

THE gene dosage balance hypothesis (GDBH) proposes, in a narrow sense, that stoichiometric imbalances in macromolecular complexes can be a source of dominant phenotypes. Gene dosage balance in such complexes is required as a “here and now” condition, as a partial aneuploid carrying a deletion or duplication of a dosage-sensitive gene will have a fitness defect (Birchler et al. 2001; Veitia 2002, 2003). Global evidence supporting the GDBH has been found in yeast. Focusing on essential genes (i.e., their homozygous deletion is lethal), Papp et al. (2003) have shown that dosage-sensitive genes (low heterozygote fitness) are at least two times more likely to encode proteins involved in complexes than are genes with low dosage sensitivity. Furthermore, a statistically significant proportion of genes whose overexpression is lethal encodes proteins involved in complexes. The concept of dosage balance is old. Consider, for instance, dosage compensation of the X chromosome. In Drosophila the transmission of the X from the female to the male is operationally equivalent to an entire chromosomal “deletion.” Balance is achieved by making the single X in the male about twice as active, transcriptionally, as either of the two X's in the female. In mammals, compensation is achieved by inactivating one X chromosome in the female (Marin et al. 2000). This clearly implies that at least some X-linked genes must respect a certain balance with autosomal products.
The need of dosage balance also implies that single-gene duplications of certain types of subunits can be harmful. Consider a complex A-B-C. Under irreversible conditions, increasing the concentration of the bridge B can be detrimental, as inactive subcomplexes AB and BC may form (lowering the yield of ABC). Subunit B exerts a titrating power on A and C when overexpressed. On the contrary, increasing A and C is neutral, apart from the selective cost of their overproduction. Thus, some genes encoding interacting pairs should remain as single copies (case of B) or otherwise undergo coduplication with genes encoding their partners. Indeed, it can be shown that coduplication of B with A or C can overcome titration by excess of B (Teichmann and Veitia 2004; Veitia 2004). Accordingly, pairs of genes encoding interacting subunits tend to have the same number of paralogs and genes belonging to huge families seldom encode components of complexes (Papp et al. 2003). Recently, the validity of the GDBH was corroborated in yeast (Yang et al. 2003). Moreover, using human data, these authors found that the gene duplication level is higher for monomers than for components of protein complexes, which is consistent with the GDBH. Besides, the proportion of unduplicated genes was found to increase with the number of subunits in a complex. Here I show, with some examples, that the dosage balance notion is applicable to other cellular dynamic systems. I focus on gene dosage increase (duplication) but a similar reasoning holds for dosage reduction. The short-term outcome of a dosage balance alteration is relevant to understanding aspects of genetic dominance, as it may induce an immediate decrease of fitness, as mentioned above in the case of protein complexes. Classical genetics regards the phenomenon of dominance as a result of intralocus interactions. Stemming from the notion of balance itself, the models sketched below show how genetic dominance can arise also from interloci interactions, in line with the arguments of Omholt et al. (2000).
Let us first consider a system displaying adaptation to a signal, which is the basis of a chemotactic mechanism (Macnab and Koshland 1972). A signal S controls (i) the translation of an RNA, constitutively present at a stable concentration, to produce a protein R and (ii) the synthesis of the protease X that degrades R (see the sniffer of Tyson et al. 2003). In response to changes in S, R undergoes transient changes but will go back to a steady state where its concentration is constant and independent of S (i.e., Rss in Figure 1). A transient increase of R above a threshold RThr may trigger an action that ceases when R comes back to Rss. The dynamics of the system can be represented by two simple differential equations (Figure 1). A 1.5-fold increase of the dosage of R, as in partial triploidy, implies increasing the steady-state concentration of its RNA and will be represented in the differential equations by 1.5 × k1. In such a case the steady-state R′ss would also increase by 1.5-fold, which can be above the threshold RThr. This is obviously a problem. However, a parallel increase of the rate of synthesis of X's mRNA (i.e., k3 becomes 1.5 × k3) will restore the normal amount of Rss. Thus, coduplication of X and R is harmless and the only visible effect is a faster adaptation.
Figure 1.—

A simple network displaying adaptation to a stimulus (a) and the corresponding differential equations (b). All reactions have been assumed to be first order (linear) with respect to all reactants. The k's are specific rates. Positive/negative terms correspond to synthesis/degradation. Rss is [R] at steady state (i.e., when dR/dt = dX/dt = 0). Obviously, co-increase of R and X (represented, to simplify the notation, by 1.5 × k1 and 1.5 × k3, respectively) leads to the same Rss.
To explain dominance of the normal phenotype, Kacser and Burns (1981) showed that changing the amounts of most enzymes does not affect the visible phenotype. However, the concentration of an intermediate can vary a lot if the activity of an appropriate enzyme is raised or lowered substantially. So, even in a “Kacserian” context, the GDBH may apply to reactions of the form → A → Y → B → when the absolute level of Y (for instance) influences or determines a phenotype. Enzymes involved in signal transduction are expected to be particularly dosage sensitive. To illustrate this point, consider a system containing two converting enzymes E1 and E2 (for instance, a protein kinase and a phosphatase) that interconvert substrates W and W* (Goldbeter and Koshland 1981; Figure 2). This topology [Goldbeter-Koshland (GK)] is common in signaling. In the GK system, the molar fraction of W* (i.e., W*/WTotal) is a function of the ratio of active E1/E2 (or more exactly of k1E1/k2E2; the k's are the catalytic constants of each enzyme; Goldbeter and Koshland 1981, 1984). E1 and E2 can be inducible or constitutive, but activated/deactivated differentially. One can be regulated and the other constitutive and even promiscuous (i.e., interacting with several partners). A curve representing the ratio of active k1E1/k2E2 vs. the molar fraction of W* ranges from a hyperbola to a sigmoid whose steepness is governed by the ratios Km1/WTotal and Km2/WTotal (Km's are the Michaelian constants of the enzymes). High values of these parameters (i.e., 1) yield a hyperbola. For low values of the parameters (i.e., 0.01), which means saturation of the converters, there is a threshold E1/E2 for which a jump from W → W* appears. Under these conditions, a gradual input is transformed into a switch-like response (Goldbeter and Koshland 1981). This amplification of the response to a stimulus that alters the ratio E1/E2 is called “zero-order ultrasensitivity.” The modular nature of the GK switch is explained by the fact that as E1 and E2 are saturated by their substrates (W and W*, respectively), the corresponding reaction rates do not depend on substrate concentration but only on the relative amounts of active converting enzymes.
Figure 2.—

A schematic Goldbeter-Koshland switch. The curves of the mole fraction of W* at the steady state as a function of stimulus strength (duration) were established from Equation 7 of Goldbeter and Koshland (1981)(corrected form in Goldbeter and Koshland 1984), for the parameters Km1/WTotal = Km2/WTotal = 0.01. The solid black sigmoid corresponds to gene dosage of E1 = E2. The gray curve corresponds to doubling E2, while the dotted curve represents doubling E1. For simplicity, activation of E1 is assumed to depend linearly on enzyme concentration [E] and stimulus strength (duration) for constant E2.
In the context of gene dosage balance, if we assume for simplicity that activation/deactivation of E1 and E2 depend linearly on the strength or duration of the stimulus, a parallel change (increase or decrease) of total E1 and E2 will change neither the position of the threshold nor the general shape of the sigmoid (Figure 2). This co-increase cannot lead E1 (E2) to become unsaturated by W (W*) because ultrasensitivity might vanish. Increasing the amount of one convertase alone (i.e., 1.5× in a triploid or 2× in a partial tetraploid) will be problematic as it shifts the position of the threshold. As shown in Figure 2, increasing E1 shifts the threshold to the left and the contrary for E2. Consider now that E2 participates in several reactions (promiscuous) and that the amount allocated to counteract the effect of E1 is fairly constant. To give rise to a GK switch, this fraction of E2 must be saturated by W*. Increasing only E1 is problematic; co-increase of E1 and E2 restores the normal activity of the switch E1/E2 but will perturb other switches in which E2 might be involved. Thus, even promiscuity imposes limits for duplication of dosage-sensitive genes.
Goldbeter (1991) proposed an elementary mitotic clock with a chain of two GK switches responsive to cyclin. The output is the activation of a cyclin-degrading protease. To generate periodic changes of cyclin levels, the circuit requires delays introduced by the accumulation of cyclin itself and of a protease-activating enzyme, both of which must trespass their corresponding thresholds. Changing the dosage of one element arbitrarily may prevent cycling due to a shift in the threshold positions. A similar phenomenon is expected to arise according to more complex models of the cell cycle. In the model of Chen et al. (2000), including also two GK switches, the activity of cyclin B-dependent kinases (that defines the start and finish points of the cycle) depends explicitly on the ratio Cln2/Cdc20.
The mitogen-activated protein kinase (MAPK) pathways are well-known intracellular signaling modules in eukaryotes. MAPKs are serine-threonine protein kinases that are activated by diverse stimuli ranging from cytokines, growth factors, neurotransmitters, hormones, cellular stress, and cell adherence. These cascades contain three levels: MAPKKK → MAPKK → MAPK with the corresponding deactivating enzymes (for review see Widmann et al. 1999). Each layer has the GK topology but they do not seem to be GK switches (Bluthgen and Herzel 2003). Modularity, to avoid cross-talk among the pathways, is ensured by tethering the kinases to scaffold proteins as well as by direct interaction between the former.
Simulations show that the MAPK pathway can convert a gradual input into a switch-like output. This property makes the cascade suitable for mediating processes like mitogenesis, cell fate induction, and oocyte maturation, where a cell switches from one discrete state to another. However, sigmoidicity depends on the assumptions of the current models [Huang and Ferrell 1996 (HF); Bhalla and Iyengar 1999 (BI)]. Sigmoidicity can be studied by fitting the curves to the Hill sigmoid (y = xn/(K + xn)), where n is the Hill coefficient (the higher it is, the steeper the sigmoid, the sharper the threshold). According to the HF model, dosage alterations of either MAPKK or MAPK-phosphatase induce important effects on sigmoidicity but according to the BI model, changes in almost all components individually lead to striking changes in sigmoidicity (Figures 3 and 4 of Bluthgen and Herzel 2003). Remarkably, co-increase or co-decrease of all components at once translates into minor changes in the position of the stimulus threshold and sigmoidicity. After doubling or halving all the components, HF curves have similar sigmoidicity and the threshold position (loosely defined as the x corresponding to y = 0.5 for a steep sigmoid) changes by only ∼25% with respect to the reference system described by Bluthgen and Herzel (2003). For the BI curves, the threshold changes by only ∼30% upon halving or doubling all components. On the basis of this evidence, it is safe to consider the whole pathway as the functional unit. Therefore, if the selectable property is a switch-like behavior, the whole MAPK module is likely to be duplicated or retained after a global duplication.
Figure 3.—

A genetic toggle. Gene u encodes a repressor of v, which is in turn a repressor of u. I1 and I2 are the inducers. Presence of I1 will trigger synthesis of u and repression of v, which persists even after removal of the inducer. (a) The null clines (containing the loci of du/dt = 0 and dv/dt = 0) intersect at three points when the repressive activities of u and v are balanced and in the presence of cooperative repression. This translates into the existence of two stable and one unstable steady states (stable state 1/high v, state 2/high u). (b) When there is an imbalance in the amounts of the repressors (i.e., relative excess of u) the system is not bistable anymore. (c) Phase diagram of the system. The lines mark the transition between bistability (the systems can flip between state 1 and state 2) and monostability (the system is unable to flip). The bistable region lies inside of each pair of curves. The small horizontal arrow represents an increase of dosage of v for the same dosage of u. The system crosses the bifurcation line and crashes (becomes monostable, permanently in state 1). The small diagonal arrow (OK) represents a co-increase of u and v: the system remains in the bistability region. Increase of cooperativity (β and γ) leads to a broader region of bistability increasing the robustness of the system. This figure is courtesy of Gardner et al. (2000) and Nature (Macmillan Magazines; modified and reproduced with permission).
Figure 4.—

Doubling the genomic content associated with an increase in nuclear and cellular volume can warrant successful duplication of certain dosage-sensitive genes. Consider that M is an inactive monomer and that Mn is the active multimer composed of n monomers. Synthesis (k1), degradation (k2), and interaction of the monomers are represented with chemical (a) and differential equations (b). For simplicity, let the association/dissociation reactions between monomers and oligomers be faster than synthesis/degradation. We can define a pseudo-equilibrium constant K in the steady state (i.e., when dM/dt = 0). As usual, increasing dosage of M (i.e., by 1.5×) will be represented by 1.5 × k1. If the initial volume does not change, this will imply a (1.5)n-fold increase of active Mn! Thus, maintaining a balance with another oligomer, say Nx, after strict coduplication in the same volume is possible only if the number of monomers involved in Mn and Nx are identical (n = x), for similar KM and KN. A polyploidization event increasing cell volume proportionally is more likely to restore balance whatever the K's, n, or x are. Similar arguments explain imbalance after a heterozygous deletion of M or N.
Simulations also show that if MAPK accumulates in the nucleus, so that as much as 50% of cytoplasmic MAPK is sequestered after phosphorylation by active nuclear MAPKK (for constant concentrations of MAPK-pase), these converters operate at saturation and a true GK-switch behavior will appear (Ferrell 1998). To keep the proper balance, coduplication of MAPKK/MAPK-pase is required, as explained above. Note that in case of whole-genome duplication, the nucleus will enlarge, leading very likely to similar intranuclear concentrations of the relevant proteins as before duplication (see below).
Finally, let us examine a minimal genetic toggle based on two repressible promoters arranged in a mutually inhibitory network. This system can be represented with the following rate equations (Gardner et al. 2000 and references therein):
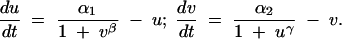 |
Here u and v are the concentrations of the repressors, and α1 and α2 represent the effective rates of synthesis of u and v, respectively. Parameters β and γ represent the cooperativity of repression of promoters 2 and 1, respectively (β and γ > 1 imply cooperativity). Such a circuit can display bistability, as it can be flipped between two stable steady states (either high u or high v) using transient inductive signals. Each state is associated with the expression of different sets of genes responsive to the repressors. Moreover, after the removal of the inducing signal, the system remains in the state where it was. This is an epigenetic mechanism to ensure stability of a developmental decision that can be subsequently reverted: a kind of conditional cellular memory.
The stability of the system can be explored by studying the curves corresponding to du/dt = 0 and dv/dt = 0 or null clines. Their intersections define points corresponding to steady states. Here, when there is cooperativity (β and γ > 1) and the repressive activities of u and v are balanced the null clines, being sigmoids, intersect three times. This translates into the existence of two stable and one unstable steady states (Figure 3a). Thus, bistability depends on the cooperative repression of transcription. If the rates of synthesis of the two repressors are not balanced, the null clines will intersect only once, producing a single stable steady state (Figure 3b, monostable system). One can draw a phase diagram to represent whether the system lies in a region of bistability (the system works) or in a region of monostability (the system does not work). Changing gene dosage of u and/or v is represented by a change of α (i.e., α1 and/or α2, respectively). The outcome of this operation will critically depend on where, in the phase diagram, the system is (Figure 3c). When α1 and α2 are high, the system operates in a wide region of bistability. For instance, an increase of α2 (by 1.5×) may be inconsequential as this means shifting the point (log α2, log α1) by ∼log 1.5 units rightward. However, high α1 and α2 imply a selective disadvantage, as synthesis of lot of repressor is costly. Natural systems are more likely to work in a region of bistability with limited amounts of u and v. In such a case, a small shift to the right can imply trespassing the (bifurcation) line from a region of bistability to a region of monostability. However, co-increasing of u and v authorizes the system to remain in the bistable region.
Before closing, note how difficult coduplication of unlinked dosage-sensitive genes can be. For instance, equate the trimer ABC to X (from Figure 1) or to E1 (from a GK switch). Let component B be titrating and genes A, B, and C be unlinked. Genes A and C can duplicate independently to yield A′ and C′ without stoichiometric interference. Both A′ and C′ should be present before “coduplication” of B (to get the right concentration of trimer) and its partner, say R (as in Figure 1). However, the increase in frequency of A′ and C′ alone and their coexistence in the same individuals is not justified by any advantage and they will tend to disappear. Thus, a scenario involving small duplications to get as few as five genes coduplicated, partly sequentially and partly concertedly, seems virtually impossible. This points toward an underestimated role of global duplications to justify the existence of paralogs of dosage-sensitive genes. In principle, an imbalance caused by changing dosage of one gene could be rescued by a parallel change of expression of the interacting partners. However, in the context of cellular pathways, this is clearly so only if both partners act as monomers (case of X and R in the example of Figure 1) or, at most, if they are homo-oligomers having the same number of monomers and similar apparent pseudo-equilibrium constants, which seems unlikely. Otherwise, a co-increase of both partners would translate into a disproportionate change in the concentration of active oligomers. This is so because oligomerization reactions take place in the same volume with a higher input of monomers. It is difficult to provide a general proof of this conjecture but an illustrative example, directly applicable to the models outlined here, is discussed in Figure 4. This merely biochemical argument points to the need for a whole-genome duplication that implies an increase of cell volume (Gregory 2001), which tends to restore the concentration of monomers and multimers as before duplication. Even yeast during its life cycle respects this constraint: the diploid cellular volume is about two times the haploid volume (Walker 1998; Sherman 2002).
There are detectable relics of whole-genome duplication events in many eukaryotes including yeast, plants, and vertebrates (Makalowski 2001; Blanc and Wolfe 2004; Kellis et al. 2004). This process is expected to be followed by deletion or rearrangements leading in part to preferential retention of “interacting” genes to avoid imbalances. After genome duplication, the retained paralogs may diverge concertedly in sequence and pattern of expression, producing new paralogous network modules unable to interfere stoichiometrically. Indeed, Blanc and Wolfe (2004) have found many groups of paralogs in Arabidopsis bearing this signature. In line with the ideas presented here, they suggest that the impact of polyploidy on the evolution of networks may be more important than the cumulative effect of the duplication of individual genes. Consider also that yeast alone contains five different MAPK pathways: the haploid mating, invasive growth, and cell wall remodeling pathways and two pathways involved in stress responses (Widmann et al. 1999). They share a common evolutionary origin (Caffrey et al. 1999) but according to the perspective outlined above, all of them cannot result from sequential single-gene duplications. Multiple rounds of genome duplication associated with preferential retention of dosage-sensitive interacting partners might explain the existence of paralogous networks and also the tendency toward nonrandom gene association in the eukaryotic genome. The perspective outlined here is compatible with, and does not diminish the evolutionary importance of, segmental duplications. Indeed, Teichmann and Veitia (2004) have shown the existence of an excess of linked gene pairs encoding subunits of stable protein complexes in yeast. We speculated that these pairs may be modules (perhaps generated by the nonrandom retention of dosage-sensitive genes) that may maintain the right stoichiometry of complexes upon segmental duplication.
The examples discussed above show that the appearance of dominant phenotypes may have simple physiological explanations in terms of dosage imbalances and that the resilience of a system to such alterations can be adjusted by selection. They illustrate also key points concerning duplicability of dosage-sensitive genes. The fact that there are rules governing changes of gene dosage does not exclude the possibility of compensation by up- or downregulation of partners in the same pathway as long as fitness is not severely compromised by the initial dosage alteration. Indeed, coevolution of cis-regulatory sequences is commonplace (Wray et al. 2003), which may explain gene synexpression (coregulation within modules in time and space; Nierhs and Pollet 1999), as a way to ensure balance. A scenario involving massive duplications is crucial to produce paralogous pathways (probably more complex than those studied by Teichmann and Babu 2004) and raw material for evolution in cases where single-gene duplication is difficult or impossible.
Acknowledgments
I thank Nils Bluthgen for his kind help concerning simulations of the MAPK pathway and for helpful comments on the manuscript. I thank Jim Collins for his comments concerning both the original and the revised manuscript and Sandrine Caburet, Indrani Bose, and Vidya Nanjundiah for their interesting suggestions.
References
- Bhalla, U., and R. Iyengar, 1999. Emergent properties of networks of biological signaling pathways. Science 283: 381–387. [DOI] [PubMed] [Google Scholar]
- Birchler, J. A., U. Bhadra, M. P. Bhadra and D. L. Auger, 2001. Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev. Biol. 234: 275–288. [DOI] [PubMed] [Google Scholar]
- Blanc, G., and K. Wolfe, 2004. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthgen, N., and H. Herzel, 2003. How robust are switches in intracellular signaling cascades? J. Theor. Biol. 225: 293–300. [DOI] [PubMed] [Google Scholar]
- Caffrey, D. R., L. A. O'Neill and D. C. Shields, 1999. The evolution of the MAP kinase pathways: coduplication of interacting proteins leads to new signaling cascades. J. Mol. Evol. 49: 567–582. [DOI] [PubMed] [Google Scholar]
- Chen, K. C., A. Csikasz-Nagy, B. Gyorffy, J. Val, B. Novak et al., 2000. Kinetic analysis of a molecular model of the budding yeast cell cycle. Mol. Biol. Cell 11: 369–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell, J. E., Jr., 1998. How regulated protein translocation can produce switch-like responses. Trends Biochem. Sci. 23: 461–465. [DOI] [PubMed] [Google Scholar]
- Gardner, T. S., C. R. Cantor and J. J. Collins, 2000. Construction of a genetic toggle switch in Escherichia coli. Nature 403: 339–342. [DOI] [PubMed] [Google Scholar]
- Goldbeter, A., and D. E. Koshland, Jr., 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA 78: 6840–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldbeter, A., and D. E. Koshland, Jr., 1984. Ultrasensitivity in biochemical systems controlled by covalent modification. Interplay between zero-order and multistep effects. J. Biol. Chem. 259: 14441–14447. [PubMed] [Google Scholar]
- Goldbeter, A., 1991. A minimal cascade model for the mitotic oscillator involving cyclin and cdc2 kinase. Proc. Natl. Acad. Sci. USA 88: 9107–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, T. R., 2001. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol. Rev. Camb. Philos. Soc. 76: 65–101. [DOI] [PubMed] [Google Scholar]
- Huang, C. Y., and J. E. Ferrell, Jr., 1996. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 93: 10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser, H., and J. A. Burns, 1981. The molecular basis of dominance. Genetics 97: 639–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., B. W. Birren and E. S. Lander, 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617–624. [DOI] [PubMed] [Google Scholar]
- Macnab, R. M., and D. E. Koshland, Jr., 1972. The gradient-sensing mechanism in bacterial chemotaxis. Proc. Natl. Acad. Sci. USA 69: 2509–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makalowski, W., 2001. Are we polyploids? A brief history of one hypothesis. Genome Res. 11: 667–670. [DOI] [PubMed] [Google Scholar]
- Marin, I., M. L. Siegal and B. S. Baker, 2000. The evolution of dosage-compensation mechanisms. BioEssays 22: 1106–1114. [DOI] [PubMed] [Google Scholar]
- Niehrs, C., and N. Pollet, 1999. Synexpression groups in eukaryotes. Nature 402: 483–487. [DOI] [PubMed] [Google Scholar]
- Omholt, S. W., E. Plahte, L. Oyehaug and K. Xiang, 2000. Gene regulatory networks generating the phenomena of additivity, dominance and epistasis. Genetics 155: 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp, B., C. Pal and L. D. Hurst, 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197. [DOI] [PubMed] [Google Scholar]
- Sherman, F., 2002. Getting started with yeast. Methods Enzymol. 350: 3–41. [DOI] [PubMed] [Google Scholar]
- Teichmann, S. A., and M. M. Babu, 2004. Gene regulatory network growth by duplication. Nat. Genet. 36: 492–496. [DOI] [PubMed] [Google Scholar]
- Teichmann, S. A., and R. A. Veitia, 2004. Genes encoding subunits of stable complexes are clustered on the yeast chromosomes: an interpretation from a dosage balance perspective. Genetics 167: 2121–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson, J. J., K. C. Chen and B. Novak, 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15: 221–231. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A., 2002. Exploring the etiology of haploinsufficiency. BioEssays 24: 175–184. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A., 2003. Nonlinear effects in macromolecular assembly and dosage sensitivity. J. Theor. Biol. 220: 19–25. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A. (Editor), 2004 Clusters of functionally related genes in eukaryotes, dosage balance and evolvability, in The Biology of Genetic Dominance. Ron Landes, Georgetown, TX (in press).
- Walker, G. M., 1998 Yeast Physiology and Biotechnology, pp. 1–60. John Wiley & Sons, Chichester, UK.
- Widmann, C., S. Gibson, M. B. Jarpe and G. L. Johnson, 1999. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79: 143–180. [DOI] [PubMed] [Google Scholar]
- Wray, G. A., M. W. Hahn, E. Abouheif, J. W. Balhoff, M. Pizer et al., 2003. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20: 1377–1419. [DOI] [PubMed] [Google Scholar]
- Yang, J., R. Lusk and W. H. Li, 2003. Organismal complexity, protein complexity, and gene duplicability. Proc. Natl. Acad. Sci. USA 100: 15661–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]