

Abstract
The analysis of the haplotype-phenotype relationship has become more and more important. We have developed an algorithm, using individual genotypes at linked loci as well as their quantitative phenotypes, to estimate the parameters of the distribution of the phenotypes for subjects with and without a particular haplotype by an expectation-maximization (EM) algorithm. We assumed that the phenotype for a diplotype configuration follows a normal distribution. The algorithm simultaneously calculates the maximum likelihood (L0max) under the null hypothesis (i.e., nonassociation between the haplotype and phenotype), and the maximum likelihood (Lmax) under the alternative hypothesis (i.e., association between the haplotype and phenotype). Then we tested the association between the haplotype and the phenotype using a test statistic, −2 log(L0max/Lmax). The above algorithm along with some extensions for different modes of inheritance was implemented as a computer program, QTLHAPLO. Simulation studies using single-nucleotide polymorphism (SNP) genotypes have clarified that the estimation was very accurate when the linkage disequilibrium between linked loci was rather high. Empirical power using the simulated data was high enough. We applied QTLHAPLO for the analysis of the real data of the genotypes at the calpain 10 gene obtained from diabetic and control subjects in various laboratories.
IN many cases, haplotypes or diplotype configurations but not genotypes are associated with phenotypes. A diplotype configuration is defined as a combination of two haplotype copies possessed by an individual, and an ordered diplotype configuration denotes an ordered list of two haplotypes arranged according to the derivation (father and mother). Since recent analyses disclosed many linked polymorphic loci within a gene, the multiple loci often have to be treated together rather than separately. A haplotype and a haplotype copy have distinct definitions in this manuscript since when a subject is homozygous for a haplotype, he (or she) is interpreted to have a single haplotype but two haplotype copies.
There are several common methods for haplotype inference using genotype SNP data. For example, the Clark algorithm (Clark 1990), the EM algorithm (Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995; Schneider et al. 2000; Kitamura et al. 2002), PHASE (Stephens et al. 2001), the PL algorithm (Niu et al. 2002), and the PL-EM algorithm (Qin et al. 2002) have been used. We also proposed an algorithm to estimate haplotypes by use of pooled genotype data (Ito et al. 2003). However, the methods to relate such inferred haplotypes to the phenotypes are still to be developed. We have recently proposed an algorithm (PENHAPLO) to test the association between qualitative phenotypes (e.g., affection status) and the presence of a haplotype by EM algorithm (Ito et al. 2004). Thus far, we have considered disease status as a qualitative trait with two outcomes, affected and unaffected, and penetrances as the conditional probabilities of phenotypes given genotypes or diplotype configurations. In practice, however, the disease phenotype often consists of a quantitative measurement such as blood sugar level. The locus for a trait concerning the above disease phenotypes is referred to as the quantitative trait locus (QTL) and the associated phenotypes as quantitative phenotypes. Such quantitative phenotypes often follow continuous distributions, and the quantitative phenotypes should be handled separately from the qualitative phenotypes. Thus, the program PENHAPLO cannot be directly applied to quantitative phenotypes.
We developed an algorithm to estimate simultaneously the diplotype configurations for the subjects and the distribution of quantitative phenotypes for different diplotype configurations and to test the association between the phenotypes and the presence of a haplotype. Note that the following considerations apply to this article. First, rather than defining the probability of a phenotype (penetrance), probability density for a value of a quantitative phenotype was defined. Second, the phenotypes were considered to depend on diplotype configurations rather than on genotypes at single loci.
Recent studies have reported that, in some cases, drug responses and other phenotypes were associated not with genotypes but with haplotypes or diplotype configurations (Judson et al. 2000; Bader 2001; Urano et al. 2002; Tanaka et al. 2002). Tanck et al. (2003) presented a method to estimate multilocus haplotype effects using a weighted penalized log-likelihood model. Schaid et al. (2002) proposed methods to test the association between ambiguous haplotypes and a variety of traits (binary, ordinal, and quantitative traits), which were based on score equations for generalized linear models (GLMs).
Therefore, it is important to develop a method for testing the association between quantitative phenotypes and different diplotype configurations. One of the problems in haplotype inference is that the diplotype configurations for some subjects are not uniquely determined (ambiguous diplotype configurations). This is because more than one diplotype configuration is possible for a subject even when the genotypes at all the relevant loci are observed. We could regard the diplotype configuration with the highest probability as the true configuration and perform the test using the inferred data; however, such a test may inflate the type I error rates.
To overcome this problem, we developed an algorithm to simultaneously estimate parameters of the phenotype distributions, haplotype frequencies, and diplotype configurations given observed genotypes and the phenotype data.
As the simplest model, we assumed that the phenotype conditional on a diplotype configuration follows a normal distribution. Thus, the distribution of the phenotype for subjects with a specific haplotype follows N(μ1, σ2), while the distribution of the same phenotype without it follows N(μ2, σ2). We estimate haplotype frequencies, diplotype configurations, and parameters of the phenotype distribution by an EM algorithm using genotype and phenotype data. Ambiguous diplotype configurations are treated as multiple diplotype configurations for each subject with different probabilities. Using simulation data and real data, we demonstrate that our approach can be used to detect the association between quantitative phenotypes and the presence of a haplotype and to estimate the distribution of the phenotypes. Although we assumed the normality for the distribution of phenotypes in the standard model, the methods to cope with the violation of the normality are discussed in this manuscript.
METHODS
Analysis of real data:
As the real data, we used the data from three linked SNP loci of the calpain (CAPN)10 gene and the quantitative phenotypes. Haplotypes at the CAPN10 gene have been reported to be associated with type 2 diabetes (Horikawa et al. 2000). We selected body mass index (BMI), blood sugar level, and insulin level as the quantitative phenotypes. We applied the real data of the genotypes at the three linked loci as well as one of the phenotypes from the subjects to QTLHAPLO. By this method we tested the association between the haplotypes and the phenotypes and, at the same time, estimated the parameters of the distribution of the phenotypes.
Algorithm:
Sample space:
In this study, the sample space is defined as a set of outcomes from the following experiment. Let us assume that there are l linked SNP loci. The number of all the possible haplotypes will be L = 2l. We set up a collection of an infinite number of haplotype copies. The relative haplotype frequencies in the collection are Θ = (θ1, … , θj, … , θL), where θj is the relative frequency of jth haplotype, and θj ≥ 0, 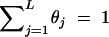 . According to the haplotype frequencies, an ordered combination of two haplotype copies is given to each of N individuals by randomly drawing them from the collection of the haplotype copies. A diplotype configuration is defined, in this article, as an ordered combination of two haplotype copies. Let a1, a2, … , aL2 be possible diplotype configurations. The probability that the ith subject has the diplotype configuration ak given Θ is P(di = ak|Θ) = θlθm, where di is a diplotype configuration for the ith subject, and l, m are the labels (1, 2, … , L) of the haplotypes that constitute ak. The ith subject develops quantitative phenotype ψi, following a probability density function. Let us assume that the phenotype for a diplotype configuration follows a normal distribution with a fixed variance but with a mean that depends on the diplotype configuration. An outcome from the experiment is defined by (Θ, D, Ψ), where D = (d1, … , dN) denotes the vectors of the diplotype configurations and Ψ = (ψ1, … , ψN) denotes the vectors of the phenotypes. The mean μ of the distribution of a phenotype is assumed to differ between the subjects with and without haplotype hb. hb is the haplotype that has a different effect from the others. Let D+ denote a set of diplotype configurations that contain the haplotype hb. We then define two normal distributions for a phenotype, one, N(μ1, σ2), for the subjects with di ∈ D+ and the other, N(μ2, σ2), for those with di∉ D+. Let fμ1x denote the probability density function that the ith individual develops a phenotype x when di ∈ D+, and let fμ2x denote the probability density function that the ith individual develops x when di∉ D+.
. According to the haplotype frequencies, an ordered combination of two haplotype copies is given to each of N individuals by randomly drawing them from the collection of the haplotype copies. A diplotype configuration is defined, in this article, as an ordered combination of two haplotype copies. Let a1, a2, … , aL2 be possible diplotype configurations. The probability that the ith subject has the diplotype configuration ak given Θ is P(di = ak|Θ) = θlθm, where di is a diplotype configuration for the ith subject, and l, m are the labels (1, 2, … , L) of the haplotypes that constitute ak. The ith subject develops quantitative phenotype ψi, following a probability density function. Let us assume that the phenotype for a diplotype configuration follows a normal distribution with a fixed variance but with a mean that depends on the diplotype configuration. An outcome from the experiment is defined by (Θ, D, Ψ), where D = (d1, … , dN) denotes the vectors of the diplotype configurations and Ψ = (ψ1, … , ψN) denotes the vectors of the phenotypes. The mean μ of the distribution of a phenotype is assumed to differ between the subjects with and without haplotype hb. hb is the haplotype that has a different effect from the others. Let D+ denote a set of diplotype configurations that contain the haplotype hb. We then define two normal distributions for a phenotype, one, N(μ1, σ2), for the subjects with di ∈ D+ and the other, N(μ2, σ2), for those with di∉ D+. Let fμ1x denote the probability density function that the ith individual develops a phenotype x when di ∈ D+, and let fμ2x denote the probability density function that the ith individual develops x when di∉ D+.
Thus, if ψi denotes the phenotype of ith subject, the probability density is
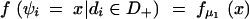 |
and
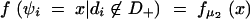 |
Note that ψi is independent of Θ conditional on di.
Likelihood function:
The observed data are the genotypes and the quantitative phenotypes of the subjects. Let Gobs = (g1, g2, … , gN) and Ψobs = (w1, w2, … , wN) denote the vectors of the observed genotypes and the quantitative phenotypes, respectively, where gi and wi denote the observed genotypes and the quantitative phenotype of the ith subject.
As the first step, we consider a general case in which the distributions differ between all the diplotype configurations. Let 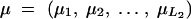 denote the vector of the means for the distributions for all possible diplotype configurations. Note that, in this context, the distributions of a phenotype are assumed to be potentially different between different diplotype configurations. Then the likelihood function is
denote the vector of the means for the distributions for all possible diplotype configurations. Note that, in this context, the distributions of a phenotype are assumed to be potentially different between different diplotype configurations. Then the likelihood function is
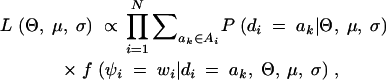 |
where Ai denotes the set of ak for the ith subject that are consistent with gi and f is the probability density function for N(μk, σ2).
Since di is independent of μ, σ and ψi is independent of Θ conditional on di,
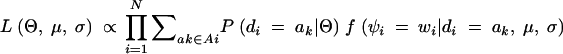 |
1 |
Under the null hypothesis that the distribution of the phenotype is independent of the diplotype configuration, the likelihood function is
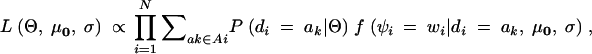 |
2 |
where, under the null hypothesis, the mean on the distribution of the phenotype for the diplotype configurations is invariable, and μ0 denotes the vectors of the means, μ0 = (μ0, μ0, … , μ0). Then again, Ai denotes the set of diplotype configurations for the ith subject that are consistent with gi.
It is not realistic, however, to assign different distributions for all different diplotype configurations for the alternative hypothesis. We then set up only two normal distributions, N(μ1, σ2) and N(μ2, σ2), for the alternative hypothesis. For the null hypothesis, we set up only one normal distribution, N(μ0, σ2). Thus, under the alternative hypothesis, the ith subject develops the phenotype x at the probability density function:
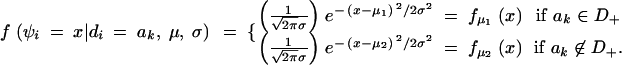 |
EM algorithm:
Our algorithm is an extension of the EM algorithm for estimating marker haplotype frequencies to the association studies. However, our likelihood function, unlike previous algorithms, includes the information about the phenotypes.
Equation 1 is maximized over Θ, μ, and σ, and the maximum likelihood thus obtained is denoted Lmax. Then Equation 2 is maximized over Θ, μ0, and σ, and the maximum likelihood thus obtained is denoted L0max. The likelihood ratio L0max/Lmax is used to test the association between the presence of the haplotype and the distribution of the phenotypes.
In the maximization for Lmax, the parameters to be estimated are Θ = (θ1, θ2, … , θL), μ1, μ2, and σ, while in the maximization for L0max, the parameters to be estimated are Θ = (θ1, θ2, … , θL), μ0, and σ. Under the null hypothesis, −2 log(L0max/Lmax) is expected to follow the χ2 distribution with 1 d.f. (Wilks 1962; Serfling 1981).
If the complete data of d1, d2, … , dN and ψ1, ψ2, … , ψN were available, the maximum-likelihood estimates of θ1, θ2, … , θL and μ, σ would be easily obtained as θ̂j = nj/(2N) for j = 1, 2, … , L and 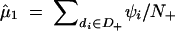 ,
, 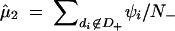 ,
,
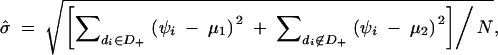 |
where nj is the number of the copies of the jth haplotype in the N subjects, N+ denotes the number of subjects who possess haplotype hb, and N− denotes the number of subjects who do not possess haplotype hb.
However, the complete data are not available, and we observe only genotypes and phenotypes of the subjects. Therefore, we substitute the expected values of nj/(2N), ∑di∈D+ψi/N+, ∑di∉D+ψi/N−, and
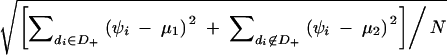 |
for the real values in the following EM algorithm.
For n = 0, initial values are given to
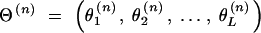 , where
, where 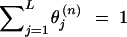 and θnj ≥ 0.
and θnj ≥ 0.For n = 0, initial values are given to
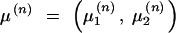 .
.For n = 0, initial values are given to σ(n).
- For all i and for all ak consistent with the genotype gi, calculate
where Ai denotes the set of am consistent with gi. Note that we examine only ak consistent with gi. In addition, since di is independent of μ(n) and σ(n), and ψi is independent of Θ(n) conditional on di, Equation 3 becomes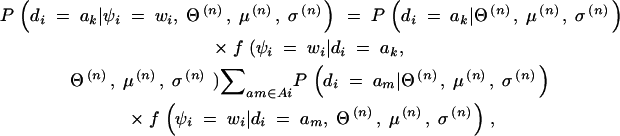
3
v. Since nj, the number of jth haplotype copies possessed by N subjects is a random variable, we can define the expected number of jth haplotype copies conditional on the observed data as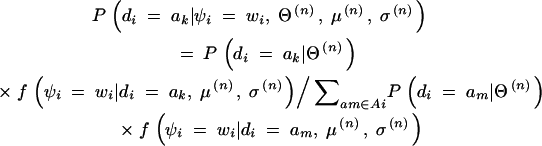
4
where gj(ak) denotes the number of jth haplotype copies in ak, and Ai again denotes the set of diplotype configurations for the ith subject that is consistent with gi. Note that gj(ak) is 0, 1, or 2. The expected values are calculated for all j.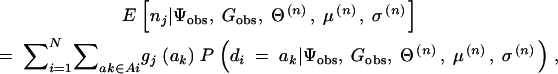
- Here, ∑di∈D+ψi/N+, ∑di∉D+ψi/N−, and
are random variables and, therefore, expected values conditional on the observed data can be defined as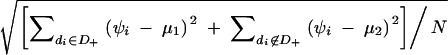
In the above equation,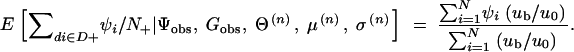
5
and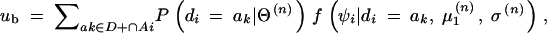
where the denominator and the numerator in Equation 5 are the summed probability densities of the observed data for the ith subject consistent with gi and those consistent with gi and ak ∈ D+ ∩ Ai: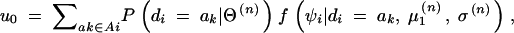
In the above equation,
6
and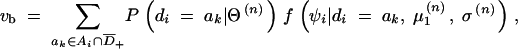
where the denominator and the numerator in Equation 6 are the summed probability densities of the observed data for the ith subject consistent with gi and those consistent with gi and ak ∈ Ai ∩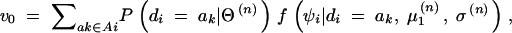
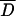 +,
+,
where n denotes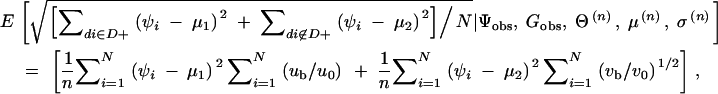
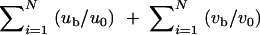 .
. - From the result of step v, Θ is updated for the next step as follows:
From the result of step vi, μ and σ are updated for the next step as follows:
viii. Steps iv–vii are repeated until the values converge. The values when converged are considered as the maximum-likelihood estimates Θ̂ = (θ̂1, θ̂2, … , θ̂L), μ̂1, μ̂2, and σ̂.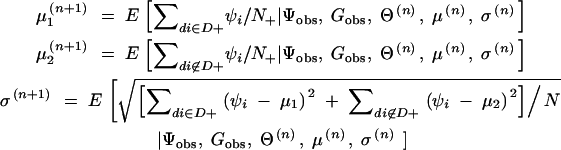
To avoid the local maximum, different sets of values for
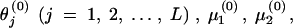 and σ(0) are tested.
and σ(0) are tested.
Here, Equation 1, given the values Θ̂, μ̂, and σ̂, is the maximum-likelihood Lmax for the alternative hypothesis. If we give the condition μ0 = (μ0, μ0) and repeat steps iv–vii, then we get the maximum-likelihood L0max for the null hypothesis. The present algorithm can handle missing data in both the observed genotypes and the phenotypes. Thus, when the genotype data were missing in some loci for the ith subject, gi, the observed genotypes for i, were interpreted as the set of all possible genotypes consistent with the observed genotypes excluding the loci where the data were missing. When the phenotype was missing for the ith subject, the likelihood of only the observed genotype data but not that of the phenotype data was included in the calculation. Even when the phenotype data were missing for some subjects, the inclusion of their observed genotype data for the analysis has increased the accuracy of the estimation of the population haplotype frequencies.
Under the null hypothesis, the statistic −2 log(L0max/Lmax) is expected to follow, asymptotically, χ2 distribution with 1 d.f.. The above algorithm is implemented as a computer program QTLHAPLO.
Designs of simulations:
The QTL parameter estimations:
The purpose of the simulation was to verify the accuracy of the estimation of the parameters for the distribution of the phenotype. A sample was generated by the experiment defined above. Then an ordered combination of two haplotypes was randomly selected from a collection of haplotype copies and given to each of the N subjects according to the given haplotype frequencies. We obtained haplotype frequencies for the SAA1 gene from a previous study (Moriguchi et al. 2001). SNP data at six loci were included in the haplotype data of the SAA1 gene. We performed two types of simulations, one using the data from six loci and the other from four loci. The latter set of loci (four loci) was obtained by excluding the first and the fourth loci, which were in only weak linkage disequilibrium with the other loci. Haplotype frequencies used in the two types of simulations are shown in Table 1. We assumed that one of the haplotypes is associated with the phenotype, and the phenotype of the subject with that haplotype follows N(μ1, σ2). The phenotype of the subject without that haplotype was assumed to follow N(μ2, σ2). Thereafter, we removed the phase information and ran our algorithm to estimate parameters.
TABLE 1.
Haplotype frequencies for theSAA1 gene
| Six-locus data
|
Four-locus data
|
||||
|---|---|---|---|---|---|
| Haplotype | Frequency | Haplotype | Frequency | Haplotype | Frequency |
| ACTGCC | 0.394 | AGCACT | 0.018 | CTCC | 0.391 |
| ACCGTCa | 0.214 | GGCGCT | 0.017 | GCCT | 0.267 |
| AGCGCT | 0.210 | ACTGTC | 0.013 | CCTCa | 0.258 |
| GCCGTC | 0.036 | ACCGCC | 0.006 | CTCT | 0.061 |
| GCTGCT | 0.035 | ACCATC | 0.006 | CTTC | 0.013 |
| GGCACT | 0.023 | AGCGCC | 0.003 | CCCC | 0.007 |
| ACTGCT | 0.023 | GCCC | 0.003 | ||
The haplotype that was assigned as the “quantitative phenotypes-associated haplotype.”
Behavior of the statistic −2 log(L0max/Lmax) under the null hypothesis:
The purpose of this simulation was to examine the distribution of the likelihood-ratio test statistic −2 log(L0max/Lmax) under the null hypothesis μ1 = μ2. The null hypothesis was equivalent to the assumption of no association between the phenotype and the presence of the haplotype. The test statistic was determined for each sample. The distribution of the test statistic was empirically estimated from samples generated by the simulation.
The estimation of power:
The purpose of this simulation was to estimate the power under the alternative hypothesis. With varying values of μ1, μ2, and σ, the empirical power was determined.
Bootstrap method to calculate standard errors of the estimated parameters:
To evaluate the reliability of estimated parameters μ̂1 , μ̂2, and σ̂, we used the bootstrap method (nonparametric bootstrap method) to calculate means and standard errors. From the original real sample, an artificial sample was generated by drawing the same number of the subjects at random. A single subject in the original sample may be repetitively drawn. It means that a new sample was drawn from the population in which the subjects in the original sample were uniformly distributed. Using the new artificial sample, the parameters were estimated using QTLHAPLO. The above procedure was repeated 10,000 times and the values of the estimated parameters were used to calculate the mean and the standard error.
Extension of the algorithm:
The present algorithm was extended so that it can handle dominant, recessive, and additive modes of inheritance. Let A denote the haplotype for a genetic region R that is related to the phenotype, and let B denote the complement of A, i.e., the set of all haplotypes other than A. We gave the following mean variables for different diplotype configurations. Thus, we gave μ1 for both AA and AB and μ2 for BB in the dominant mode, while we gave μ1 for AA and μ2 for both AB and BB in the recessive mode. In the additive mode, we gave μ1 and μ2 for AA and BB, respectively, and (μ1 + μ2)/2 for AB. In addition, we have implemented the mode in which the three different means μ1, μ2, and μ3 were given to the three different diplotype configurations.
Another extension is to define A not as a single haplotype but as a set of multiple haplotypes. For example, we can denote D+ as the set of diplotype configurations that contain either of the two phenotype-associated haplotypes. More generally, we can define a set Q as a set of all phenotype-associated haplotypes and D+ as the set of diplotype configurations with at least one member of Q. We implemented dominant, recessive, and additive modes for the analysis using such sets of haplotypes. In this way, we can test the association between a set of haplotypes and a phenotype. Since a SNP can be defined as a set of haplotypes, we could test the association between a SNP and a phenotype in this way. This extension was also implemented in QTLHAPLO.
RESULTS
Analysis of real data:
We analyzed the data from the diabetic patients. The data included the genotypes at CAPN10 and quantitative phenotypes such as BMI, blood glucose level (BS), and immunoreactive insulin level (IRI). The precise data will be published elsewhere (N. Iwasaki, Y. Horikawa, Y. Kitamura, Y. Nakamura, Y. Tanizawa, Y. Oka, K. Hara, T. Kadowaki, T. Awata, M. Honda, K. Yamashita, M. Ogata, N. Kamatani, N. J. Cox, G. I. Bell and Y. Iwamoto). These quantitative phenotypes are expected to follow asymptotically normal distributions (data not shown). Table 2 also shows the haplotype frequencies Θ inferred under the hypothesis of no linkage disequilibrium. Table 3 shows that the pairwise linkage disequilibrium measures D, D′, and r2 estimated under the presence of linkage disequilibrium. These results showed that there was considerable linkage disequilibrium between each pair of the loci of CAPN10.
TABLE 2.
Estimated haplotype frequencies for theCAPN10 gene
| Haplotypea | Frequencyb | Frequency under linkage equilibriumc |
|---|---|---|
| 121 | 0.5696 | 0.4285 |
| 112 | 0.2588 | 0.0974 |
| 111 | 0.1078 | 0.2557 |
| 221 | 0.0468 | 0.0250 |
| 122 | 0.0087 | 0.1632 |
| 212 | 0.0070 | 0.0057 |
| 222 | 0.0012 | 0.0095 |
Genotypes were determined at three SNP sites in the CAPN10 gene in 281 diabetic subjects. These three SNP sites have been reported to be associated with the development of type II diabetes (Horikawa et al. 2000). Using the genotype but not the phenotype data in 281 subjects, the haplotype frequencies were estimated by QTLHAPLO using only the genotype data.
Haplotype involving the three SNP sites within the CAPN10 gene.
The haplotype frequencies Θ were estimated from the genotype data by assuming the presence of the linkage disequilibrium using QTLHAPLO.
The haplotype frequencies Θ were estimated from the genotype data by assuming the linkage equilibrium. Note that the frequencies of a haplotype are expressed as the product of the allele frequencies in the case of linkage equilibrium.
TABLE 3.
Estimated linkage disequilibrium measures for theCAPN10 gene
| Locus 1 | Locus 2 | Disequilibrium parameter: D | Standardized disequilibrium: D′ |
r2 |
|---|---|---|---|---|
| 1 | 2 | −0.0138 | −0.6709 | 0.0157 |
| 1 | 3 | −0.0094 | −0.6148 | 0.0084 |
| 2 | 3 | 0.1628 | 0.9424 | 0.5669 |
The haplotype frequencies Θ were estimated from the genotype data from 281 subjects at the three loci within the CAPN10 gene under the assumption of the presence of linkage disequilibrium, and the pairwise linkage disequilibrium measures D, D′, and r2 were calculated from the estimated Θ̂.
Then, we incorporated the quantitative phenotype data in addition to the genotype data into the analysis. Thus, one of the following quantitative phenotypes was selected: BMI, BS at 0 min (BS 0′), BS 30′, BS 60′, BS 120′, IRI at 0 min (IRI 0′), IRI 30′, IRI 60′, or IRI 120′ (Table 4). The results indicate that there were significant associations between the presence of the haplotype 112 and both BS 30′ and BS 60′ (Table 4). Table 5 shows that when the haplotype 112 was assumed to be the phenotype-associated haplotype, μ̂1 > μ̂2, suggesting that the subjects with the 112 haplotype exhibit higher blood glucose levels at 30 and 60 min after the glucose ingestion than those without the haplotype. SE by the bootstrap method were (μ1 ± SE, μ2 ± SE, σ ± SE) = (147.1 ± 2.6, 138.9 ± 2.2, 28.2 ± 1.1) in the blood glucose level at 30 min, and (138.5 ± 3.7, 129.0 ± 3.0, 38.8 ± 1.5) in the blood glucose level at 60 min. In addition, haplotype 122 was significantly associated with BS 0′ (Table 4). However, this may not necessarily indicate that the subjects with the 122 haplotype exhibit lower fasting glucose levels than those without the haplotype. In fact, the frequency of the 122 haplotype was 0.0087, a value too low to evaluate (Table 2). Although such problems as multiple testing should be kept in mind, these results suggest an association between the 112 haplotype and blood glucose levels.
TABLE 4.
Results of the test of association between the possession of a haplotype within theCAPN10 gene and a phenotype
| Haplotypea
|
||||||
|---|---|---|---|---|---|---|
| Quantitative phenotypeb |
111 | 112 | 121 | 122 | 212 | 221 |
| BMI | 0.6945c | 0.8070 | 0.8212 | 0.2023 | 0.6404 | 0.6388 |
| BS 0′ | 0.1359 | 0.9367 | 0.3346 | 0.0202 | 0.3308 | 0.7343 |
| BS 120′ | 0.1629 | 0.3311 | 0.7492 | 0.8296 | 0.7076 | 0.3930 |
| BS 30′ | 0.3446 | 0.0140 | 0.6959 | 0.9199 | 0.9823 | 0.2765 |
| BS 60′ | 0.5855 | 0.0406 | 0.3630 | 0.4207 | 0.6450 | 0.6953 |
| IRI 0′ | 0.8445 | 0.8333 | 0.6737 | 0.3340 | 0.4997 | 0.6336 |
| IRI 120′ | 0.5277 | 0.5698 | 0.2823 | 0.3505 | 0.9530 | 0.7354 |
| IRI 30′ | 0.8457 | 0.4698 | 0.5068 | 0.2656 | 0.8750 | 0.7758 |
| IRI 60′ | 0.8581 | 0.0589 | 0.3135 | 0.3548 | 0.8576 | 0.7383 |
Each haplotype was assumed to be the phenotype-associated haplotype.
One of the various quantitative phenotypes was selected for the test. The genotype data for 281 subjects were combined with the phenotype data and analyzed by QTLHAPLO to test the association between the possession of a haplotype and the quantitative phenotype. BMI, body mass index; BS, blood glucose level; IRI, immunoreactive insulin level; 0′, 30′, 60′, and 120′: 0, 30, 60, and 120 min, respectively.
The results show P-values that exclude the null hypothesis that the quantitative phenotype is not associated with the haplotype. The underlined values indicate P-values <0.05.
TABLE 5.
Estimates of the parameters for the distributions of the phenotypes under various conditions
| Haplotypea
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 111
|
112
|
121
|
|||||||
| Quantitative phenotypeb |
μ̂1 | μ̂2 | σ̂ | μ̂1 | μ̂2 | σ̂ | μ̂1 | μ̂2 | σ̂ |
| BMI | 22.4c | 22.3 | 3.01 | 22.4 | 22.26 | 3.01 | 22.3 | 22.4 | 3.01 |
| BS 0′ | 91.0 | 93.2 | 9.35 | 92.8 | 92.74 | 9.39 | 93.0 | 91.7 | 9.37 |
| BS 30′ | 139.2 | 143.3 | 28.5 | 147.1 | 138.9 | 28.2 | 142.8 | 141.2 | 28.5 |
| BS 60′ | 130.5 | 133.8 | 39.0 | 138.5 | 129.0 | 38.8 | 134.2 | 128.9 | 39.0 |
| BS 120′ | 102.1 | 105.9 | 18.2 | 106.4 | 104.2 | 18.2 | 105.3 | 104.5 | 18.2 |
| IRI 0′ | 1.78 | 1.77 | 0.423 | 1.77 | 1.78 | 0.424 | 1.78 | 1.75 | 0.424 |
| IRI 30′ | 3.49 | 3.48 | 0.540 | 3.51 | 3.46 | 0.539 | 3.47 | 3.52 | 0.539 |
| IRI 60′ | 3.58 | 3.60 | 0.562 | 3.67 | 3.54 | 0.559 | 3.61 | 3.53 | 0.561 |
| IRI 120′ | 3.25 | 3.30 | 0.545 | 3.31 | 3.27 | 0.545 | 3.31 | 3.22 | 0.544 |
| Haplotypea
| |||||||||
| 122
|
212
|
221
|
|||||||
| Quantitative phenotypeb |
μ̂1 | μ̂2 | σ̂ | μ̂1 | μ̂2 | σ̂ | μ̂1 | μ̂2 | σ̂ |
| BMI | 20.3 | 22.3 | 3.00 | 21.6 | 22.3 | 3.01 | 22.6 | 22.3 | 3.01 |
| BS 0′ | 82.8 | 92.9 | 9.30 | 88.6 | 92.8 | 9.37 | 92.2 | 92.8 | 9.38 |
| BS 30′ | 141.2 | 142.5 | 28.5 | 141.9 | 142.5 | 28.5 | 136.8 | 143.1 | 28.5 |
| BS 60′ | 118.7 | 133.3 | 39.0 | 125.1 | 133.3 | 39.0 | 130.3 | 133.4 | 39.0 |
| BS 120′ | 103.4 | 105.2 | 18.2 | 102.0 | 105.2 | 18.2 | 102.3 | 105.5 | 18.2 |
| IRI 0′ | 1.58 | 1.78 | 0.423 | 1.91 | 1.77 | 0.423 | 1.74 | 1.78 | 0.424 |
| IRI 30′ | 3.76 | 3.48 | 0.539 | 3.52 | 3.48 | 0.540 | 3.51 | 3.48 | 0.540 |
| IRI 60′ | 3.35 | 3.60 | 0.561 | 3.64 | 3.60 | 0.562 | 3.56 | 3.60 | 0.562 |
| IRI 120′ | 3.05 | 3.29 | 0.545 | 3.27 | 3.29 | 0.545 | 3.26 | 3.29 | 0.545 |
The genotype data at the three loci within the CAPN10 gene were combined with the data of one of the quantitative phenotypes and analyzed by QTLHAPLO under the alternative hypothesis, assuming that one of the haplotypes was the phenotype-associated haplotype. The underlined data indicate that the difference was considered significant (P-values <0.05) by the test described in methods.
One of the haplotypes was assumed to be the phenotype-associated haplotype.
One of the quantitative phenotypes was selected as the phenotype to be tested.
Maximum-likelihood estimates of the parameters under the alternative hypothesis.
The accuracy of estimated values of parameters:
We used the simulation to generate samples under either the null or the alternative hypothesis and analyzed the data in the sample using QTLHAPLO.
First, haplotype frequencies Θ were employed from the four-locus data at the SAA1 gene, as shown in Table 1. The CCTC haplotype was considered to be the phenotype-associated haplotype. Note that all of the four loci were in tight linkage disequilibrium with each other. Two haplotype copies were selected using the haplotype frequencies and assigned to each subject. The phenotype of the subject was determined stochastically using two normal distributions. N(μ1, σ2) was used when the subject possessed the phenotype-associated haplotype, while N(μ2, σ2) was used when the subject did not possess the haplotype. The parameters μ1, μ2, and σ were given to each simulation as described in Table 6. A sample consisted of a total of N subjects.
TABLE 6.
Accuracy of estimation of the parameters for the distribution of a quantitative phenotype in the analysis of simulated four-locus data for theSAA1 gene
| Sampleb
|
Estimatedc
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Populationa (μ1, μ2, σ) |
N | Mean 1 | Mean 2 | SD | μ̂1 ± SE | μ̂2 ± SE | σ̂ ± SE | P-valuesd |
| (160, 160, 5.0)e | 100 | 160.31 | 159.03 | 5.142 | 160.31 ± 0.72 | 159.03 ± 0.72 | 5.142 ± 0.319 | 0.223 |
| 200 | 159.00 | 159.94 | 5.148 | 159.00 ± 0.57 | 159.94 ± 0.47 | 5.148 ± 0.243 | 0.197 | |
| 400 | 159.87 | 159.78 | 4.860 | 159.87 ± 0.36 | 159.78 ± 0.33 | 4.860 ± 0.143 | 0.862 | |
| 1000 | 160.18 | 159.92 | 4.883 | 160.18 ± 0.23 | 159.92 ± 0.21 | 4.883 ± 0.114 | 0.404 | |
| (161, 160, 5.0) | 100 | 161.17 | 159.75 | 5.271 | 161.17 ± 0.80 | 159.75 ± 0.71 | 5.271 ± 0.328 | 0.188 |
| 200 | 161.22 | 160.00 | 4.795 | 161.22 ± 0.46 | 160.00 ± 0.49 | 4.795 ± 0.191 | 0.0739 | |
| 400 | 160.89 | 160.01 | 4.788 | 160.89 ± 0.36 | 160.01 ± 0.32 | 4.788 ± 0.156 | 0.0661 | |
| 1000 | 161.38 | 160.16 | 5.091 | 161.38 ± 0.24 | 160.16 ± 0.22 | 5.091 ± 0.120 | 0.000159 | |
| (163, 160, 5.0) | 100 | 163.27 | 160.23 | 4.925 | 163.27 ± 0.80 | 160.23 ± 0.63 | 4.926 ± 0.352 | 0.00312 |
| 200 | 162.90 | 159.38 | 5.020 | 162.90 ± 0.56 | 159.38 ± 0.47 | 5.020 ± 0.223 | 1.61 × 10−6 | |
| 400 | 162.74 | 159.70 | 4.858 | 162.74 ± 0.36 | 159.70 ± 0.33 | 4.859 ± 0.167 | 1.23 × 10−9 | |
| 1000 | 163.10 | 159.68 | 4.933 | 163.10 ± 0.23 | 159.68 ± 0.20 | 4.934 ± 0.107 | 3.86 × 10−26 | |
| (165, 160, 5.0) | 100 | 163.80 | 160.07 | 5.159 | 163.80 ± 0.80 | 160.08 ± 0.69 | 5.159 ± 0.304 | 0.000598 |
| 200 | 164.99 | 160.17 | 4.953 | 164.99 ± 0.48 | 160.17 ± 0.51 | 4.953 ± 0.248 | 8.66 × 10−11 | |
| 400 | 165.16 | 160.67 | 4.894 | 165.16 ± 0.37 | 160.67 ± 0.33 | 4.895 ± 0.169 | 3.32 × 10−18 | |
| 1000 | 165.12 | 160.21 | 4.875 | 165.12 ± 0.23 | 160.21 ± 0.21 | 4.875 ± 0.113 | 1.30 × 10−50 | |
A sample of size N was obtained by simulation using a set of given parameters, and the data obtained were analyzed, after removing the phase information, using QTLHAPLO for both the estimation of parameters and the test of the association between the presence of a haplotype and the quantitative phenotype.
Values described in parentheses were given to the parameters μ1, μ2, and σ. Two haplotypes were selected from the population haplotype pool according to the haplotype frequencies (Θ) obtained from the four-locus data of the SAA1 gene (see Table 1) and given to each subject. The quantitative phenotype was determined stochastically for each subject depending on whether the phenotype-associated haplotype (CCTC haplotype was assumed to be the phenotype-associated haplotype in this case) was present (μ1 was used) or absent (μ2 was used) using the two normal distributions N(μ1, σ2) and N(μ2, σ2).
For each sample, the means of the quantitative phenotypes for the subjects with the phenotype-associated haplotype (mean 1) and that for the subjects without the haplotype (mean 2) were determined. SDs of the quantitative phenotypes for all the subjects were calculated as follows: 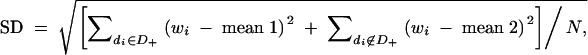 where D+ is a set of diplotype configurations with the phenotype-associated haplotype, while di is the diplotype configuration for the ith subject. wi is the observed quantitative phenotype of the ith subject.
where D+ is a set of diplotype configurations with the phenotype-associated haplotype, while di is the diplotype configuration for the ith subject. wi is the observed quantitative phenotype of the ith subject.
From the sample, phase information was removed. The genotype information and the phenotype information were used for the estimation of the parameters using QTLHAPLO. SEs of the estimated parameters were calculated as described in Designs of simulations.
At the same time, the sample statistic −2 log(L0max/Lmax) was calculated for each sample, and the P-value was determined by QTLHAPLO assuming that, under the null hypothesis, the sample statistic followed a χ2 distribution with 1 d.f.
This parameter set is equivalent to the null hypothesis.
After diplotype configurations and phenotypes were determined for all the subjects, the phase information was removed. Using the genotype information and the phenotypes of the subjects, we used QTLHAPLO to estimate the parameters Θ, μ1, μ2, and σ and, at the same time, calculated P-values for excluding the null hypothesis.
The results showed that our algorithm is highly accurate for estimating the parameters μ1, μ2, and σ, whether the simulation is performed under the null hypothesis or the alternative hypothesis under the given conditions (Table 6). As expected, the P-value to exclude the null hypothesis was high when the simulation was under the null hypothesis, while it was low when the simulation was performed under the alternative hypothesis (Table 6).
Next, Θ was employed from the six-locus data at the SAA1 gene, shown in Table 1. Note that two of the six loci were in only weak linkage disequilibrium with the other loci. In this case, the ACCGTC haplotype was considered to be the phenotype-associated haplotype. The simulation and the analysis of the data generated by the simulation were performed exactly as in the case of the four-locus data as described above except for the number of loci.
Table 7 again shows that the estimation of the parameters was accurate. The risk to exclude the null hypothesis (P-value) was high when the data were simulated under the null hypothesis, while it was very low when they were simulated under the alternative hypothesis (Table 7).
TABLE 7.
Accuracy of estimation of the parameters for the distribution of a quantitative phenotype in the analysis of simulated six-locus data for theSAA1 gene
| Sampleb
|
Estimatedb
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Populationa (μ1, μ2, σ) |
N | Mean 1 | Mean 2 | SD | μ̂1 ± SE | μ̂2 ± SE | σ̂ ± SE | P-valuesb |
| (160, 160, 5.0)b | 100 | 159.33 | 159.65 | 5.178 | 159.20 ± 0.80 | 159.72 ± 0.69 | 5.174 ± 0.312 | 0.637 |
| 200 | 159.88 | 159.36 | 5.164 | 159.87 ± 0.70 | 159.36 ± 0.44 | 5.164 ± 0.239 | 0.517 | |
| 400 | 159.51 | 159.98 | 4.855 | 159.39 ± 0.40 | 160.05 ± 0.30 | 4.851 ± 0.143 | 0.194 | |
| 1000 | 159.94 | 160.10 | 4.884 | 159.95 ± 0.26 | 160.09 ± 0.19 | 4.884 ± 0.113 | 0.642 | |
| (161, 160, 5.0) | 100 | 159.99 | 160.44 | 5.229 | 160.16 ± 0.88 | 160.34 ± 0.67 | 5.233 ± 0.329 | 0.870 |
| 200 | 160.99 | 160.12 | 4.737 | 160.95 ± 0.55 | 160.13 ± 0.47 | 4.739 ± 0.234 | 0.220 | |
| 400 | 160.65 | 159.99 | 4.781 | 160.63 ± 0.40 | 160.02 ± 0.30 | 4.782 ± 0.170 | 0.209 | |
| 1000 | 160.77 | 159.84 | 5.056 | 160.78 ± 0.27 | 159.84 ± 0.20 | 5.055 ± 0.115 | 0.00345 | |
| (163, 160, 5.0) | 100 | 162.81 | 160.49 | 4.915 | 162.90 ± 0.79 | 160.51 ± 0.63 | 4.911 ± 0.350 | 0.0219 |
| 200 | 162.55 | 159.90 | 5.235 | 162.50 ± 0.59 | 159.87 ± 0.49 | 5.236 ± 0.285 | 0.000794 | |
| 400 | 163.20 | 159.83 | 5.188 | 163.06 ± 0.39 | 159.95 ± 0.34 | 5.229 ± 0.205 | 6.47 × 10−9 | |
| 1000 | 162.76 | 159.89 | 4.873 | 162.65 ± 0.25 | 159.98 ± 0.20 | 4.900 ± 0.109 | 2.90 × 10−17 | |
| (165, 160, 5.0) | 100 | 165.15 | 159.23 | 5.178 | 165.04 ± 0.90 | 159.30 ± 0.64 | 5.227 ± 0.319 | 5.58 × 10−7 |
| 200 | 164.47 | 159.17 | 4.541 | 164.26 ± 0.50 | 159.27 ± 0.44 | 4.623 ± 0.189 | 3.95 × 10−13 | |
| 400 | 164.98 | 160.03 | 5.021 | 164.95 ± 0.42 | 160.09 ± 0.31 | 5.047 ± 0.171 | 1.72 × 10−19 | |
| 1000 | 164.89 | 160.11 | 4.957 | 164.81 ± 0.24 | 160.19 ± 0.20 | 4.996 ± 0.112 | 9.62 × 10−44 | |
A sample of size N was obtained by the simulation using a set of given parameters, and the data obtained were analyzed, after removing the phase information, using QTLHAPLO for both the estimation of parameters and the test of the association between the presence of a haplotype and the quantitative phenotype.
The conditions of the simulations were the same as in Table 6 except for the following two points: the haplotype frequencies (Θ) obtained from the six-locus data of the SAA1 gene (see Table 1) were used and the ACCGTC haplotype was assumed to be the phenotype-associated haplotype.
The methods for the calculations of the values in these categories are the same as those in Table 6.
Accuracy of estimated diplotype configuration:
Using the simulated data, the posterior probability distribution of the diplotype configuration (diplotype distribution) for each subject conditional on the observed genotype and phenotype data [P(di = ak|Gobs, Ψobs)] was determined by QTLHAPLO. The diplotype distribution for each subject conditional on only the observed genotype data [P(di = ak|Gobs)] was also determined using the same program. It is of interest to examine whether the diplotype distribution changes when the observed phenotype data are added to the observed genotype data. Another question is whether the inference becomes more accurate when the observed phenotype data are incorporated. Table 8 shows the comparison of the diplotype distribution for each subject inferred by only the genotype data and inferred by both the genotype and the phenotype data. In this case, the simulation was performed under the conditions μ1 = 165, μ2 = 160, σ = 5.0, and N = 1000. The results were shown only for individuals i = 1, 2, … , 10. For the subjects 1, 3, 4, 5, 6, 8, and 9, the diplotype configurations were concentrated on single events whether or not the observed phenotype data were incorporated (Table 8). For subjects 2, 7, and 10, the diplotype configurations were not concentrated on single events; however, the distributions were almost identical between the two inferences, one made by incorporating the observed phenotype data and the other without them (Table 8).
TABLE 8.
Posterior probability distribution of diplotype configuration for each subject: four-locus data
| Subject (i) | Quantitative phenotype |
Diplotype configuration |
True or falsea | Posterior distributionb |
Posterior distributionc |
|---|---|---|---|---|---|
| 1 | 157.1 | GCCT GCCT | True | 1.0000 | 1.0000 |
| 2 | 170.3 | CCTC CTCC | True | 0.9993 | 0.9999 |
| CCCC CTTC | False | 0.0007 | 0.0001 | ||
| 3 | 173.4 | CCTC GCCT | True | 1.0000 | 1.0000 |
| 4 | 158.2 | CTCT GCCT | True | 1.0000 | 1.0000 |
| 5 | 170.6 | CTCT GCCT | True | 1.0000 | 1.0000 |
| 6 | 162.4 | CTCC CTCC | True | 1.0000 | 1.0000 |
| 7 | 161.6 | CTCC GCCT | True | 0.9975 | 0.9975 |
| CTCT GCCC | False | 0.0025 | 0.0025 | ||
| 8 | 149.4 | CTCC CTCC | True | 1.0000 | 1.0000 |
| 9 | 164.3 | CCTC GCCT | True | 1.0000 | 1.0000 |
| 10 | 165.1 | CTCC GCCT | True | 0.9975 | 0.9975 |
| CTCT GCCC | False | 0.0025 | 0.0025 |
Simulations were started by assigning diplotype configurations to N = 1000 number of subjects according to the haplotype frequencies employed from the four-locus data for the SAA1 gene. Depending on whether the subject possessed the phenotype-associated haplotype CCTC, a quantitative phenotype was drawn from N(μ1, σ2) or N(μ2, σ2), where μ1 = 165, μ2 = 160, and σ = 5. After removing the phase information, QTLHAPLO was used to determine the posterior probability distribution of the diplotype configuration (diplotype distribution) for each subject either by using only genotype data or by using both genotype and phenotype data.
Possible diplotype configurations for each subject were compared with the diplotype configuration before the phase information was removed. “True” means that the diplotype configuration before the removal of the phase information was the same as the estimated diplotype configuration, while “False” means that they were different.
Posterior probability distribution of the diplotype configuration given only the observed genotype data [P(di = ak|Gobs)].
Posterior probability distribution of the diplotype configuration given the observed genotype and phenotype data [P(di = ak|Gobs, Ψobs)].
The comparison of the diplotype distribution between the two inferences was done using the six-locus data for the SAA1 gene. Part of the results are shown in Table 9. In this case, the diplotype distributions were not concentrated on single events in the subjects i = 53, 55, 57, 58, and 59. For the subject i = 55, the diplotype distribution differed significantly between the two inferences. Since this subject has a quantitative phenotype of 167.8, the subject is likely to possess the phenotype-associated haplotype ACCGTC because μ1 = 165 and μ2 = 160. The incorporation of the phenotype data changed the diplotype distribution of the subject i = 55 so that the probability of the diplotype configuration occurring with the phenotype-associated haplotype (ACCGTC GCTGCT) increased. Thus the inclusion of the phenotype data changed the diplotype distribution for each subject and seemed to improve the accuracy of the inference of the diplotype configurations.
TABLE 9.
Estimated probability distribution of diplotype configuration for each subject: six-locus data
| Subject (i) | Quantitative phenotype |
Diplotype configuration |
True or false | Posterior distribution |
Posterior distribution |
|---|---|---|---|---|---|
| 51 | 157.3 | AGCGCT AGCACT | True | 1.0000 | 1.0000 |
| 52 | 172.0 | AGCGCT AGCGCT | True | 1.0000 | 1.0000 |
| 53 | 152.6 | ACTGCC AGCGCT | True | 0.9993 | 0.9993 |
| ACTGCT AGCGCC | False | 0.0007 | 0.0007 | ||
| 54 | 155.5 | AGCGCT AGCGCT | True | 1.0000 | 1.0000 |
| 55 | 167.8 | ACCGTC GCTGCT | True | 0.8871 | 0.9619 |
| ACTGCT GCCGTC | False | 0.1129 | 0.0381 | ||
| 56 | 161.7 | ACTGCC ACTGCC | True | 1.0000 | 1.0000 |
| 57 | 165.7 | ACTGCC AGCGCT | True | 0.9993 | 0.9993 |
| ACTGCT AGCGCC | False | 0.0007 | 0.0007 | ||
| 58 | 153.9 | ACTGCC AGCGCT | True | 0.9993 | 0.9993 |
| ACTGCT AGCGCC | False | 0.0007 | 0.0007 | ||
| 59 | 157.0 | ACTGCC AGCGCT | True | 0.9993 | 0.9993 |
| ACTGCT AGCGCC | False | 0.0007 | 0.0007 | ||
| 60 | 166.7 | ACTGCC ACTGCC | True | 1.0000 | 1.0000 |
We have intensively addressed this issue by the simulation; i.e., we asked whether the inference of the diplotype configurations becomes more accurate by incorporating the phenotype data in addition to the genotype data. Thus, the inference of the diplotype configuration for each subject was performed using only the genotype data or using both genotype and phenotype data from the simulated samples. Then, we counted how many of the subjects' inferred diplotype configurations became more accurate and how many became less accurate by incorporating the phenotype data. When the posterior probability of the true diplotype configuration became higher by incorporating the phenotype data, as was the case with the subject i = 55 in Table 9, we judged that the inference became more accurate. On the other hand, when the posterior probability of the true diplotype configuration became lower by the incorporation of the phenotype data, we judged that the inference became less accurate. When the six-locus haplotype frequencies were used, the proportions of the subjects whose inference of the diplotype configurations became more and less accurate by the incorporation of the phenotype data were 0.1957 ± 0.0429 and 0.1061 ± 0.0413 (results from simulations under the same conditions as in Table 9), respectively. On the other hand, when the four-locus haplotype frequencies were used, the proportions of the subjects whose inference of the diplotype configurations became more and less accurate by the incorporation of the phenotype data were 0.1387 ± 0.0111 and 0.0589 ± 0.0075 (results from simulations under the same conditions as in Table 8), respectively.
Distribution of the statistic −2 log(L0max/Lmax) under the null hypothesis:
The null hypothesis is that the distribution of the quantitative phenotype is independent of the diplotype configurations. It is equivalent to the assumption of μ1 = μ2. The samples were simulated under the null hypothesis using various values of μ1 = μ2, and σ. For Θ, the four-locus data (Table 1) were used and the 10,000 independent samples were generated. Figure 1 shows the histogram for the statistic −2 log(L0max/Lmax) obtained by the analysis of the simulated data using QTLHAPLO. Thus, when the parameters were μ1 = μ2 = 160 and σ = 5, the statistic followed asymptotically the χ2 distribution with 1 d.f. Similar simulations were performed under various conditions followed by the analysis of the data using QTLHAPLO. Table 10 shows the results of the estimated values of the parameters and the type I error rates using two different Θ data sets (four-locus and six-locus data sets in Table 1) and two sample sizes (100 and 1000). The estimated parameters were accurate for both Θ data sets and two sample sizes. In addition, when the value of 3.841, where the cumulative distribution function for χ2 distribution with 1 d.f. becomes 0.95, was set as the threshold, the proportion of the samples that generated statistic values over the threshold (empirical type I error rate) was very close to the expected value of 0.05 (Table 10).
Figure 1.—

Histograms of the statistic −2 log(L0max/Lmax) produced under the null hypothesis. Simulation was performed under the null hypothesis, μ1 = μ2 = 160, σ = 5.0. Sample size N was either (A) 100 or (B) 1000, and number of repeats for a simulation was 10,000. The histograms of the statistic are shown with bars. The probability density function of χ2 distribution with 1 d.f. is shown with curves.
TABLE 10.
Estimated parameters and empirical type I error rates for analysis of the simulated data under the null hypothesis
| Θ | μ1 = μ2 | Sample size N | No. of samples |
μ̂1a (mean ± SD) | μ̂2a (mean ± SD) | σ̂a (mean ± SD) | Type I error rate |
|---|---|---|---|---|---|---|---|
| Four-locus model | 160 | 100 | 10,000 | 160.01 ± 0.753 | 160.01 ± 0.676 | 4.936 ± 0.353 | 0.0496 |
| Four-locus model | 160 | 1000 | 10,000 | 160.00 ± 0.237 | 160.00 ± 0.213 | 4.995 ± 0.112 | 0.0514 |
| Six-locus model | 160 | 100 | 10,000 | 160.00 ± 0.822 | 159.99 ± 0.635 | 4.933 ± 0.352 | 0.0606 |
| Six-locus model | 160 | 1000 | 10,000 | 160.00 ± 0.256 | 160.00 ± 0.201 | 4.994 ± 0.110 | 0.0541 |
Each simulation was performed under the null hypothesis, μ1 = 160, μ2 = 160, σ = 5, with N = 100 or 1000. Every simulation was repeated 10,000 times for each condition.
Mean ± SD of the estimates for parameters of the distribution of the quantitative phenotype obtained by the analysis.
Power of the test:
We determined the empirical powers of the present test using various conditions. First, samples were simulated under the alternative conditions and the data were analyzed by QTLHAPLO. The proportions of the samples that generated the statistic over the threshold value of 3.841 were considered as empirical powers. The results show that the power increases as a function of |μ1 − μ2| and sample size (Figure 2). Additional simulation experiments with different parameters followed by the analysis of the data show that the power was a function of |μ1 − μ2|/σ as expected (data not shown). Thus, our algorithm has a sufficient power when |μ1 − μ2|/σ and sample size are large.
Figure 2.—

Power of the test with regard to sample size and |μ1 − μ2|/σ. The solid line is for N = 100, and the dashed line is for N = 1000.
DISCUSSION
In this investigation, we developed an algorithm to estimate the parameters of the distribution of a quantitative phenotype and to test the association between the presence of a haplotype and the quantitative phenotype. The data used are genotype data at linked loci as well as the data of a quantitative phenotype in multiple subjects.
We examined whether our algorithm could accurately estimate the parameters. Samples of genotypes and phenotypes for multiple subjects were generated using various sets of parameters, and the data were analyzed by the maximum-likelihood method. The maximum-likelihood estimates thus obtained were very close to the values of the parameters that had been given before the simulation, indicating that our algorithm could accurately estimate the parameters.
Then we examined the distribution of the generalized likelihood-ratio statistic, obtained by analyzing the data derived under the null hypothesis. Under various conditions, the statistic was found to follow an asymptotically χ2 distribution with 1 d.f. In addition, the analysis of the data simulated under the alternative hypothesis indicated that the power was considerably high when |μ1 − μ2|/σ and sample size N were sufficiently large; i.e., (μ1 − μ2)/σ = 0.2, N = 1000 and (μ1 − μ2)/σ = 0.6, N = 100.
The importance of |μ1 − μ2|/σ for the power of the test is easily understood as follows. Let A denote the haplotype for a genetic region R that is related to the phenotype, and let B denote the complement of A, i.e., the set of all haplotypes other than A. In fact, both A and B may be sets of haplotypes rather than single haplotypes. In our model, the distribution of the phenotype was assumed to be different between the subjects with the (unordered) diplotype configurations AA, AB, and BB. This means that we divided the phenotype into two parts, i.e., the part due to the effect of the diplotype configurations in region R and the part independent of that effect. The latter part contains both environmental and genetic elements unrelated to region R. Thus, we assumed covariance neither between the effects of region R and other genetic loci nor between the effects of region R and the environment. It means that no epistasis was assumed in our model.
The impact of the effect on the phenotype is evaluated by comparing the variances (Fisher 1918). Thus, the impact of the effect of region R on the phenotype is evaluated by the ratio of the variance of the effect of region R to the total phenotypic variance (Amos 1994; Almasy and Blangero 1998; Pratt et al. 2000; Sham et al. 2000).
The mathematical modeling of this kind was initiated by Fisher (1918) many years ago, although it was not about the diplotype configurations but about the genotypes. Let σ2t and σ2r denote the total phenotypic variance and the variance due to region R, respectively. The ratio σ2r/σ2t is an indicator of the impact of region R in the total phenotypic variation. The difference 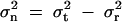 contains elements from both the environment and the genetic loci other than region R. If region R is the only genetic region relevant to the phenotype, then
contains elements from both the environment and the genetic loci other than region R. If region R is the only genetic region relevant to the phenotype, then  where σ2e denotes the variance due to environment. Note that, in this case,
where σ2e denotes the variance due to environment. Note that, in this case, 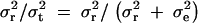 equals to heritability.
equals to heritability.
According to our model, the means of the phenotypes for the subjects with the diplotype configurations of AA and BB are μ1 and μ2. Then, if Hardy-Weinberg equilibrium can be assumed, the phenotypic variance due to region R is written when μ1 ≥ μ2 as
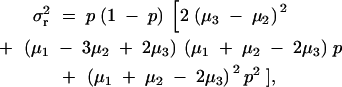 |
7 |
where μ3 denotes the mean of the phenotypes for the subjects with the diplotype configuration of AB, and p denotes the population frequency of the haplotype A.
In our model, σ2n, the variance unrelated to region R is equal to σ2, the variance of the phenotypes for the subjects with the same diplotype configurations for region R. Note that we assumed no difference in the variance for different normal distributions between the phenotypes for different diplotype configurations.
In the dominant model (the phenotypes for AA and AB are the same), μ3 = μ1 and Equation 7 becomes
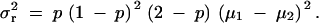 |
8 |
In the recessive model (the phenotypes for AB and BB are the same), μ3 = μ2 and Equation 7 becomes
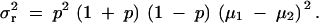 |
9 |
In the additive model, μ3 = (μ1 + μ2)/2 and Equation 7 becomes
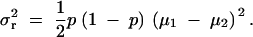 |
10 |
Therefore, in dominant (8), recessive (9), and additive (10) modes, σ2r has the form of f(p)(μ1 − μ2)2, and 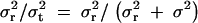 has the form of
has the form of
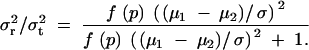 |
Thus, the ratio of the phenotypic variance due to the difference in the diplotype configurations for region R to the total phenotypic variance is positively correlated with |μ1 − μ2|/σ. Note that f(p) ≥ 0 for 0 ≤ p ≤ 1. This ratio σ2r/σ2t is equivalent to the heritability when region R is the only genetic region influencing the phenotype.
One of the problems in the haplotype inference is that the diplotype configurations of some subjects are not unequivocally determined. Such subjects with ambiguous diplotype configurations should be treated in the analysis. If one attempts to test the association between the diplotype configurations and a phenotype, the subjects with ambiguous diplotype configurations cannot be unequivocally categorized. As is often done, they can be classified into some categories according to the most likely diplotype configurations. However, such forced categorization may cause inflation of type I errors. In fact, our simulation studies have shown that the algorithm presented here is superior to such methods in that it allows the presence of ambiguous diplotype configurations when testing the association between the presence of a haplotype and a quantitative phenotype.
The problems of ambiguous diplotype configurations are amplified when the linkage disequilibrium of the loci to be analyzed is weak. We analyzed two cases in detail. In one case, all the four loci were in tight linkage disequilibrium, while two of the six loci were in weak linkage disequilibrium in the other. When all the four loci were in tight linkage disequilibrium, the percentage of the subjects with ambiguous diplotype configurations was low and the degree of the ambiguity was minimal. However, when two of the six loci were in weak linkage disequilibrium, the problems of the ambiguous diplotype configurations became large. Interestingly, the estimated probability of the true diplotype configuration was often larger when the phenotype data in addition to the genotype data were incorporated for the analysis than when only the genotype data were used. This indicates that the inference of the diplotype configurations becomes more accurate by incorporating the phenotype data when there is a true association between the presence of a haplotype and a quantitative phenotype.
Wu et al. (2002) proposed the joint linkage and linkage disequilibrium mapping strategy for estimating allelic frequencies, recombination fractions, and linkage disequilibria for multiallelic markers in natural populations using the Fisher-scoring algorithm. The genomic region within which the linkage disequilibrium is tight is denoted the haplotype block or linkage disequilibrium (LD) block. Within the haplotype block, the problem of ambiguous diplotype configurations is not large; however, it is likely to emerge when polymorphic loci outside the haplotype blocks or those in the region that includes the border(s) of the block(s) are the targets of the study. The value of the present algorithm may be high especially when the involved loci are not within a block.
We then applied this algorithm to the analysis of the data from diabetic patients. The data were composed of the genotypes at three SNP loci within the CAPN10 gene as well as the quantitative phenotypes. The three loci were in moderate linkage disequilibrium (|D′| > 0.6). The analysis has shown that there were significant associations between certain haplotypes and some quantitative phenotypes.
We modeled the test of the association between haplotypes and quantitative phenotypes in a way similar to that employed by Chiano and Clayton (1998), Fallin et al. (2001), Zaykin et al. (2002), and Lou et al. (2003). Thus, Chiano and Clayton (1998) developed the linear logistic regression model, which not only tests for association but also determines how far the haplotype harboring the putative disease gene extends, and estimated haplotype frequencies by the EM algorithm. Zaykin et al. (2002) have also developed a statistical method to test the association of haplotype frequencies with phenotypes in samples of unrelated individuals. They estimated haplotype frequencies using the EM algorithm and then related the inferred haplotype probabilities for each individual to the phenotype using regression-based analysis. Fallin et al. (2001) devised a method to test the association between haplotypes inferred by the EM algorithm and the disease phenotype using the chi-square statistic for contingency tables. They applied their method for testing the association between multiple SNPs in the APOE gene region and Alzheimer's disease and showed that it was useful even when the linkage disequilibrium was weak and the effect of the gene was rather small. The proposed framework by Lou et al. (2003) can accommodate genetic effects of different kinds for the QTL. Our model is easily extendable to estimate the interactions of two haplotypes and between haplotypes and environment (Chiano and Clayton 1998). There are some similarities between the above methods and our algorithm; however, our algorithm differs from each of them. For example, epistasis is not assumed in our algorithm while that by Lou et al. (2003) assumed its presence. In the extended phase of our algorithm, sets of haplotypes rather than single haplotypes can be handled. In addition, the sample size of 100 is sufficient for our algorithm while their algorithm needs larger sizes (Lou et al. 2003).
Our algorithm can be applied to real data only when the quantitative phenotypes are expected to follow the normal distributions. Indeed, many quantitative phenotypes may follow asymptotically normal distributions; however, there are certainly phenotypes that do not. One of the solutions to such problems may be to use the transformed value of the quantitative phenotype that is expected to follow, under the null hypothesis, a normal distribution. Some of the mathematical transformations that convert the phenotype include the logarithm transformation for the skewed trait (Schaid et al. 2002; Wrigth 1968) and the power transformation (Hoaglin et al. 1983). The nonparametric method may be another approach.
Although our algorithm is useful for cohort studies, it may be extended in other types of studies. One extension is the application of our algorithm to case-control studies. In principle, this algorithm can be applied to the data from cohort studies and clinical trials but not to those from case-control studies. The reason is that the estimated parameters Θ, μ1, μ2, and σ do not indicate the population parameters when this algorithm is applied to data from case-control studies. However, the test of the association between the presence of a haplotype and a quantitative phenotype may be possible by this algorithm even for the data from case-control studies. In this case, however, the maximum-likelihood estimates obtained by the algorithm are not the real estimates of the parameters. We are now extensively analyzing this issue by simulations to examine whether the application of the present algorithm to data from case-control studies is plausible. The test of the association between a phenotype and the diplotype configurations using subjects with “extreme” phenotypic values will be useful. If we can obtain two samples, one with high phenotypic values and the other with low phenotypic values, the test of the association will become very powerful. In this case, however, the same problem as stated above in the case of case-control studies will emerge. Although the data from such samples can be submitted to our algorithm and the outputs will be obtained, the estimated parameters (for example, Θ) do not indicate population parameters. We are now extensively analyzing this issue by simulations and have found that our algorithm can be used to analyze the data from the subjects with extreme phenotypes in some cases. It means that although the estimated parameters were incorrect, the type I errors did not inflate very much. However, it is still to be clarified under what conditions such application is plausible.
Recently, several groups have proposed algorithms and methods to study the association between haplotypes and quantitative phenotypes. Lou et al. (2003) proposed a haplotype-based algorithm for multilocus linkage disequilibrium mapping of quantitative trait loci with epistasis. Thus, the likelihood approach to the estimation of haplotype frequencies is useful; however, there are some limitations (Fallin and Schork 2000; Tishkoff et al. 2000). Fallin and Schork (2000) reported that the accuracy of haplotype estimation increases as the amount of linkage disequilibrium between loci increases using the likelihood approach. In this respect, Tanck et al. (2003) developed the weighted penalized log-likelihood model and compared it with the different log-likelihood models.
Although there have been several proposals for studies of the association between quantitative phenotypes and haplotypes, procedures that are both reliable and accurate still need to be developed. Such sophisticated methods will be necessary because a number of different quantitative phenotypes are expected to be studied in the near future at the population basis. Thus, not only quantitative phenotypes obtained by simple clinical examinations but also multiple clinical tests as well as the results from DNA microarray studies can be used. Even quantitative data from proteomics studies can be used as quantitative phenotypes.
In conclusion, we developed an algorithm to simultaneously estimate, by the maximum-likelihood method, the population haplotype frequencies and the parameters of the distributions of quantitative phenotypes that are different between subjects with different diplotype configurations using both genotype and phenotype data from multiple subjects. Using a test statistic, −2 log(L0max/Lmax), we could construct a method to test the association between the presence of a haplotype and a quantitative phenotype. We implemented this algorithm in a computer program, QTLHAPLO. The analysis of the simulated and real data using this program indicated that this method can accurately estimate the parameters and reliably test the association between the haplotypes and the phenotypes.
Acknowledgments
This study was supported by a grant from the New Energy and Industry Technology Development Organization.
References
- Almasy, L., and J. Blangero, 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62: 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos, C. I., 1994. Robust variance-components approach for assessing genetic linkage in pedigrees. Am. J. Hum. Genet. 54: 535–543. [PMC free article] [PubMed] [Google Scholar]
- Bader, J. S., 2001. The relative power of SNPs and haplotype as genetic markers for association tests. Pharmacogenomics 2: 11–24. [DOI] [PubMed] [Google Scholar]
- Chiano, M. N., and D. G. Clayton, 1998. Fine genetic mapping using haplotype analysis and the missing data problem. Ann. Hum. Genet. 62: 55–60. [DOI] [PubMed] [Google Scholar]
- Clark, A. G., 1990. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol. Biol. Evol. 7: 111–122. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., and M. Slatkin, 1995. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 12: 921–927. [DOI] [PubMed] [Google Scholar]
- Fallin, D., and N. Schork, 2000. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization-algorithm for unphased diploid genotype data. Am. J. Hum. Genet. 67: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin, D., A. Cohen, L. Essioux, I. Chumakov, M. Blumenfeld et al., 2001. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res. 11: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1918. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 52: 399–433. [Google Scholar]
- Hoaglin, D. C., F. Mosteller and J. W Tukey (Editors), 1983 Understanding robust and exploratory data analysis. John Wiley & Sons, New York.
- Horikawa, Y., N. Oda, N. J. Cox, X. Li, M. Orho-Melander et al., 2000. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 26: 163–175. [DOI] [PubMed] [Google Scholar]
- Ito, T., S. Chiku, E. Inoue, M. Tomita, T. Morisaki et al., 2003. Estimation of haplotype frequencies, linkage-disequilibrium measures, and combination of haplotype copies in each pool by use of pooled DNA data. Am. J. Hum. Genet. 72: 384–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T., E. Inoue and N. Kamatani, 2004. Association test algorithm between a qualitative phenotype and a haplotype or haplotype set using simultaneous estimation of haplotype frequencies, diplotype configurations and diplotype-based penetrances. Genetics (in press). [DOI] [PMC free article] [PubMed]
- Judson, R., J. C. Stephens and A. Windemuth, 2000. The predictive power of haplotypes in clinical response. Pharmacogenomics 1: 5–16. [DOI] [PubMed] [Google Scholar]
- Kitamura, Y., M. Moriguchi, H. Kaneko, H. Morisaki, T. Morisaki et al., 2002. Determination of probability distribution of diplotype configuration (diplotype distribution) for each subject from genotypic data using the EM algorithm. Ann. Hum. Genet. 66: 183–193. [DOI] [PubMed] [Google Scholar]
- Long, J. C., R. C. Williams and M. Urbanek, 1995. An E-M algorithm and testing strategy for multiple locus haplotypes. Am. J. Hum. Genet. 56: 799–810. [PMC free article] [PubMed] [Google Scholar]
- Lou, X. Y., G. Casella, R. C. Littell, M. C. K. Yang, J. A. Johnson et al., 2003. A haplotype-based algorithm for multilocus linkage disequilibrium mapping of quantitative trait loci with epistasis. Genetics 163: 1533–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi, M., C. Terai, H. Kaneko, Y. Koseki, H. Kajiyama et al., 2001. A novel single-nucleotide polymorphism at the 5′-flanking region of SAA1 associated with risk of type AA amyloidosis secondary to rheumatoid arthritis. Arthritis Rheum. 44: 1266–1272. [DOI] [PubMed] [Google Scholar]
- Niu, T., Z. S. Qin, X. Xu and J. S. Liu, 2002. Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am. J. Hum. Genet. 70: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, S. C., M. J. Daly and L. Kruglyak, 2000. Exact multipoint quantitative-trait linkage analysis in pedigrees by variance components. Am. J. Hum. Genet. 66: 1153–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Z. S., T. Niu and A. S. Liu, 2002. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 71: 1242–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid, D. J., C. M. Rowland, D. E. Tines, R. M. Jacobson and G. A. Poland, 2002. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 70: 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, S., D. Roessli and L. Excoffier, 2000 Arlequin: A Software for Population Genetics Data Analysis: Ver. 2.000. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva, Geneva.
- Serfling, R. J., 1981 Approximation Theorems of Mathematical Statistics. John Wiley & Sons, New York.
- Sham, P. C., S. S. Cherny, S. Purcell and J. K. Hewitt, 2000. Power of linkage versus association analysis of quantitative traits, by use of variance-components models, for sibship data. Am. J. Hum. Genet. 66: 1616–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., N. J. Smith and P. Donnelly, 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, E., A. Taniguchi, W. Urano, H. Nakajima, Y. Matsuda et al., 2002. Adverse effects of sulfasalazine in patients with rheumatoid arthritis are associated with diplotype configuration at the N-acetyltransferase 2 gene. J. Rheumatol. 29: 2492–2499. [PubMed] [Google Scholar]
- Tanck, M. W. T., A. H. E. M. Klerkx, J. W. Jukema, P. DeKnijff, J. J. P. Kastelein et al., 2003. Estimation of multilocus haplotype effects using weighted penalised log-likelihood: analysis of five sequence variations at the cholesteryl ester transfer protein gene locus. Ann. Hum. Genet. 67: 175–184. [DOI] [PubMed] [Google Scholar]
- Tishkoff, S., A. Pakstis, G. Ruano and K. Kidd, 2000. The accuracy of statistical methods for estimation of haplotype frequencies: an example from the CD4 locus. Am. J. Hum. Genet. 56: 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano, W., A. Taniguch, H. Yamanaka, E. Tanaka, H. Nakajima et al., 2002. Polymorphisms in the methylenetetrahydrofolate reductase gene were associated with both the efficacy and the toxicity of methotrexate used for the treatment of rheumatoid arthritis, as evidenced by single locus and haplotype analysis. Pharmacogenomics 12: 183–190. [DOI] [PubMed] [Google Scholar]
- Wilks, S. S., 1962 Mathematical Statistics. John Wiley & Sons, New York.
- Wrigth, S., 1968 Evolution and the Genetics of Population, Vol. 1: Genetics and Biometric Foundations. University of Chicago, Chicago.
- Wu, R., M. Chang-Xing and G. Casella, 2002. Joint linkage and linkage disequilibrium mapping of quantitative trait loci in natural populations. Genetics 160: 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin, D. V., P. H. Westfall, S. S. Young, M. A. Karnoub, M. J. Wagner et al., 2002. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum. Hered. 53: 79–91. [DOI] [PubMed] [Google Scholar]