

Abstract
We constructed male and female consensus linkage maps for the Pacific oyster Crassostrea gigas, using a total of 102 microsatellite DNA markers typed in 11-day-old larvae from three families. We identified 11 and 12 linkage groups in the male and female consensus maps, respectively. Alignment of these separate maps, however, suggests 10 linkage groups, which agrees with the haploid chromosome number. The male linkage map comprises 88 loci and spans 616.1 cM, while the female map comprises 86 loci and spans 770.5 cM. The male and the female maps share 74 loci; 2 markers remain unlinked. The estimated coverages for the consensus linkage maps are 79% for the male and 70–75% for the female, on the basis of two estimates of genome length. Ninety-five percent of the genome is expected to lie within 16 and 21 cM of markers on the male and female maps, respectively, while 95% of simulated minimum distances to the male and female maps are within 10.1 and 13.6 cM, respectively. Females have significantly more recombination than males, across 118 pairs of linked markers in common to the parents of the three families. Significant differences in recombination and orders of markers are also evident among same-sex parents of different families as well as sibling parents of opposite sex. These observations suggest that polymorphism for chromosomal rearrangements may exist in natural populations, which could have profound implications for interpreting the evolutionary genetics of the oyster. These are the first linkage maps for a bivalve mollusc that use microsatellite DNA markers, which should enable them to be transferred to other families and to be useful for further genetic analyses such as QTL mapping.
ACCORDING to the “Elm-Oyster Model” (Williams 1975), high fecundity and early mortality of trees and bivalve molluscs favor individual variation in fitness and in traits correlated with fitness. Consistent with a prediction of this model, recent breeding experiments have uncovered a large load of highly deleterious recessive mutations in the Pacific oyster Crassostrea gigas (Launey and Hedgecock 2001; Bucklin 2002). Large mutational loads in oysters and other bivalves likely explain previous reports of correlation between fitness-related traits, such as growth, and heterozygosity at allozyme-coding loci (Zouros and Pogson 1994; Britten 1996; David 1998), growth heterosis and inbreeding depression (Lannan 1980; Hedgecock et al. 1995, 1996; Bayne et al. 1999; Evans et al. 2003), and negative correlation between growth and degree of somatic cell aneuploidy (Thiriot-Quievreux et al. 1992; Zouros et al. 1996; Leitão et al. 2001). The source and maintenance of mutational loads and whether balancing selection plays any role in maintaining genetic diversity and fitness variation in these highly fecund species remain open questions that will likely be resolved only through further experiments. A genetic linkage map would facilitate these studies.
Oysters are economically important. The Pacific oyster has had the highest worldwide production of any cultured aquatic species since 1993; in 2001, world production of this species was 4.1 million metric tons (Food and Agriculture Organization 2003). Still, oysters are in an early stage of domestication and only a few large-scale breeding programs have been initiated (e.g., Hedgecock et al. 1997; Langdon et al. 2003). A linkage map would facilitate marker-assisted selection, QTL-mapping, and functional genomic approaches to improving yield of farmed oysters.
A moderately dense linkage map can be made rapidly using amplified fragment length DNA polymorphisms (RAPDs and AFLP markers), as recently demonstrated for several aquatic species, including Eastern and Pacific oysters (Yu and Guo 2003; Li and Guo 2004). Such markers may transfer poorly to new populations or even to crosses from the same population (Y. Li et al. 2003), however, so we have elected to construct a less dense but more portable linkage map of codominant microsatellite DNA polymorphisms (Love et al. 1990). Microsatellite DNA markers have high frequencies of null alleles in oysters, which poses problems for studies of natural populations but not for mapping in pedigreed populations (McGoldrick et al. 2000; Launey and Hedgecock 2001; G. Li et al. 2003).
Widespread distortions of segregation ratios have hampered many previous studies of inheritance and linkage in various bivalve molluscs (reviewed by McGoldrick et al. 2000). This phenomenon is explained by a large load of recessive deleterious mutations and low survival of identical-by-descent homozygotes (Launey and Hedgecock 2001). Much of the selective mortality occurs around the time of metamorphosis from the larval to the juvenile stage (Launey and Hedgecock 2001; Bucklin 2002). To reduce or eliminate segregation distortion caused by selection, we made a linkage map by genotyping 11-day-old larvae from double-hybrid crosses. The 11-day-old larva maximizes the amount of template DNA while minimizing the chances that selective mortality has already distorted segregation ratios. The double-hybrid cross reduces the probability of identity by descent.
A primary genetic linkage map is an essential prerequisite to detailed genetic studies in any organism. We present microsatellite linkage maps for the Pacific oyster C. gigas based on analysis of three different, double-hybrid reference families developed for the study. We report map locations for 100 microsatellite markers, composing 10 linkage groups, noting significant differences in recombination and marker order between male and female parents and also among parents of the same sex.
MATERIALS AND METHODS
Oyster families:
Double-hybrid crosses were made among four filial lines (F3 7 × 6; F2 5 × 2; F2 2 × 5; F2 7 × 9), which were derived by inbreeding and crossbreeding from a naturalized population of Pacific oysters in Dabob Bay, Washington (Hedgecock 1994; Launey and Hedgecock 2001). While families 2 × 5 and 5 × 2 share great-grandparents (from inbred lines 92-2 and 89-5, respectively), families 7 × 6 and 7 × 9 are descended from lines 89-7 and 93-7, respectively, and are unrelated. Each mapping family was obtained from a single, biparental cross, following standard methods for artificial fertilization and larval rearing (Hedgecock et al. 1995). The three mapping families, designated 1, 2, and 3, male parent listed first, are (7 × 9) × (2 × 5), (2 × 5) × (7 × 9), and (7 × 6) × (5 × 2). Families 1 and 2 produced by reciprocal crosses between the same two F2 families are more closely related to each other than either is to family 3. Larvae from these families were harvested at 11 days, killed with buffered formalin (3 drops/15 ml seawater), rinsed, and preserved in 70% ethanol.
DNA extraction, microsatellite markers, and PCR procedures:
DNA from the parents, as well as from the grand- and great-grandparents of the mapping families, was extracted from adductor muscle using a CTAB extraction (G. Li et al. 2003). DNA from 11-day-old larvae was extracted with proteinase-K, at 1 mg/ml of buffer (1.5 ml thermophilic DNA polymerase 10× buffer; Promega, Madison, WI), 75 μl Tween 20, adjusted to 15 ml with distilled water). Individual larvae were extracted with 200 μl of this solution, in 96-well trays, and then incubated at 55° for 3 hr and at 95° for 30 min, using a Perkin-Elmer 9600 thermocycler. Aliquots of 50 μl of the extract were frozen to minimize DNA degradation. DNA was successfully extracted from larvae that were kept in 70% ethanol for >1 year.
We tested 115 microsatellite loci for this study, finding 102 informative. We used the 79 microsatellites (ucdCgi107-204) recently described by G. Li et al. (2003), as well as the 11 loci previously cloned in this laboratory (ucdCgi001, -002, -003, -006, -014, -017, -018, -021, -022, -024, and -028; McGoldrick 1997; McGoldrick et al. 2000). We used 12 loci from the literature [CG44, CG49, and CG108 from Magoulas et al. (1998)(), renamed here for consistency imbCgi44, imbCgi49, and imbCgi108; L10, L16, and L48 from Huvet et al. (2000)(), renamed here um2Cgi10, um2Cgi16, and um2Cgi48; and cmrCgi1, cmrCgi3, cmrCgi5, cmrCgi61, cmrCgi141, and cmrCgi151 from McGoldrick et al. (2000)].
Amplification of microsatellite DNA by polymerase chain reaction (PCR) was performed in a 7.5-μl reaction, using 96-well plates containing 1.5 μl of template DNA, 1 mm Taq buffer, varying concentrations of MgCl2, depending on the locus, 125 μm of dNTP, 400 μm of tetramethylrhodamine-6-dATP (NEL 470; New England Nuclear, Boston), 1.0 μm of each primer, and 0.325 units Taq polymerase (Promega). PCR was done with an initial denaturing at 92° for 2 min, followed by 30 cycles each of 30 sec denaturing at 92°, 30 sec annealing at locus-specific temperatures, 45 sec extension at 72°, and concluding with a final extension at 72° for 5 min. Products were separated on 8% acrylamide gels (acrylamide:bisacrylamide, 29:1, 7 m urea), using 1× Tris borate EDTA (TBE) buffer, and visualized using a Hitachi FMBIO II scanner (MiraiBio, Alameda, CA).
Distortion of segregation ratios:
We selected 11-day-old larvae for analysis, to minimize segregation distortion (Launey and Hedgecock 2001; Bucklin 2002) while providing sufficient DNA. A preliminary test of segregation distortion for 18 loci in family (7 × 6) × (5 × 2) showed only one distortion significant at the 5% level (the number of individuals tested ranged from 47 to 242 with an average of 114), so we judged our strategies for avoiding segregation distortion to be successful. For the remainder of the data, we tested segregation distortion locus by locus, for each extraction, and over all extractions, using chi-square tests, with the significance of the individual tests adjusted for multiple testing (Bonferroni correction; Rice 1989). The adjustment for multiple testing was done within types of segregation, depending on whether a pair of parents had two alleles (cross type AA × AB, A⊘ × AB, or AB × AB), three alleles (AA × BC, A⊘ × BC, or AB × AC) or four alleles (AB × CD).
Linkage analysis:
Grandparents and parents were genotyped for 115 microsatellite markers to determine polymorphism and linkage phase. Because of the small volume of template DNA available from a larva (∼200 μl), four trays of individuals were extracted for each family to test linkage of all loci. Each extraction tray consisted of both parents and 94 larvae. We tested a maximum number of markers with the first extraction to determine provisional linkage groups. A second extraction was used to test the linkage of untested markers, along with a couple of loci from each of the provisional linkage groups. The third and fourth extractions were used to confirm linkage groups, by genotyping simultaneously all loci belonging to each linkage group. Maps obtained in these studies are based on the third and the fourth extractions. The third extraction was used to order loci in the first five or six linkage groups and the fourth extraction was used to order loci in the remaining linkage groups.
Linkage was assessed separately for the male and female parents of each mapping cross, using the backcross design of Mapmaker 3.0 (Lander et al. 1987). First, the paternal and maternal alleles were separated in the progeny, where permitted by the allelic diversity of the grandparents at a locus, and then progeny genotypes were coded A, if the parental allele was derived from the grandfather (i.e., this genotype was coded as the homozygote in a backcross to a paternal inbred line), and H, if the parental allele was derived from the grandmother (i.e., this genotype was coded as the heterozygote in the backcross). When grandparental alleles could not be unambiguously assigned, alternative linkage phases were used to determine the correct parental haplotype. Coded male and female data were processed separately, yielding separate male and female maps. An initial grouping of markers was performed with a LOD cutoff of 3.0. The sequence of markers in each group was determined with the group compare command, and the Kosambi distances were obtained by the map command. The automatic error detection command (Lincoln and Lander 1992) was used to search for potential double recombinants. Once errors were corrected, by removing double recombinants, map distances were recalculated. The final order was confirmed using the ripple command in MAPMAKER, which compares the likelihood of the original map order with those generated by changing the order of the neighboring loci. A consensus map for each sex was built by compiling data for the three families. The family with the largest number of loci was used as a framework, to which information from the other two families was added. Results from the heterogeneity G-tests (below) were used to adjust distances between markers; i.e., if two or three families showed no significant difference for a pair of loci, the distance between this pair was obtained from the pooled data and put on the map. Maps were drawn using MapChart software (Voorrips 2002).
Genome size and coverage:
We estimated the genome lengths from the consensus male and female linkage maps in two ways. First, we calculated the average spacing, s, between markers, dividing the total length of all linkage groups by the number of intervals (number of markers minus number of linkage groups, 11 in the male and 12 in the female). We estimated genome length by adding 2s to the length of each linkage group to account for terminal chromosome regions (Fishman et al. 2001). Second, after method 4 of Chakravarti et al. (1991), we multiplied the length of each linkage group by a factor of (m + 1)/(m − 1), where m is the number of loci on the linkage group (i.e., disregarding markers mapping to the same place). We estimated the coverage of the male and female consensus maps by simulating the addition of 10,000 genes to each linkage group, assigning relative positions with a random number from the interval [0, 1]. The relative positions of mapped markers were calculated after adding s to both ends of the linkage group and dividing the distance of each marker from one end by the total length of the linkage group (map length plus 2s). We then calculated the mean and standard deviation of the minimum distance of simulated genes to the framework of existing markers (in map units). These statistics were then compared to the expected distance of a gene from the closest of n random markers, E(m) = L[2(n + 1)]−1, with its upper 95% confidence interval of (L/2)(1 − 0.051/n) (Lynch and Walsh 1998). When there are C linear chromosomes, the proportion of a genome of length L that is within m map units of n randomly distributed markers is
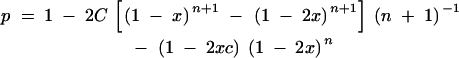 |
(where x = m/L) (Bishop et al. 1983).
Differences among families:
Significance of recombination within families and heterogeneity of recombination among extractions and among families were tested by hierarchical G-statistics (Liu 1997). We first compared segregation from male and female parents separately, by linkage group, for each pair of loci in common. We then tested the heterogeneity of recombination between male and female parents, using pooled data from the previous step, when recombination was statistically homogeneous among extractions or parents.
RESULTS
Extractions and number of loci tested:
Of the 115 microsatellite DNA markers typed, 102 markers were informative in at least one family and were available for mapping. The total number of loci tested was 68 for family 1 (7 × 9) × (2 × 5), 67 for family 2 (2 × 5) × (7 × 9), and 78 for family 3 (7 × 6) × (5 × 2). Of the 102 segregating markers, 73 (72%) were assayed in two or more families; of the 29 markers segregating in only one family, 22 are in family 3, the most distantly related. The mean numbers of individuals tested per locus for all extractions were 169.1, 118.5, and 177.0 for families 1, 2, and 3, respectively (Table 1).
TABLE 1.
Number of loci and individuals tested, by extraction, for three mapping families
| Extraction
|
|||||
|---|---|---|---|---|---|
| Family | 1 | 2 | 3 | 4 | Total |
| 1. (7 × 9) × (2 × 5) | |||||
| No. of loci | 22 | 38 | 36 | 48 | 68 |
| Mean no. of larvae per locus | 74.5 | 73.0 | 86.1 | 83.7 | 169.1 |
| N minimum | 55 | 49 | 49 | 67 | 55 |
| N maximum | 83 | 89 | 94 | 94 | 346 |
| 2. (2 × 5) × (7 × 9) | |||||
| No. of loci | 23 | 49 | 25 | — | 67 |
| Mean no. of larvae per locus | 82.7 | 83.9 | 84.9 | — | 118.5 |
| N minimum | 62 | 43 | 62 | — | 52 |
| N maximum | 94 | 93 | 94 | — | 240 |
| 3. (7 × 6) × (5 × 2) | |||||
| No. of loci | 36 | 48 | 66 | 20 | 78 |
| Mean no. of larvae per locus | 86.9 | 79.1 | 82.4 | 87.4 | 177.0 |
| N minimum | 37 | 48 | 52 | 76 | 67 |
| N maximum | 94 | 89 | 93 | 91 | 348 |
Segregation distortion:
Distortions of observed genotypic ratios from those expected under Mendelian inheritance were found at only seven loci across the three mapping families; the chi-square test results are given in supplementary tables at http://www.genetics.org/supplemental/. In family 2, four loci (ucdCgi181, ucdCgi190, ucdCgi200, and imbCig44) showed significant deviation from Mendelian segregation ratios. For each of the two loci, ucdCgi181 and ucdCgi200, which form linkage group X in family 1, one of four expected genotypes was absent or present only once in sample sizes of 85 and 75, respectively. These same loci show no segregation distortion in family 1. At the imbCgi44 locus, the departure from Mendelian expectations is attributable to a significant imbalance in the representation of the dam's alleles (a ratio of 53:133 for the 090 and null alleles, respectively). At the ucdCgi190 locus, the parental cross of type A⊘ × AB yields a 71:22:64 ratio for the dominant-A, null heterozygote, and codominant categories AB, respectively, suggesting a deficiency of null heterozygotes. In family 3, ucdCgi150 and cmrCgi161 loci were distorted; although both are A⊘ × AB-type crosses, the former shows a deficiency of the dominant category (22 AA + A⊘:26 B⊘:19 AB), suggesting selection against AA homozygotes (Launey and Hedgecock 2001), while the latter shows a deficiency of null heterozygotes (39 AA + A⊘:8 B⊘:35 AB).
Linkage maps:
The genotypes of grandparents, parents, and progeny for the three mapping families are given in supplementary tables at http://www.genetics.org/supplemental/. On the basis of these data, we present consensus maps for male and female parents, representing, when aligned to each other, 10 linkage groups (Figure 1). Of the 102 markers segregating in at least one family, 100 (98%) show detectable linkage. The two unlinked markers (ucdCgi179, ucdCgi180) were segregating only in family 3. We did not test ucdCgi008 in our families but added it to the first linkage group, on the basis of previous evidence that it is linked to ucdCgi018 (Launey and Hedgecock 2001). Solid segments and underlined locus names in Figure 1 indicate linkages supported by observations from more than one family.
Figure 1.—


Consensus linkage maps for the Pacific oyster Crassostrea gigas, based on genotypes for 100 microsatellite DNA markers in the three F2 mapping families. The 10 linkage groups are ordered by mean total length, from largest to smallest. Maps based on recombination from female parents are on the left; male maps are on the right. Intervals, for which the recombination rate is statistically homogeneous among families, are indicated by solid segments and underlining of flanking loci names. The “i” is omitted from marker names; asterisk next to ucdCgi008 denotes locus location taken from Launey and Hedgecock (2001). Dotted brackets indicate that gene orders were significantly different (LOD > 3.0) between two families.
The male map comprises 88 loci, spanning 616.1 cM, with an average spacing of 8.0 cM. The number of loci per linkage group varies from 2 to 13 with a mean of 8 (Table 2). The female map comprises 86 loci, covering 770.5 cM with an average spacing of 10.4 cM. The number of loci per linkage group varies from 2 to 20 with a mean of 7.2. The two consensus maps share 74 loci. We arbitrarily ordered the 10 linkage groups from longest to shortest, on the basis of average length in centimorgans of the male and female maps (Table 2).
TABLE 2.
Statistics for 10 linkage groups composing a genetic map for the Pacific oyster
| Male
|
Female
|
Marker spacing
|
Loci
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| LG | No. loci | cM | No. loci | CM | Mean length |
Male | Female | Common | Specific |
| I | 13 | 124.4 | 20 | 147.9 | 136.2 | 10.4 | 7.8 | 12 | 10 |
| II | 12 | 94.4 | 11 | 108.3 | 101.4 | 8.6 | 10.8 | 9 | 5 |
| III | 9 | 66.6 | 8 | 131.1 | 98.8 | 8.3 | 18.7 | 8 | 1 |
| IV | 13 | 67.6 | 12 | 96 | 81.8 | 5.6 | 8.7 | 12 | 1 |
| V | 11 | 84.6 | 6 | 62.5 | 72.6 | 8.5 | 15.6 | 6 | 3 |
| VI | 7 | 37.3 | 7 | 87.9 | 62.6 | 6.2 | 14.7 | 7 | 0 |
| VII | 10 | 52.3 | 10 | 60.4 | 56.4 | 5.8 | 6.7 | 9 | 2 |
| VIII | 5 | 50.4 | 3 | 21.1 | 35.8 | 12.6 | 10.6 | 3 | 2 |
| IX | 6 | 31.6 | 7 | 35.8 | 33.7 | 6.3 | 7.2 | 6 | 1 |
| X | 2 | 6.9 | 2 | 19.5 | 13.2 | 6.9 | 19.5 | 2 | 0 |
Length in centimorgans for the male and female map, mean number of loci per linkage group for the male and the female, and number of loci shared between both maps and specific to one or the other map are shown.
Distribution of markers:
Distributions of loci from four microsatellite libraries (A–D, enriched for CA, GA, ATG, and TAGA repeats, respectively; G. Li et al. 2003), over the 10 linkage groups, are shown in Figure 2. Library A provides 11 loci, mapping to 7 linkage groups; library B provides 31 loci, mapping to 8 linkage groups; library C provides 22 loci, mapping to 8 linkage groups; library D provides 15 loci, mapping to 7 linkage groups. A contingency chi-square test for the 10-linkage-group × 4-library table is significant (χ2 = 41.056, 27 d.f., P = 0.023, by the pseudo-probability test; Zaykin and Pudovkin 1993) because 5 loci from library A appear on LG VI where only 1 is expected.
Figure 2.—

Distribution of recently cloned microsatellite DNA markers (G. Li et al. 2003) across 10 linkage groups. Libraries A, B, C, and D are CA-, GA-, ATG-, and TAGA-enriched libraries, respectively.
The 24 previously published markers map to 7 of the 10 linkage groups. Six markers from CSIRO Marine Research, Tasmania (McGoldrick et al. 2000), map 1 each to LG III (cmrCgi001) and LG V (cmrCgi151) and 2 each to LG I (cmrCgi005 and cmrCgi061) and LG VII (cmrCgi003 and cmrCgi141). The position of cmrCgi061 is provisional, as it shows a small distortion from Mendelian segregation ratios. The 3 markers published by (Magoulas et al. 1998) map to 3 different linkage groups, LG I (imbCgi44), LG III (imbCgi49), and LG VII (imbCgi108), while the 3 markers published by Huvet et al. (2000), um2Cgi10, um2Cgi16, and um2Cgi48, map to the groups IX, II, and VII, respectively. Of the markers previously published by McGoldrick (1997), McGoldrick et al. (2000), and Launey and Hedgecock (2001), 4 are located on LG I (ucdCgi003, -006, -008, and -018), 3 on LG V (ucdCgi014, -017, and -021), 2 on LG VII (ucdCgi022 and ucdCgi024), and 1 on each of LG II (ucdCgi001), LG III (ucdCgi002), and LG VI (ucdCg028).
Markers may be classified by size of the repeat element (di-, tri-, or tetranucleotide) or by crossed categories of motif complexity (simple or compound vs. pure or interrupted by non-motif nucleotides; following Chambers and MacAvoy (2000), with compound and complex and motifs combined). The 99 markers comprise 64 dinucleotide repeats, 20 trinucleotide repeats, and 15 tetranucleotide repeats. Loci with different repeat motifs are randomly distributed over the 10 linkage groups (χ2 = 15.582, 18 d.f., P = 0.62), as are the 83 mapped markers, for which information on motif complexity is available (χ2 = 33.046, 27 d.f., P = 0.192).
Genome length and coverage:
Adding twice the average spacing of markers on the male and female map (8.0 and 10.4 cM, respectively) to the lengths of each linkage group (11 for the male and 12 for the female) and summing across linkage groups, we estimate lengths of 776 and 1020 cM for the male and female genomes, respectively. The percentages of the genome covered by the consensus male and female linkage maps, on the basis of these estimated genome lengths, are 79 and 75%, respectively. A second estimate of genome length, in which the length of each linkage group is expanded by (m + 1)/(m − 1), where m is the number of unique loci mapped, yields lengths of 783 and 1094 cM for male and female genomes, respectively. The coverages of male and female consensus maps, on the basis of these estimates, are 79 and 70%, respectively.
Assuming random distribution of markers and 10 linkage groups, we estimate that 95% of the genome is within 16.1 cM of a marker on the male map, using either estimate of male genome length, and within 21 or 22 cM of a marker on the female map, depending on genome length estimate. The distributions of minimum distances of 10,000 randomly assigned genes to mapped male and female markers are nonnormal; median minimum distances are 3.1 cM for the male and 4.0 cM for the female. The minimum distances, below which 95% of simulated values lie, are 10.1 cM for the male map and 13.6 cM for the female map. On the other hand, a theoretical expected distance E(m) of a gene from the closest marker on the male map is ∼4.6 cM with an upper limit of 13.6–13.7 cM, depending on which estimate of genome length is used. For the female map, E(m) is 5.9 cM with an upper limit of 17.5 cM, using the first estimate of genome length, and 6.3 cM with an upper 95% confidence limit of 18.7 cM, using the second estimate of genome length.
Differences in recombination within and among families:
We use three or four sets of 94 progeny for marker testing in the three families. Although recombination data are taken only from the third or fourth extractions for the maps, data from earlier extractions allow tests of the heterogeneity of recombination within and among families. Within-family heterogeneity would presumably reflect artifacts, scoring errors, or sampling error, whereas heterogeneity among families might indicate biological variation in recombination rate. An example of these hierarchical G-tests for heterogeneity of recombination rate is presented in Table 3 for ucdCgi001 and ucdCgi158, a pair of markers on LG II. In this example, recombination rates for different extractions of progeny are homogeneous for male 1 but slightly heterogeneous for male 3 (Table 3C). Across 223 such tests of heterogeneity among extractions within family, 17 (7.6%) are significant at the α0.05 level, corrected for simultaneous multiple testing.
TABLE 3.
Recombination data forucdCgi001-ucdCgi158 on LG II from three male parents
| Cross | AB | Ab | aB | ab | r′ | θ = r′ | θ = 0.5 | G | P | LOD |
|---|---|---|---|---|---|---|---|---|---|---|
| A. Heterogeneity of recombination among male parents | ||||||||||
| M1 | 83 | 1 | 1 | 75 | 0.013 | −10.752 | −110.904 | 200.304 | 0.00 | 43.5 |
| M2 | 21 | 19 | 14 | 13 | 0.493 | −46.433 | −46.441 | 0.015 | 0.90 | 0.0 |
| M3 | 61 | 17 | 23 | 58 | 0.252 | −89.685 | −110.210 | 41.051 | 0.00 | 8.9 |
| Total | 241.370 | 52.4 | ||||||||
| Pool | 165 | 32 | 43 | 146 | 0.194 | −190.066 | −267.555 | 154.978 | 0.00 | 33.6 |
| Heterogeneity | 86.392 | 0.00 | 18.8 | |||||||
| Totals | AB | Ab | aB | ab | r′ | θ = r′ | θ = 0.5 | G | P | LOD |
| B. Heterogeneity of recombination among pairs of male parents | ||||||||||
| M1 + M2 | 200.319 | 43.5 | ||||||||
| Pool | 104 | 15 | 20 | 88 | 0.154 | −97.587 | −157.344 | 119.514 | 0.00 | 26.0 |
| Heterogeneity | 80.805 | 0.00 | 17.5 | |||||||
| M1 + M3 | 241.355 | 52.4 | ||||||||
| Pool | 144 | 18 | 24 | 133 | 0.132 | −124.261 | −221.114 | 193.706 | 0.00 | 42.1 |
| Heterogeneity | 47.649 | 0.00 | 10.3 | |||||||
| M2 + M3 | 41.066 | 8.9 | ||||||||
| Pool | 82 | 31 | 42 | 71 | 0.323 | −142.180 | −156.651 | 28.942 | 0.00 | 6.3 |
| Heterogeneity | 12.124 | 0.00 | 2.6 | |||||||
| Extraction | AB | Ab | aB | ab | r′ | θ = r′ | θ = 0.5 | G | P | LOD |
| C. Heterogeneity of recombination among extractions | ||||||||||
| M1, ext 2 | 41 | 0 | 0 | 32 | 0.000 | −0.023 | −50.603 | 101.159 | 0.00 | 22.0 |
| M1, ext 4 | 42 | 1 | 1 | 43 | 0.023 | −9.522 | −60.304 | 101.563 | 0.00 | 22.0 |
| Total | 202.722 | 44.0 | ||||||||
| Pool | 83 | 1 | 1 | 75 | 0.013 | −10.752 | −110.904 | 200.304 | 0.00 | 43.5 |
| Heterogeneity | 2.418 | 0.12 | 0.5 | |||||||
| M3, ext 2 | 20 | 10 | 15 | 26 | 0.352 | −46.061 | −49.213 | 6.305 | 0.01 | 1.4 |
| M3, ext 3 | 41 | 7 | 8 | 32 | 0.170 | −40.181 | −60.997 | 41.631 | 0.00 | 9.0 |
| Total | 47.936 | 10.4 | ||||||||
| Pool | 61 | 17 | 23 | 58 | 0.252 | −89.685 | −110.210 | 41.051 | 0.00 | 8.9 |
| Heterogeneity | 6.885 | 0.01 | 1.5 | |||||||
Haplotype frequencies are given in the columns headed AB, Ab, aB, and aa, with AB and ab being the parental types and Ab and aB being recombinant types. The likelihood of the observed recombination fraction, r′, is given in the column headed θ = r′, while the likelihood that the markers are unlinked is given in the column headed θ = 0.5. The G-test statistic, probability (P), and LOD score associated with difference in log likelihoods for the two hypotheses are given in the last three columns. ext, extraction.
Significant heterogeneity in recombination between a given pair of loci is observed among families. For example, ucdCgi001 and ucdCgi158 are tightly linked in male 1 (r = 0.013), unlinked in male 2 (r = 0.493), and moderately linked in male 3 (r = 0.252); the heterogeneity of recombination among males is highly significant, as is the heterogeneity in all pair comparisons of males (Table 3, A and B). Of the 172 tests of heterogeneity of recombination rate among families, 49 (28.5%) are significant at the adjusted α0.05 level, 3.7 times greater than the proportion of heterogeneous tests within family. Significant tests are distributed over linkage groups in proportion to the numbers of heterogeneity tests per linkage group. There is no difference in the proportions of significant tests, either within or between families, for the male and female segregation data (tests not shown).
Differences in order of markers on linkage groups exist among families, but few of them are significant at LOD score >3.0. Comparing the order of markers for the three families, by male and female parents, we find 71 cases, in which loci order is conserved, and 29 cases, in which the order of markers is significantly different. However, of the alternative marker orders given by Mapmaker, only 15 are significantly different (LOD > 3.0), as illustrated in Figure 3.
Figure 3.—

Examples of difference in gene order between male and female parents, for linkage groups II, IV, and V. The LOD scores for imposing the first gene order on the second linkage group are, from left to right, 11.5, 15.0, and 3.1.
Differences in recombination between sexes:
The sexes show substantial differences in recombination rates, both in general and for specific pairs of linked markers. In general, there is less recombination and genetic distance in the male linkage map (Table 2). One hundred and eighteen pairs of markers, representing all linkage groups, are segregating in both the male and female parents of the three mapping families and show significant linkage for at least one parent (48 for family 1, 21 for family 2, and 49 for family 3). The plot of male on female recombination fractions for these pairs of markers shows greater recombination in the female (Figure 4). Forty-nine pairs of markers show statistically significant differences in recombination values between the male and female parents; of these, only 5 pairs of markers have significantly more recombination in the male than in the female. Regression of the data in Figure 4 reveals both a significant intercept (0.205, P < 0.001) and a slope that is significantly <1 (0.479, upper 95% confidence limit = 0.737). Analysis using a separate-slopes model, however, reveals significantly different slopes and intercepts among families (P < 0.000 for family and P = 0.0003 for slope within family; r = 0.826, 0.396, and 0.391, for families 1, 2, and 3, respectively).
Figure 4.—

Male vs. female recombination fraction, for 118 pairs of markers segregating from both parents, by family. Squares are data from family 1, diamonds are data from family 2, and triangles are data from family 3; open symbols are cases in which the recombination fraction θ is statistically homogeneous between the female and male parents; solid symbols are cases in which θ is significantly heterogeneous between parents.
The difference in recombination between the sexes is especially dramatic between the sibling male and female parents from lines 2 × 5 and 7 × 9 (Figure 5), which are the sire and dam and dam and sire, respectively, of mapping families 1 and 2. Over both sets of sibling parents, there are 68 pairs of markers that were segregating in both parents and showed significant linkage in at least one parent. Of the 68 pairs of loci, 31 show significant heterogeneity of recombination between the sibling parents. For the first pair of siblings from family 2 × 5, the 19 cases of significant heterogeneity are spread over seven linkage groups; although 3 of these cases involve only a single pair of markers, the other 4 cases show heterogeneity in recombination for 2 or more pairs of markers. Nine cases of significant heterogeneity are observed for linkage group II. For the second pair of siblings from family 7 × 9, the 12 cases of significant heterogeneity occur on four linkage groups, with 8 of these occurring on linkage group VI.
Figure 5.—

Male vs. female recombination fraction, for 68 pairs of markers segregating from sibling male and female parents from two families. Diamonds are data for sibs from family 2 × 5, and squares are data for sibs from family 7 × 9; open symbols are cases in which the recombination fraction θ is statistically homogeneous between the female and male siblings, and solid symbols are cases in which θ is significantly heterogeneous.
DISCUSSION
Numbers of markers and linkage groups, genome size, and coverage:
Although moderately dense linkage maps have previously been produced for marine shrimp and oysters, using AFLPs (Wilson et al. 2002; Yu and Guo 2003; Li and Guo 2004), this linkage map for the Pacific oyster is the first for a marine invertebrate made exclusively from microsatellite DNA markers. The expected advantage of a microsatellite-based linkage map, in contrast to an AFLP-based map, is portability to other families. The large number of null alleles segregating for microsatellite markers in the Pacific oyster may reduce this expected advantage within species and will likely diminish its applicability to congeneric species (McGoldrick et al. 2000; G. Li et al. 2003).
The strategy of genotyping 11-day-old larvae from double-hybrid crosses, which we employed in response to evidence that selection acts early in the life cycle against homozygotes for recessive deleterious mutations (Launey and Hedgecock 2001; Bucklin 2002), appears to have been effective in reducing distortion of segregation ratios in our mapping families. The resulting consensus male and female linkage maps constructed from a total of 100 microsatellite DNA markers appear to provide low-density resolution of the Pacific oyster genome. The male and female consensus maps have 11 and 12 linkage groups, respectively, but alignment of these two maps indicates 10 linkage groups, the same as the number of haploid chromosomes (Leitão et al. 1999). The ratios of longest to shortest linkage groups, 147.9:19.5 cM or 7.6:1 in the female and 94.4:6.9 cM or 13.7:1 in the male, are much greater than the 2:1 ratio of the longest and shortest of the meiotic chromosomes. This observation, together with observations of >10 linkage groups, small linkage groups, and two unlinked microsatellite markers, suggests that gaps remain to be filled by adding more markers. Nevertheless, the estimated coverages for the consensus linkage maps are 79% for the male and 70 and 75% for the female, on the basis of the two estimates of genome length.
We estimate from theory that 95% of the genome is located within 16 and 22 cM of a locus on the male and female maps, respectively. The distributions of minimum distance for 10,000 simulated gene additions to the male and female maps are nonnormal, with modes at ∼0.5 cM, maxima of 15.2 and 20.5 cM, and medians of 3.1 and 4.0 cM, for male and female maps, respectively. Ninety-five percent of minimum distances are within 10.1 and 13.6 cM of markers on the male and female maps. The smaller expected distances in simulations may reflect a nonrandom distribution of markers across linkage groups. We do observe a nonrandom distribution of markers from library A over linkage groups, for example.
Average numbers of chiasmata per chromosome in the Pacific and Eastern oysters are ∼1.1–1.2, which yield theoretical map lengths of 550–600 cM (X. Guo, H. Yang and Z. Wang, unpublished data cited in Li and Guo 2004). Observed map lengths, 616.1 cM for the male and 770.5 cM for the female, are close to those expected on the basis of cytological observations, but estimated, not actual genome lengths should be compared to the cytological observations. For the male, the expected genome length of 783 cM is 42% greater than the minimum theoretical length; for the female, the expected genome length of 1094 is 99% greater than the minimum theoretical length. The AFLP male and female linkage maps developed by Li and Guo (2004) for the Pacific oyster are 32–79% longer than their theoretical sizes on the basis of cytological observation of chiasmata frequency. These authors attribute the discrepancy between observed and theoretical sizes to low marker density, which likely applies to our results as well. A more saturated, mixed map of AFLP and microsatellite markers should give more information about the genome length in C. gigas and is presently under construction.
Map differences between sexes:
In human, mouse, cattle, pig, fish, and indeed most vertebrates studied so far, recombination rates show a significant difference between sexes. Female maps are usually longer than male maps (Barendse et al. 1994; Ellegren et al. 1994; Dib et al. 1996; Dietrich et al. 1996; Sakamoto et al. 2000; Waldbieser et al. 2001; Singer et al. 2002). Higher recombination rates near the centromere in the female and near the telomere in the male were observed in rainbow trout and zebrafish (Knapik et al. 1998; Sakamoto et al. 2000). The molecular mechanism responsible for differences in recombination rates between the two sexes is not currently well understood. In C. gigas, as in other species, the female map is longer than the male map, although we cannot localize differences in recombination rate to centromeres or telomeres. Nevertheless, the difference is remarkable since the Pacific oyster is a protandric species, generally maturing first as a male and able to switch sex in subsequent spawning seasons (Guo et al. 1998). It should be possible with oysters, therefore, to observe recombination in the same individual acting as a male or a female. Here, we report that recombination rates and even gene orders can be significantly different between full siblings of different sexes.
Map differences between families:
Most published linkage maps are based on a single family. The rainbow trout map, having been constructed using two families, is an exception that reveals significant differences in map distances between families. Because we use three families for linkage mapping, we examine variation in recombination rates and gene orders among parents. Moreover, because markers are tested on different sets of progeny from each parent (although mapping is based on just the last extraction tested for all the markers in a linkage group), we can contrast the heterogeneity of recombination among parents to that among different sets of progeny from the same parent. Within family, recombination between pairs of markers is significantly heterogeneous in 17 of 223 (7.6%) cases, slightly more than the 5% expected by chance; this level of heterogeneity may be ascribed to sampling errors and technical artifacts. Between families, however, the level of heterogeneity in recombination rate is nearly four times greater, 49 of 172 (28.5%) cases. This additional heterogeneity in recombination rate, together with evidence for significantly different gene orders, suggests polymorphism for chromosomal rearrangements in the natural population of Pacific oysters from which our lines were derived. Some of the differences in gene order between the sexes, especially between sibling parents of opposite sex, may thus be attributable to a population rather than a sex-specific cause.
Polymorphism for chromosomal rearrangements is unknown from cytological observations of meiotic chromosomes in oysters (Longwell et al. 1967). Genetic linkage studies will be an important means of documenting the extent of such chromosomal variation in natural populations, the evolutionary consequences of which are well understood from studies in Drosophila and plants (Dobzhansky 1970). In the oyster, chromosomal rearrangements could be a potent factor reducing crossovers and genetic recombination, allowing blocks of the genome to retain linkage disequilibria for longer than might be expected in putatively large, well-mixed populations. This reduction in genetic recombination could have profound consequences for understanding population genetic structure, the causes of phenotypic variation, and the process of adaptation in this and likely many other marine bivalve molluscs.
Future uses:
The scaffold of microsatellite DNA markers presented here should enable the rapid construction of the next generation of higher-density linkage maps, although the precise linkage and order of markers will likely vary from family to family. Adding AFLP or SNP markers to microsatellite DNA markers will yield high-density linkage maps for the oyster, which will have many uses in experimental settings. They will be indispensable tools for mapping QTL for growth heterosis (Hedgecock et al. 1995; McGoldrick 1997), mapping the location of lethal genes (Launey and Hedgecock 2001; Bucklin 2002), and locating expressed genes that are economically important for aquaculture. High-density linkage maps will also be necessary to map more precisely the extent in oyster populations of the chromosomal rearrangements and polymorphisms revealed by this study.
The number of mapped microsatellite DNA markers presently available can be used effectively, now, to understand the behavior of meiotic chromosomes in diploid and polyploid oysters. Centromeres can be added to the linkage map by studies of triploid or gynogenetic progeny produced by inhibition of the second meiotic division (Guo and Gaffney 1993). Owing to their reduced reproductive allocation and near sterility, triploid oysters are being farmed commercially worldwide (Nell 2002) and being considered for introduction into the Chesapeake Bay (National Research Council 2004). Understanding the numbers and kinds of gametes produced by triploids, crucial information for evaluating the risks of an intentional introduction, could be greatly advanced by the use of mapped microsatellite DNA markers. Tetraploid Pacific oysters are now being produced commercially for the propagation of triploid seed via crosses of tetraploid males and diploid females (Guo and Allen 1995; Eudeline et al. 2000). At present, we have little understanding of how meiosis works in a tetraploid oyster and what the consequences might be for further breeding. Finally, mapped markers could be useful in determining which chromosomes are missing in aneuploids and, thus, explaining the relationship between growth and degree of aneuploidy (Thiriot-Quievreux et al. 1992; Zouros et al. 1996; Leitão et al. 2001).
Acknowledgments
We thank Vanessa Ribes, Gang Li, and Katherine Bucklin for help in molecular analysis. We thank Olivier Le Provost for drawing the map. This work was supported by a grant for U.S. Department of Agriculture National Research Initiative Competitive Grant Program, agreement no. 99-35205-8260.
We dedicate this study to the memory of Will Borgenson, who reared and cared for the parents of the mapping families.
References
- Barendse, W., S. M. Armitage, L. M. Kossarek, A. Shalom, B. W. Kirkpatrick et al., 1994. A genetic linkage map of the bovine genome. Nat. Genet. 6: 227–235. [DOI] [PubMed] [Google Scholar]
- Bayne, B. L., D. Hedgecock, D. McGoldrick and R. Rees, 1999. Feeding behaviour and metabolic efficiency contribute to growth heterosis in Pacific oysters. [Crassostrea gigas (Thunberg)]. J. Exp. Mar. Biol. Ecol. 233: 115–130. [Google Scholar]
- Bishop, D. T., C. Cannings, M. Skolnick and J. A. Williamson, 1983 The number of polymorphic DNA clones required to map the human genome, pp. 181–200 in Statistical Analysis of DNA Sequence Data, edited by B. S. Weir. Marcel Dekker, New York.
- Britten, H., 1996. Meta-analysis of the association between multilocus heterozygosity and fitness. Evolution 50: 2158–2164. [DOI] [PubMed] [Google Scholar]
- Bucklin, K., 2002 Analysis of the causes of inbreeding in the Pacific oyster Crassostrea gigas. Ph.D. Dissertation, University of California, Davis, CA.
- Chakravarti, A., L. K. Lasher and J. E. Reefer, 1991. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, G. K., and E. S. MacAvoy, 2000. Microsatellites: consensus and controversy. Comp. Biochem. Physiol. B 126: 455–476. [DOI] [PubMed] [Google Scholar]
- David, P., 1998. Heterozygosity-fitness correlations: new perspectives on old problems. Heredity 80: 531–537. [DOI] [PubMed] [Google Scholar]
- Dib, C., S. Faure, C. Fizames, D. Samson, N. Drouot et al., 1996. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380: 152–154. [DOI] [PubMed] [Google Scholar]
- Dietrich, W. F., J. Miller, R. Steen, M. A. Merchant, D. Damron-Boles et al., 1996. A comprehensive genetic map of the mouse genome. Nature 380: 149–152. [DOI] [PubMed] [Google Scholar]
- Dobzhansky, Th., 1970 Genetics of the Evolutionary Process. Columbia University Press, New York.
- Ellegren, H., B. P. Chowdhary, M. Johansson, L. Marklund, M. Fredholm et al., 1994. A primary linkage map of the porcine genome reveals a low rate of genetic recombination. Genetics 137: 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eudeline, B., S. K. Allen, Jr. and X. Guo, 2000. Optimization of tetraploid induction in Pacific oysters, Crassostrea gigas, using first polar body as a natural indicator. Aquaculture 187: 73–84. [Google Scholar]
- Evans, F., S. Matson, J. Brake and C. Langdon, 2003. The effects of inbreeding on performance traits of adult Pacific oysters (Crassostrea gigas). Aquaculture 230: 89–98. [Google Scholar]
- Fishman, L., A. J. Kelly, E. Morgan and J. H. Willis, 2001. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159: 1701–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Agriculture Organization, 2003 World aquaculture production of fish, crustaceans, molluscs, etc., by principal species (http://www.fao.org/fi/statist/summtab/default.asp).
- Guo, X. M., and S. K. Allen, 1995. The successful induction of tetraploidy in the Pacific oyster Crassostrea gigas (Thunberg). Aquaculture 137: 152–153. [Google Scholar]
- Guo, X., and P. M. Gaffney, 1993. Artificial gynogenesis in the Pacific oyster, Crassostrea gigas. 2. Allozyme inheritance and early growth. J. Hered. 84: 311–315. [Google Scholar]
- Guo, X., D. Hedgecock, W. K. Hershberger, K. Cooper and S. K. Allen, Jr., 1998. Genetic determinants of protandric sex in the Pacific oyster, Crassostrea gigas Thunberg. Evolution 52: 394–402. [DOI] [PubMed] [Google Scholar]
- Hedgecock, D., 1994 Does variance in reproductive success limit effective population sizes of marine organisms?, pp. 122–134 in Genetics and Evolution of Aquatic Organisms, edited by A. R. Beaumont. Chapman & Hall, London.
- Hedgecock, D., D. J. McGoldrick and B. L. Bayne, 1995. Hybrid vigor in Pacific oysters: an experimental approach using crosses among inbred lines. Aquaculture 137: 285–298. [Google Scholar]
- Hedgecock, D., D. J. McGoldrick, D. T. Manahan, J. Vavra, N. Appelmans et al., 1996. Quantitative and molecular genetic analyses of heterosis in bivalve molluscs. J. Exp. Mar. Biol. Ecol. 203: 49–59. [Google Scholar]
- Hedgecock, D., C. Langdon, M. Blouin and S. K. Allen, 1997 Genetic Improvement of Cultured Pacific Oysters by Selection. Agricultural Experiment Station, Oregon State University, Corvallis, OR.
- Huvet, A., P. Boudry, M. Ohresser, C. Delsert and F. Bonhomme, 2000. Variable microsatellites in the Pacific oyster Crassostrea gigas, and other cupped oysters species. Ann. Genet. 31: 68–79. [DOI] [PubMed] [Google Scholar]
- Knapik, E. W., A. Goodman, M. Ekker, M. Chevrette, J. Delgado et al., 1998. A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 18: 338–343. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., P. Green, J. Abrahamson, A. Barlow, M. J. Daly et al., 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181. [DOI] [PubMed] [Google Scholar]
- Langdon, C., F. Evans, D. Jacobson and M. Blouin, 2003. Yields of cultured Pacific oysters Crassostrea gigas Thunberg improved after one generation of selection. Aquaculture 220: 227–244. [Google Scholar]
- Lannan, J. E., 1980. Broodstock management of Crassostrea gigas I. Genetic and environmental variation in survival in the larval rearing system. Aquaculture 21: 323–336. [Google Scholar]
- Launey, S., and D. Hedgecock, 2001. High genetic load in the Pacific oyster Crassostrea gigas. Genetics 159: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitão, A., C. Thiriot-Quievreux, P. Boudry and I. Malheiro, 1999. A ‘G’ chromosome banding study of three cupped oyster species: Crassostrea gigas, Crassostrea angulata and Crassostrea virginica (Mollusca:Bivalvia). Genet. Sel. Evol. 31: 519–527. [Google Scholar]
- Leitão, A., P. Boudry and C. Thiriot-Quievreux, 2001. Negative correlation between aneuploidy and growth in the Pacific oyster, Crassostrea gigas: ten years of evidence. Aquaculture 193: 39–48. [Google Scholar]
- Li, G., S. Hubert, K. Bucklin, V. Ribes and D. Hedgecock, 2003. Characterization of 79 microsatellite DNA markers in the Pacific oysters Crassostrea gigas. Mol. Ecol. Notes 3: 228–232. [Google Scholar]
- Li, L., and X. Guo, 2004. AFLP-based genetic linkage maps of the Pacific oyster Crassostrea gigas Thunberg. Mar. Biotech. 6: 26–36. [DOI] [PubMed] [Google Scholar]
- Li, Y. T., K. Byrne, E. Miggiano, V. Whan, S. Moore et al., 2003. Genetic mapping of the kuruma prawn Penaeus japonicus using AFLP markers. Aquaculture 219: 143–156. [Google Scholar]
- Lincoln, S., and E. S. Lander, 1992. Systematic detection of errors in genomics linkage data. Genomics 14: 604–610. [DOI] [PubMed] [Google Scholar]
- Liu, B.-H., 1997 Statistical Genomics: Linkage, Mapping, and QTL Analysis. CRC Press, Boca Raton, FL.
- Longwell, A. C., S. S. Stiles and D. G. Smith, 1967. Chromosome complement of the American oyster Crassostrea virginica, as seen in meiotic and cleaving eggs. Can. J. Genet. Cytol. 9: 845–856. [DOI] [PubMed] [Google Scholar]
- Love, J. M., A. M. Knight, M. A. Mcaleer and J. A. Todd, 1990. Towards construction of high resolution map of the mouse genome using PCR-analyzed microsatellites. Nucleic Acids Res. 18: 4123–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998 Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Magoulas, A., B. Ghjetvaj, V. Terzoglou and E. Zouros, 1998. Three polymorphic microsatellites in the Japanese oyster, Crassostrea gigas (Thunberg). Ann. Genet. 29: 69–70. [Google Scholar]
- McGoldrick, D. J., 1997 An experimental investigation of the genetic basis of heterosis in the Pacific oyster Crassostrea gigas. Ph.D. Dissertation, University of California, Davis, CA.
- McGoldrick, D., D. Hedgecock, L. J. English, P. Baoprasertkul and R. D. Ward, 2000. The transmission of microsatellite alleles in Australian and North American stocks of the Pacific oyster (Crassostrea gigas): selection and null alleles. J. Shellfish Res. 19: 779–788. [Google Scholar]
- National Research Council, 2004 Non-Native Oysters in the Chesapeake Bay. National Academies Press, Washington, DC.
- Nell, J. A., 2002. Farming triploid oysters. Aquaculture 210: 69–88. [Google Scholar]
- Rice, W., 1989. Analysis tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- Sakamoto, T., R. G. Danzmann, K. Gharbi, P. Howard, A. Ozaki et al., 2000. A microsatellite linkage map of Rainbow trout (Oncorhynchus mykiss) characterized by large sex-specific differences in recombination rates. Genetics 155: 1331–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, A., H. Perlman, Y.-L. Yan, C. Walker, G. Corley-Smith et al., 2002. Sex-specific recombination rates in zebrafish (Danio rerio). Genetics 160: 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiriot-Quievreux, C., G. H. Pogson and A. E. Zouros, 1992. Genetics of growth rate variation in bivalves: aneuploidy and heterozygosity effects in a Crassostrea gigas family. Genome 35: 39–45. [Google Scholar]
- Voorrips, R. E., 2002. MapChart: software for the presentation of linkage maps and QTLs. J. Hered. 93: 77–78. [DOI] [PubMed] [Google Scholar]
- Waldbieser, G. C., B. G. Bosworth, D. J. Nonneman and W. R. Wolters, 2001. A microsatellite-based genetic linkage map for channel catfish, Ictalurus punctatus. Genetics 158: 727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, G. C., 1975 Sex and Evolution. Princeton University Press, Princeton, NJ.
- Wilson, K., Y. Li, V. Whan, S. Lehnert, K. Byrne et al., 2002. Genetic mapping of the black tiger shrimp Penaeus monodon with amplified fragment length polymorphism. Aquaculture 204: 297–309. [Google Scholar]
- Yu, Z., and X. Guo, 2003. Genetic linkage map of the Eastern oyster Crassostrea virginica Gmelin. Biol. Bull. 204: 327–338. [DOI] [PubMed] [Google Scholar]
- Zaykin, D. V., and A. I. Pudovkin, 1993. 2 programs to estimate significance of CHI-2 values using pseudo-probability tests. J. Hered. 84: 152. [Google Scholar]
- Zouros, E., and G. H. Pogson, 1994 Heterozygosity, heterosis and adaptation, pp. 135–146 in Genetics and Evolution of Aquatic Organisms, edited by A. R. Beaumont. Chapman & Hall, London.
- Zouros, E., C. Thiriot-Quievreux and G. Kotoulas, 1996. The negative correlation between somatic aneuploidy and growth in the oyster Crassostrea gigas and the application for the effects of induced polyploidization. Genet. Res. 68: 109–116. [Google Scholar]