the Drosophila embryo. Neuron 26: 93–106. [DOI] [PubMed] [Google Scholar]
- Bellaiche, Y., A. Radovic, D. F. Woods, C. D. Hough, M. L. Parmentier et al., 2001. The Partner of Inscuteable/Discs-large complex is required to establish planar polarity during asymmetric cell division in Drosophila. Cell 106: 355–366. [DOI] [PubMed] [Google Scholar]
- Bilder, D., 2001. PDZ proteins and polarity: functions from the fly. Trends Genet. 17: 511–519. [DOI] [PubMed] [Google Scholar]
- Bilder, D., and N. Perrimon, 2000. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 403: 676–680. [DOI] [PubMed] [Google Scholar]
- Bilder, D., M. Li and N. Perrimon, 2000. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289: 113–116. [DOI] [PubMed] [Google Scholar]
- Bilder, D., M. Schober and N. Perrimon, 2003. Integrated activity of PDZ protein complexes regulates epithelial polarity. Nat. Cell Biol. 5: 53–58. [DOI] [PubMed] [Google Scholar]
- Blanchard, J. M., 2000. Small GTPases, adhesion, cell cycle control and proliferation. Pathol. Biol. 48: 318–327. [PubMed] [Google Scholar]
- Blaumueller, C. M., and M. Mlodzik, 2000. The Drosophila tumor suppressor expanded regulates growth, apoptosis, and patterning during development. Mech. Dev. 92: 251–262. [DOI] [PubMed] [Google Scholar]
- Bolliger, M. F., K. Frei, K. H. Winterhalter and S. M. Gloor, 2001. Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem. J. 356: 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton, S. J., A. Brook, K. Staehling-Hampton, P. Heitzler and N. Dyson, 2000. A role for Ebi in neuronal cell cycle control. EMBO J. 19: 5376–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby, A., and H. Richardson, 2003. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22: 5769–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby, A. M., C. B. Zraly, J. A. Horsfield, J. Secombe, R. Saint et al., 2002. Drosophila cyclin E interacts with components of the Brahma complex. EMBO J. 21: 3377–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum, R. J., B. A. Connolly and L. A. Feig, 2003. Regulation of p70 S6 kinase by complex formation between the Rac guanine nucleotide exchange factor (Rac-GEF) Tiam1 and the scaffold spinophilin. J. Biol. Chem. 278: 18833–18841. [DOI] [PubMed] [Google Scholar]
- Campbell, G. L., and A. Tomlinson, 2000. Transcriptional regulation of the Hedgehog effector CI by the zinc-finger gene combgap. Development 127: 4095–4103. [DOI] [PubMed] [Google Scholar]
- Campbell, S. D., F. Sprenger, B. A. Edgar and P. H. O'Farrell, 1995. Drosophila Wee1 kinase rescues fission yeast from mitotic catastrophe and phosphorylates Drosophila Cdc2 in vitro. Mol. Biol. Cell 6: 1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, F., L. S. Steelman, J. G. Shelton, J. T. Lee, P. M. Navolanic et al., 2003. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (review). Int. J. Oncol. 22: 469–480. [PubMed] [Google Scholar]
- Charrasse, S., M. Meriane, F. Comunale, A. Blangy and C. Gauthier-Rouviere, 2002. N-cadherin-dependent cell-cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J. Cell Biol. 158: 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., S. W. Oh, Z. Zheng, H. W. Chen, H. H. Shin et al., 2003. Cyclin D-Cdk4 and cyclin E-Cdk2 regulate the Jak/STAT signal transduction pathway in Drosophila. Dev. Cell 4: 179–190. [DOI] [PubMed] [Google Scholar]
- Chou, M. M., J. M. Masuda-Robens and M. L. Gupta, 2003. Cdc42 promotes G1 progression through p70 S6 kinase-mediated induction of cyclin E expression. J. Biol. Chem. 278: 35241–35247. [DOI] [PubMed] [Google Scholar]
- Clark, I. E., K. C. Dobi, H. K. Duchow, A. N. Vlasak and E. R. Gavis, 2002. A common translational control mechanism functions in axial patterning and neuroendocrine signaling in Drosophila. Development 129: 3325–3334. [DOI] [PubMed] [Google Scholar]
- Crack, D., J. Secombe, M. Coombe, A. Brumby, R. Saint et al., 2002. Analysis of Drosophila cyclin EI and II function during development: identification of an inhibitory zone within the morphogenetic furrow of the eye imaginal disc that blocks the function of cyclin EI but not cyclin EII. Dev. Biol. 241: 157–171. [DOI] [PubMed] [Google Scholar]
- Danen, E. H., and K. M. Yamada, 2001. Fibronectin, integrins, and growth control. J. Cell Physiol. 189: 1–13. [DOI] [PubMed] [Google Scholar]
- Datar, S. A., H. W. Jacobs, A. F. de la Cruz, C. F. Lehner and B. A. Edgar, 2000. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 19: 4543–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, A., Y. Wei, L. E. O'Brien, T. Jou and K. E. Mostov, 2003. Beta-1 Integrin Outside-In signaling controls apical targeting of the polarity complex of PKC lambda and Cdc42 during mammalian epithelial morphogenesis. Mol. Biol. Cell (Suppl. 14): 135a. [Google Scholar]
- de Lorenzo, C., B. M. Mechler and P. J. Bryant, 1999. What is Drosophila telling us about cancer? Cancer Metastasis Rev. 18: 295–311. [DOI] [PubMed] [Google Scholar]
- de Nooij, J. C., and I. K. Hariharan, 1995. Uncoupling cell fate determination from patterned cell division in the Drosophila eye. Science 270: 983–985. [DOI] [PubMed] [Google Scholar]
- de Nooij, J. C., M. A. Letendre and I. K. Hariharan, 1996. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell 87: 1237–1247. [DOI] [PubMed] [Google Scholar]
- De Vries, L., and M. Gist Farquhar, 1999. RGS proteins: more than just GAPs for heterotrimeric G proteins. Trends Cell Biol. 9: 138–144. [DOI] [PubMed] [Google Scholar]
- Deng, W. M., M. Schneider, R. Frock, C. Castillejo-Lopez, E. A. Gaman et al., 2003. Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development 130: 173–184. [DOI] [PubMed] [Google Scholar]
- Dickson, B. J., 1998. Photoreceptor development: breaking down the barriers. Curr. Biol. 8: R90–R92. [DOI] [PubMed] [Google Scholar]
- Dong, X., K. H. Zavitz, B. J. Thomas, M. Lin, S. Campbell et al., 1997. Control of G1 in the developing Drosophila eye: rca1 regulates Cyclin A. Genes Dev. 11: 94–105. [DOI] [PubMed] [Google Scholar]
- Du, W., J. E. Xie and N. Dyson, 1996. Ectopic expression of dE2F and dDP induces cell proliferation and death in the Drosophila eye. EMBO J. 15: 3684–3692. [PMC free article] [PubMed] [Google Scholar]
- Duman-Scheel, M., L. Weng, S. Xin and W. Du, 2002. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417: 299–304. [DOI] [PubMed] [Google Scholar]
- Dumstrei, K., F. Wang, D. Shy, U. Tepass and V. Hartenstein, 2002. Interaction between EGFR signaling and DE-cadherin during nervous system morphogenesis. Development 129: 3983–3994. [DOI] [PubMed] [Google Scholar]
- Dyson, N., 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12: 2245–2262. [DOI] [PubMed] [Google Scholar]
- Eaton, S., 2003. Cell biology of planar polarity transmission in the Drosophila wing. Mech. Dev. 120: 1257–1264. [DOI] [PubMed] [Google Scholar]
- Edgar, B. A., and C. F. Lehner, 1996. Developmental control of cell cycle regulators: a fly's perspective. Science 274: 1646–1652. [DOI] [PubMed] [Google Scholar]
- Egger, B., R. Leemans, T. Loop, L. Kammermeier, Y. Fan et al., 2002. Gliogenesis in Drosophila: genome-wide analysis of downstream genes of glial cells missing in the embryonic nervous system. Development 129: 3295–3309. [DOI] [PubMed] [Google Scholar]
- Ekholm, S. V., and S. I. Reed, 2000. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 12: 676–684. [DOI] [PubMed] [Google Scholar]
- Fanto, M., L. Clayton, J. Meredith, K. Hardiman, B. Charroux et al., 2003. The tumor-suppressor and cell adhesion molecule Fat controls planar polarity via physical interactions with Atrophin, a transcriptional co-repressor. Development 130: 763–774. [DOI] [PubMed] [Google Scholar]
- Frydman, H. M., and A. C. Spradling, 2001. The receptor-like tyrosine phosphatase lar is required for epithelial planar polarity and for axis determination within drosophila ovarian follicles. Development 128: 3209–3220. [DOI] [PubMed] [Google Scholar]
- Gateff, E., U. Kurzik-Dumke, J. Wismar, T. Loffler, N. Habtemichael et al., 1996. Drosophila differentiation genes instrumental in tumor suppression. Int. J. Dev. Biol. 40: 149–156. [PubMed] [Google Scholar]
- Giles, R. H., J. H. van Es and H. Clevers, 2003. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 1653: 1–24. [DOI] [PubMed] [Google Scholar]
- Giot, L., J. S. Bader, C. Brouwer, A. Chaudhuri, B. Kuang et al., 2003. A protein interaction map of Drosophila melanogaster. Science 320: 1727–1736. [DOI] [PubMed] [Google Scholar]
- Greenwalt, D. E., R. H. Lipsky, C. F. Ockenhouse, H. Ikeda, N. N. Tandon et al., 1992. Membrane glycoprotein CD36: a review of its roles in adherence, signal transduction, and transfusion medicine. Blood 80: 1105–1115. [PubMed] [Google Scholar]
- Grigliatti, T. A., 1998 Mutagenesis, pp. 55–83 in Drosophila: A Practical Approach, edited by D. B. Roberts. Oxford University Press, London/New York/Oxford.
- Harbour, J. W., and D. C. Dean, 2000. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 12: 685–689. [DOI] [PubMed] [Google Scholar]
- Harbour, J. W., and D. C. Dean, 2001. Corepressors and retinoblastoma protein function. Curr. Top. Microbiol. Immunol. 254: 137–144. [DOI] [PubMed] [Google Scholar]
- Hay, B. A., and M. Guo, 2003. Coupling cell growth, proliferation, and death. Hippo weighs in. Dev. Cell 5: 361–363. [DOI] [PubMed] [Google Scholar]
- Hazlehurst, L. A., J. S. Damiano, I. Buyuksal, W. J. Pledger and W. S. Dalton, 2000. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 19: 4319–4327. [DOI] [PubMed] [Google Scholar]
- Hering, H., and M. Sheng, 2002. Direct interaction of Frizzled-1, -2, -4, and -7 with PDZ domains of PSD-95. FEBS Lett. 521: 185–189. [DOI] [PubMed] [Google Scholar]
- Horsfield, J., A. Penton, J. Secombe, F. M. Hoffman and H. Richardson, 1998. decapentaplegic is required for arrest in G1 phase during Drosophila eye development. Development 125: 5069–5078. [DOI] [PubMed] [Google Scholar]
- Huang, Y. Z., M. Zang, W. C. Xiong, Z. Luo and L. Mei, 2003. Erbin suppresses the MAP kinase pathway. J. Biol. Chem. 278: 1108–1114. [DOI] [PubMed] [Google Scholar]
- Humbert, P., S. Russell and H. Richardson, 2003. Dlg, Scrib and Lgl in cell polarity, cell proliferation and cancer. BioEssays 25: 542–553. [DOI] [PubMed] [Google Scholar]
- Iwai, Y., Y. Hirota, K. Ozaki, H. Okano, M. Takeichi et al., 2002. DN-cadherin is required for spatial arrangement of nerve terminals and ultrastructural organization of synapses. Mol. Cell. Neurosci. 19: 375–388. [DOI] [PubMed] [Google Scholar]
- Jaffe, A. B., and A. Hall, 2002. Rho GTPases in transformation and metastasis. Adv. Cancer Res. 84: 57–80. [DOI] [PubMed] [Google Scholar]
- Jasper, H., V. Benes, A. Atzberger, S. Sauer, W. Ansorge et al., 2002. A genomic switch at the transition from cell proliferation to terminal differentiation in the Drosophila eye. Dev. Cell 3: 511–521. [DOI] [PubMed] [Google Scholar]
- Johnston, L. A., and B. A. Edgar, 1998. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature 394: 82–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, L. A., and A. L. Sanders, 2003. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat. Cell Biol. 5: 827–833. [DOI] [PubMed] [Google Scholar]
- Johnston, L. A., D. A. Prober, B. A. Edgar, R. N. Eisenman and P. Gallant, 1999. Drosophila myc regulates cellular growth during development. Cell 98: 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallay, L. M., Y. Braiterman, A. Chen and A. L. Hubbard, 2003. scribble interacts with the pdz-binding motif of Vangl2 in vitro. Mol. Biol. Cell (Suppl. 14): 461a. [Google Scholar]
- Karim, F. D., and G. M. Rubin, 1998. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125: 1–9. [DOI] [PubMed] [Google Scholar]
- Katoh, M., 2002. Strabismus (STB)/Vang-like (VANGL) gene family (Review). Int. J. Mol. Med. 10: 11–15. [PubMed] [Google Scholar]
- Keegan, J., M. Schmerer, B. Ring and D. Garza, 2001. The 62E early-late puff of Drosophila contains D-spinophilin, an ecdysone-inducible PDZ-domain protein dynamically expressed during metamorphosis. Genet. Res. 77: 27–39. [DOI] [PubMed] [Google Scholar]
- Knoblich, J. A., K. Sauer, L. Jones, H. Richardson, R. Saint et al., 1994. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell 77: 107–120. [DOI] [PubMed] [Google Scholar]
- Lane, M. E., K. Sauer, K. Wallace, Y. N. Jan, C. F. Lehner et al., 1996. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell 87: 1225–1235. [DOI] [PubMed] [Google Scholar]
- Lane, M. E., M. Elend, D. Heidmann, A. Herr, S. Marzodko et al., 2000. A screen for modifiers of cyclin E function in Drosophila melanogaster identifies Cdk2 mutations, revealing the insignificance of putative phosphorylation sites in Cdk2. Genetics 155: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C. H., T. Herman, T. R. Clandinin, R. Lee and S. L. Zipursky, 2001. N-cadherin regulates target specificity in the Drosophila visual system. Neuron 30: 437–450. [DOI] [PubMed] [Google Scholar]
- Lehman, K., G. Rossi, J. E. Adamo and P. Brennwald, 1999. Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J. Cell Biol. 146: 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S., Y. Li, R. W. Carthew and Z. C. Lai, 1997. Photoreceptor cell differentiation requires regulated proteolysis of the transcriptional repressor Tramtrack. Cell 90: 469–478. [DOI] [PubMed] [Google Scholar]
- Lifschytz, E., and R. Falk, 1968. Fine structure of a chromosome segment in Drosophila. Drosoph. Inf. Serv. 43: 193. [Google Scholar]
- Lim, J. Y., H. Kim, S. W. Kim, P. W. Huh, K. H. Lee et al., 2003. Merlin suppresses the SRE-dependent transcription by inhibiting the activation of Ras-ERK pathway. Biochem. Biophys. Res. Commun. 302: 238–245. [DOI] [PubMed] [Google Scholar]
- Lin, H. V., D. B. Doroquez, S. Cho, F. Chen, I. Rebay et al., 2003. Splits ends is a tissue/promoter specific regulator of Wingless signaling. Development 130: 3125–3135. [DOI] [PubMed] [Google Scholar]
- Lu, B., F. Roegiers, L. Y. Jan and Y. N. Jan, 2001. Adherens junctions inhibit asymmetric division in the Drosophila epithelium. Nature 409: 522–525. [DOI] [PubMed] [Google Scholar]
- Lundberg, A. S., and R. A. Weinberg, 1998. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18: 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, L., 2000. Trio quartet in D. (melanogaster). Neuron 26: 1–2. [DOI] [PubMed] [Google Scholar]
- Malumbres, M., and M. Barbacid, 2003. Ras oncogenes: the first 30 years. Nat. Cancer Rev. 3: 7–13. [DOI] [PubMed] [Google Scholar]
- Martin, K. A., and J. Blenis, 2002. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv. Cancer Res. 86: 1–39. [DOI] [PubMed] [Google Scholar]
- Matsuo, T., K. Takahashi, S. Kondo, K. Kaibuchi and D. Yamamoto, 1997. Regulation of cone cell formation by Canoe and Ras in the developing Drosophila eye. Development 124: 2671–2680. [DOI] [PubMed] [Google Scholar]
- McCartney, B. M., R. M. Kulikauskas, D. R. LaJeunesse and R. G. Fehon, 2000. The neurofibromatosis-2 homologue, Merlin, and the tumor suppressor expanded function together in Drosophila to regulate cell proliferation and differentiation. Development 127: 1315–1324. [DOI] [PubMed] [Google Scholar]
- Mettouchi, A., S. Klein, W. Guo, M. Lopez-Lago, E. Lemichez et al., 2001. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol. Cell 8: 115–127. [DOI] [PubMed] [Google Scholar]
- Meyer, C. A., H. W. Jacobs, S. A. Datar, W. Du, B. A. Edgar et al., 2000. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19: 4533–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michl, P., C. Barth, M. Buchholz, M. M. Lerch, M. Rolke et al., 2003. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 63: 6265–6271. [PubMed] [Google Scholar]
- Moberg, K. H., D. W. Bell, D. C. Wahrer, D. A. Haber and I. K. Hariharan, 2001. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413: 311–316. [DOI] [PubMed] [Google Scholar]
- Montcouquiol, M., R. A. Rachel, P. J. Lanford, N. G. Copeland, N. A. Jenkins et al., 2003. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423: 173–177. [DOI] [PubMed] [Google Scholar]
- Moore, A. W., L. Y. Jan and Y. N. Jan, 2002. hamlet, a binary genetic switch between single- and multiple-dendrite neuron morphology. Science 297: 1355–1358. [DOI] [PubMed] [Google Scholar]
- Murdoch, J. N., D. J. Henderson, K. Doudney, C. Gaston-Massuet, H. M. Phillips et al., 2003. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum. Mol. Genet. 12: 87–98. [DOI] [PubMed] [Google Scholar]
- Murphy, T. D., 2003. Drosophila skpA, a component of SCF ubiquitin ligases, regulates centrosome duplication independently of cyclin E accumulation. J. Cell Sci. 116: 2321–2332. [DOI] [PubMed] [Google Scholar]
- Musch, A., D. Cohen, C. Yeaman, W. J. Nelson, E. Rodriguez-Boulan et al., 2002. Mammalian homolog of Drosophila tumor suppressor lethal (2) giant larvae interacts with basolateral exocytic machinery in Madin-Darby canine kidney cells. Mol. Biol. Cell 13: 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoulas, O., S. J. Beek, S. L. Moseley, C. M. McCallum, M. Sarte et al., 1998. The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development 125: 3955–3966. [DOI] [PubMed] [Google Scholar]
- Parmentier, M. L., D. Woods, S. Greig, P. G. Phan, A. Radovic et al., 2000. Rapsynoid/partner of inscuteable controls asymmetric division of larval neuroblasts in Drosophila. J. Neurosci. 20: RC84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazman, C., C. A. Mayes, M. Fanto, S. R. Haynes and M. Mlodzik, 2000. Rasputin, the Drosophila homologue of the RasGAP SH3 binding protein, functions in ras- and Rho-mediated signaling. Development 127: 1715–1725. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno, M., C. Jamora and E. Fuchs, 2003. Sticky business: orchestrating cellular signals at adherens junctions. Cell 112: 535–548. [DOI] [PubMed] [Google Scholar]
- Prober, D. A., and B. A. Edgar, 2000. Ras1 promotes cellular growth in the Drosophila wing. Cell 100: 435–446. [DOI] [PubMed] [Google Scholar]
- Prout, M., Z. Damania, J. Soong, D. Fristrom and J. W. Fristrom, 1997. Autosomal mutations affecting adhesion between wing surfaces in Drosophila melanogaster. Genetics 146: 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K., and C. J. Der, 2001. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 171: 1–10. [DOI] [PubMed] [Google Scholar]
- Rehberg, M., and R. Graf, 2002. Dictyostelium EB1 is a genuine centrosomal component required for proper spindle formation. Mol. Biol. Cell 13: 2301–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, H., L. V. O'Keefe, T. Marty and R. Saint, 1995. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development 121: 3371–3379. [DOI] [PubMed] [Google Scholar]
- Richardson, H. E., L. V. O'Keefe, S. I. Reed and R. Saint, 1993. A Drosophila G1-specific cyclin E homolog exhibits different modes of expression during embryogenesis. Development 119: 673–690. [DOI] [PubMed] [Google Scholar]
- Rogers, S. L., G. C. Rogers, D. J. Sharp and R. D. Vale, 2002. Drosophila EB1 is important for proper assembly, dynamics, and positioning of the mitotic spindle. J. Cell Biol. 158: 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls, M. M., R. Albertson, H. P. Shih, C. Y. Lee and C. Q. Doe, 2003. Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell Biol. 163: 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai, E., and C. J. Marshall, 2002. Rho-GTPases and cancer. Nat. Rev. Cancer 2: 133–142. [DOI] [PubMed] [Google Scholar]
- Sander, E. E., J. P. ten Klooster, S. van Delft, R. A. van der Kammen and J. G. Collard, 1999. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147: 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan, K., and F. Sobels, 1976 Radiation genetics, pp. 1090–1251 in The Genetics and Biology of Drosophila, Vol. 1c, edited by M. Ashburner and E. Novitski. Academic Press, London.
- Sauer, F., D. A. Wassarman, G. M. Rubin and R. Tjian, 1996. TAF(II)s mediate activation of transcription in the Drosophila embryo. Cell 87: 1271–1284. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S. M., and K. Burridge, 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11: 274–286. [DOI] [PubMed] [Google Scholar]
- Secombe, J., J. Pispa, R. Saint and H. Richardson, 1998. Analysis of a Drosophila cyclin E hypomorphic mutation suggests a novel role for cyclin E in cell proliferation control during eye imaginal disc development. Genetics 149: 1867–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan, S. K., and A. Bradley, 1998. Functional characterization of BRCA1 and BRCA2: clues from their interacting proteins. J. Mamm. Gland Biol. Neoplasia 3: 413–421. [DOI] [PubMed] [Google Scholar]
- Sharrow, M., and M. Tiemeyer, 2001. Gliolectin-mediated carbohydrate binding at the Drosophila midline ensures the fidelity of axon pathfinding. Development 128: 4585–4595. [DOI] [PubMed] [Google Scholar]
- Shen, B., H. Liu, E. Y. Skolnik and J. L. Manley, 2001. Physical and functional interactions between Drosophila TRAF2 and Pelle kinase contribute to Dorsal activation. Proc. Natl. Acad. Sci. USA 98: 8596–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr, C. J., and J. M. Roberts, 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13: 1501–1512. [DOI] [PubMed] [Google Scholar]
- Simske, J. S., S. M. Kaech, S. A. Harp and S. K. Kim, 1996. LET-23 receptor localization by the cell junction protein LIN-7 during C. elegans vulval induction. Cell 85: 195–204. [DOI] [PubMed] [Google Scholar]
- Smith, A. V., J. A. King and T. L. Orr-Weaver, 1993. Identification of genomic regions required for DNA replication during Drosophila embryogenesis. Genetics 135: 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol, N. S., and L. Cooley, 2003. Drosophila filamin is required for follicle cell motility during oogenesis. Dev. Biol. 260: 260–272. [DOI] [PubMed] [Google Scholar]
- Staehling-Hampton, K., P. J. Ciampa, A. Brook and N. Dyson, 1999. A genetic screen for modifiers of E2F in Drosophila melanogaster. Genetics 153: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, K. A., G. H. Yee, C. E. Roote, E. L. Williams, S. Zusman et al., 1997. A novel alpha integrin subunit associates with betaPS and functions in tissue morphogenesis and movement during Drosophila development. Development 124: 4583–4594. [DOI] [PubMed] [Google Scholar]
- Stossel, T. P., J. Condeelis, L. Cooley, J. H. Hartwig, A. Noegel et al., 2001. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell. Biol. 2: 138–145. [DOI] [PubMed] [Google Scholar]
- Su, L. K., M. Burrell, D. E. Hill, J. Gyuris, R. Brent et al., 1995. APC binds to the novel protein EB1. Cancer Res. 55: 2972–2977. [PubMed] [Google Scholar]
- Svoboda, Y. H., M. K. Robson and J. A. Sved, 1995. P-element-induced male recombination can be produced in Drosophila melanogaster by combining end-deficient elements in trans. Genetics 139: 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanentzapf, G., and U. Tepass, 2003. Interactions between the crumbs, lethal giant larvae and bazooka pathways in epithelial polarization. Nat. Cell Biol. 5: 46–52. [DOI] [PubMed] [Google Scholar]
- Tang, A. H., T. P. Neufeld, E. Kwan and G. M. Rubin, 1997. PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism. Cell 90: 459–467. [DOI] [PubMed] [Google Scholar]
- Tepass, U., E. Gruszynski-DeFeo, T. A. Haag, L. Omatyar, T. Torok et al., 1996. shotgun encodes Drosophila E-cadherin and is preferentially required during cell rearrangement in the neurectoderm and other morphogenetically active epithelia. Genes Dev. 10: 672–685. [DOI] [PubMed] [Google Scholar]
- Tepass, U., G. Tanentzapf, R. Ward and R. Fehon, 2001. Epithelial cell polarity and cell junctions in Drosophila. Annu. Rev. Genet. 35: 747–784. [DOI] [PubMed] [Google Scholar]
- Thomas, B. J., and D. A. Wassarman, 1999. A fly's eye view of biology. Trends Genet. 15: 184–190. [DOI] [PubMed] [Google Scholar]
- Thomas, B. J., K. H. Zavitz, X. Dong, M. E. Lane, K. Weigmann et al., 1997. roughex down-regulates G2 cyclins in G1. Genes Dev. 11: 1289–1298. [DOI] [PubMed] [Google Scholar]
- Torok, T., G. Tick, M. Alvarado and I. Kiss, 1993. P-lacW insertional mutagenesis on the second chromosome of Drosophila melanogaster: isolation of lethals with different overgrowth phenotypes. Genetics 135: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng, A. S., and I. K. Hariharan, 2002. An overexpression screen in Drosophila for genes that restrict growth or cell-cycle progression in the developing eye. Genetics 162: 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura, T., H. Oda, R. Kraut, S. Hayashi, Y. Kotaoka et al., 1996. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes Dev. 10: 659–671. [DOI] [PubMed] [Google Scholar]
- Volarevic, S., M. J. Stewart, B. Ledermann, F. Zilberman, L. Terracciano et al., 2000. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288: 2045–2047. [DOI] [PubMed] [Google Scholar]
- Walsh, E. P., and N. H. Brown, 1998. A screen to identify Drosophila genes required for integrin-mediated adhesion. Genetics 150: 791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore, C., 2003. Sonic hedgehog in normal and neoplastic proliferation: insight gained from human tumors and animal models. Curr. Opin. Genet. Dev. 13: 34–42. [DOI] [PubMed] [Google Scholar]
- Wu, S., J. Huang, J. Dong and D. Pan, 2003. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114: 445–456. [DOI] [PubMed] [Google Scholar]
- Xin, S., L. Weng, J. Xu and W. Du, 2002. The role of RBF in developmentally regulated cell proliferation in the eye disc and in Cyclin D/Cdk4 induced cellular growth. Development 129: 1345–1356. [DOI] [PubMed] [Google Scholar]
- Yang, C. H., J. D. Axelrod and M. A. Simon, 2002. Regulation of Frizzled by fat-like cadherins during planar polarity signaling in the Drosophila compound eye. Cell 108: 675–688. [DOI] [PubMed] [Google Scholar]
- Zhang, H., J. P. Stallock, J. C. Ng, C. Reinhard and T. P. Neufeld, 2000. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 14: 2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. D., J. Kassis, B. Olde, D. M. Mellerick and W. F. Odenwald, 1996. Pollux, a novel Drosophila adhesion molecule, belongs to a family of proteins expressed in plants, yeast, nematodes, and man. Genes Dev. 10: 1108–1119. [DOI] [PubMed] [Google Scholar]